
Reprogramming: A Preventive Strategy in Hypertension Focusing on the Kidney



Abstract
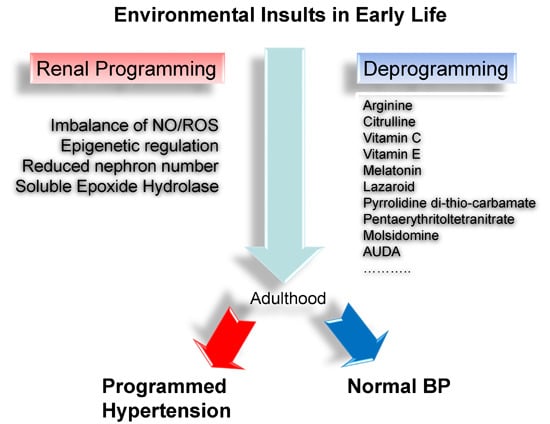
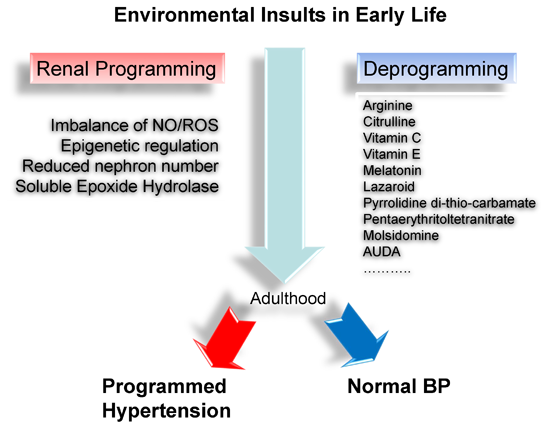
:
1. Introduction
2. Studies in Humans

{kind=link}
| Study | Offspring, n | Age Range, years | Country | Risk Factors | Reporting Findings Related to BP |
|---|---|---|---|---|---|
| Project Viva [10] | 746 M/F | 3 | United States | Maternal smoking | Pregnancy smokers had children with higher SBP |
| London Health Science Centre [11] | 658 M/F | 3–16 | United Kingdom | High maternal pre-pregnancy BMI, large birth weight | SBP correlates with maternal pre-pregnancy BMI. DBP positively correlates with birth weight, gestational age, and maternal pre-pregnancy BMI |
| ABCD [12] | 1834 M/F | 5–6 | Netherlands | Low maternal 25-hydroxyvitamin D level (25(OH)D) | ↑ 10 nmol/L maternal 25OHD ↓ 0.21 mmHg offspring DBP |
| Tohoku Study of Child Development [13] | 377 M/F | 7 | Japan | Short-term breastfeeding | BP in the long-term breastfeeding group was significantly lower than in the short-term breastfeeding group |
| ALSPAC [14] | 3062 M/F | 9–12 | United Kingdom | Pre-eclampsia or gestational hypertension | Offspring of women with pre-eclampsia or gestational hypertension had higher SBP and DBP |
| ALSPAC [15] | 3525 M/F | 9.9 | United Kingdom | Low maternal 25(OH)D | Maternal 25(OH)D was inversely associated with SBP |
| ALSPAC [16] | 2200 M/F | 16 | United Kingdom | Excessive gestational weight gain, high maternal pre-pregnancy BMI | Gestational weight gain and maternal pre-pregnancy BMI positively correlate with offspring SBP and DBP |
| POPS-19 [17] | 596 M/F | 19 | Netherlands | Increased postnatal weight gain | Higher postnatal weight gain, higher SBP |
| MUSP [18] | 2271 M/F | 21 | Australia | Excessive gestational weight gain | Greater GWG is associated with higher offspring SBP |
| JPS [19] | 1256 M/F | 32 | Jerusalem | High maternal pre-pregnancy body mass index (BMI) | High maternal pre-pregnancy BMI, high offspring SBP and DBP |
| Dutch Famine study [20] | 359 M/F | 59 | Netherlands | Undernutrition | SBP and DBP were 2.77 and 1.27 mmHg higher in offspring exposed to famine than those without exposure |
3. Studies in Rodents
| Programming Mechanism | Species | Programming Effects | Treatment | Period of Treatment | Reprogramming Effects | Age at which Effects Were Measured | Ref. |
|---|---|---|---|---|---|---|---|
| 50% caloric restriction during pregnancy | Wistar rats | Hypertension ↑ Oxidative stress ↓ EDVD | Vit. C or E | 14 to 16 weeks of age | ↓ BP ↓ Oxidative stress. Normalized EDVD | 14 to 16 weeks of age | [29] |
| 50% caloric restriction during pregnancy | Wistar rats. | ↑ BP LBW Endothelial dysfunction Impaired renal function ↓ Glomerular number | Micronutrients mix: Vit. C, E, selenium and folic acid | During pregnancy | ↓ BP Prevented LBW ↑ Vascular function | Until 14–16 weeks of age | [30] |
| Genetic hypertension/Aging | SHR and aging WKY rats | Hypertension Proteinuria | l-arginine plus antioxidants (Vit. C, E and taurine) | 2 weeks before until 4 or 8 weeks after birth | ↓ BP Prevented proteinuria. Transient ↓ of oxidative stress | Every 6 weeks until 50 weeks of age | [31] |
| Genetic hypertension. | SHR and WKY rats | Hypertension. High RVR Wide range of RBF autoregulation | l-arginine plus antioxidants (Vit. C, E and taurine) | 2 weeks before until 8 weeks after birth | ↓ Hypertension and RVR RBF autoregulation shifted towards WKY pattern | At 9 week old | [32] |
| Genetic hypertension, renal injury | FHH rats | Hypertension Progressive renal injury | l-arginine plus antioxidants (Vit. C, E and taurine) | 2 weeks before until 4 weeks after birth | Prevented hypertension. ↓ Proteinuria ↓ Glomerulosclerosis in females | Until 36 weeks of age | [33] |
| 50% caloric restriction during pregnancy and lactation | Sprague-Dawley rats | LBW, impaired renal function, renal injury | Citrulline | During pregnancy and lactation | ↑ BP ↓ Renal injury ↑ Nephron number | Until 12 weeks of age | [34] |
| Genetic hypertension. | SHR and WKY rats | Hypertension. ↓ Renal NO at 2 weeks. ↓ Renal gene expression of ASS and ASL at 2 weeks | Citrulline | 2 weeks before until 6 weeks after birth | ↓ Hypertension in females and until 20 weeks in males ↑ Renal NO at 2 weeks | Until 50 weeks of age | [35] |
| Diabetes (STZ) during pregnancy and lactation | Sprague-Dawley rats | Hypertension, renal injury | Citrulline | During pregnancy and lactation | ↓ BP ↓ Renal injury | Until 12 weeks of age | [36] |
| Dexamethasone during pregnancy | Sprague-Dawley rats | Hypertension | Citrulline | During pregnancy and lactation | ↓ BP | Until 12 weeks of age | [37] |
| L-NAME during pregnancy | Sprague-Dawley rats | Hypertension | Citrulline | During pregnancy and lactation | ↓ BP ↓ ADMA | Until 12 weeks of age | [38,39] |
| Genetic hypertension, renal injury | FHH rats | Hypertension Progressive renal injury | Molsidomine (NO donor) | 2 weeks before until 4 weeks after birth | Prevented hypertension. ↓ Glomerulosclerosis. | Until 36 and 42 weeks of age | [40] |
| Genetic hypertension | SHR | Hypertension Proteinuria | Vit. C, E and Molsidomine (NO donor) or Tempol (SOD mimetic) | 2 weeks before until 4 or 8 weeks after birth | ↓ BP Prevented proteinuria | Every 6 weeks until 50 weeks of age | [41] |
| Genetic hypertension, renal injury | FHH rats | Hypertension Progressive renal injury | Molsidomine (NO donor) | From 2 weeks before until 4 weeks after birth | Differential regulation of renal ribosome protein genes at 2 days and 2 weeks of age ↓ Renal ribosome structures at 2 weeks of age | Until 42 weeks of age | [42] |
| Genetic hypertension | SHR | Hypertension | Pentaerythritoltetranitrate (NO donor and antioxidant) | During pregnancy and lactation | ↓ BP in females, epigenetic changes relating to eNOS in the aorta | At 6 and 8 months of age | [43] |
| Dexamethasone during pregnancy | Sprague-Dawley rats | Hypertension | Melatonin | During pregnancy and lactation | ↓ BP ↑ Nephron number | Until 16 weeks of age | [44] |
| Dexamethasone during pregnancy | Sprague-Dawley rats | Hypertension | Melatonin | During pregnancy and lactation | Preserved histone deacetylase gene expression | Until 16 weeks of age | [45] |
| High fructose intake during pregnancy | Sprague-Dawley rats | Hypertension | Melatonin | During pregnancy and lactation | Prevented hypertension Increased renal NO | Until 12 weeks of age | [46] |
| Protein restricted diet during pregnancy | Wistar rats | Hypertension Vascular dysfunction Microvascular rarefaction ↑ Oxidative stress | Lazaroid (inhibitor of lipid peroxidation) | During pregnancy | Prevented hypertension. Improved vascular function and microvascular rarefaction. ↓ Oxidative stress. | Until 12 weeks of age | [47] |
| Genetic hypertension | SHR and WKY rats | Hypertension | Pyrrolidine di-thio-carbamate (NF-κB inhibitor) | 2 weeks before until 4 weeks after birth | ↓ BP ↑ Natriuresis at 4 weeks ↓ Oxidative stress markers | Until 28 weeks of age | [48] |
| Genetic hypertension | FHH rats | Hypertension Progressive renal injury | Pyrrolidine di-thio-carbamate (NF-κB inhibitor) | 2 weeks before until 4 weeks after birth | Prevented hypertension ↓ Glomerulosclerosis | Until 36 and 42 weeks of age | [49] |
4. Reprogramming the Balance of Nitric Oxide and Reactive Oxygen Species
5. Probing the Renal Transcriptome
6. Epigenetics in Programming and Reprogramming
7. Reprogramming via Inhibition of Soluble Epoxide Hydrolase
8. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- McMillen, I.C.; Robinson, J.S. Developmental origins of the metabolic syndrome: Prediction, plasticity, and programming. Physiol. Rev. 2005, 85, 571–633. [Google Scholar] [CrossRef] [PubMed]
- Hochberg, Z.; Feil, R.; Constancia, M.; Fraga, M.; Junien, C.; Carel, J.C.; Boileau, P.; le Bouc, Y.; Deal, C.L.; Lillycrop, K.; et al. Child health, developmental plasticity, and epigenetic programming. Endocr. Rev. 2011, 32, 159–224. [Google Scholar] [CrossRef] [PubMed]
- Paixão, A.D.; Alexander, B.T. How the kidney is impacted by the perinatal maternal environment to develop hypertension. Biol. Reprod. 2013, 89, 144. [Google Scholar] [CrossRef] [PubMed]
- Barker, D.J.; Osmond, C.; Golding, J.; Kuh, D.; Wadsworth, M.E. Growth in utero, blood pressure in childhood and adult life, and mortality from cardiovascular disease. BMJ 1989, 298, 564–567. [Google Scholar] [CrossRef] [PubMed]
- Kyle, U.G.; Pichard, C. The Dutch Famine of 1944–1945: A pathophysiological model of long-term consequences of wasting disease. Curr. Opin. Clin. Nutr. Metab. Care 2006, 9, 388–394. [Google Scholar] [CrossRef] [PubMed]
- Huxley, R.R.; Shiell, A.W.; Law, C.M. The role of size at birth and postnatal catch-up growth in determining systolic blood pressure: A systematic review of the literature. J. Hypertens. 2000, 18, 815–831. [Google Scholar] [CrossRef] [PubMed]
- Luyckx, V.A.; Bertram, J.F.; Brenner, B.M.; Fall, C.; Hoy, W.E.; Ozanne, S.E.; Vikse, B.E. Effect of fetal and child health on kidney development and long-term risk of hypertension and kidney disease. Lancet 2013, 382, 273–283. [Google Scholar] [CrossRef] [Green Version]
- Barker, D.J.; Bagby, S.P.; Hanson, M.A. Mechanisms of disease: In utero programming in the pathogenesis of hypertension. Nat. Clin. Pract. Nephrol. 2006, 2, 700–707. [Google Scholar] [CrossRef] [PubMed]
- Keller, G.; Zimmer, G.; Mall, G.; Ritz, E.; Amann, K. Nephron number in patients with primary hypertension. N. Engl. J. Med. 2003, 348, 101–108. [Google Scholar] [CrossRef] [PubMed]
- Oken, E.; Huh, S.Y.; Taveras, E.M.; Rich-Edwards, J.W.; Gillman, M.W. Associations of maternal prenatal smoking with child adiposity and blood pressure. Obes. Res. 2005, 13, 2021–2028. [Google Scholar] [CrossRef] [PubMed]
- Filler, G.; Yasin, A.; Kesarwani, P.; Garg, A.X.; Lindsay, R.; Sharma, A.P. Big mother or small baby: Which predicts hypertension? J. Clin. Hypertens. 2011, 13, 35–41. [Google Scholar] [CrossRef] [PubMed]
- Hrudey, E.J.; Reynolds, R.M.; Oostvogels, A.J.; Brouwer, I.A.; Vrijkotte, T.G. The association between maternal 25-hydroxyvitamin D concentration during gestation and early childhood cardio-metabolic outcomes: Is there interaction with pre-pregnancy BMI? PLoS ONE 2015, 10, e0133313. [Google Scholar] [CrossRef] [PubMed]
- Hosaka, M.; Asayama, K.; Staessen, J.A.; Ohkubo, T.; Hayashi, K.; Tatsuta, N.; Kurokawa, N.; Satoh, M.; Hashimoto, T.; Hirose, T.; et al. Breastfeeding leads to lower blood pressure in 7-year-old Japanese children: Tohoku study of child development. Hypertens. Res. 2013, 36, 117–122. [Google Scholar] [CrossRef] [PubMed]
- Fraser, A.; Nelson, S.M.; Macdonald-Wallis, C.; Sattar, N.; Lawlor, D.A. Hypertensive disorders of pregnancy and cardiometabolic health in adolescent offspring. Hypertension 2013, 62, 614–620. [Google Scholar] [CrossRef] [PubMed]
- Williams, D.M.; Fraser, A.; Fraser, W.D.; Hyppönen, E.; Davey Smith, G.; Deanfield, J.; Hingorani, A.; Sattar, N.; Lawlor, D.A. Associations of maternal 25-hydroxyvitamin D in pregnancy with offspring cardiovascular risk factors in childhood and adolescence: Findings from the avon longitudinal study of parents and children. Heart 2013, 99, 1849–1856. [Google Scholar] [CrossRef] [PubMed]
- Fraser, A.; Tilling, K.; Macdonald-Wallis, C.; Sattar, N.; Brion, M.J.; Benfield, L.; Ness, A.; Deanfield, J.; Hingorani, A.; Nelson, S.M.; et al. Association of maternal weight gain in pregnancy with offspring obesity and metabolic and vascular traits in childhood. Circulation 2010, 121, 2557–2564. [Google Scholar] [CrossRef] [PubMed]
- Keijzer-Veen, M.G.; Finken, M.J.; Nauta, J.; Dekker, F.W.; Hille, E.T.; Frölich, M.; Wit, J.M.; van der Heijden, A.J. Dutch POPS-19 collaborative study group. Is blood pressure increased 19 years after intrauterine growth restriction and preterm birth? A prospective follow-up study in The Netherlands. Pediatrics 2005, 116, 725–731. [Google Scholar] [CrossRef] [PubMed]
- Mamun, A.A.; O’Callaghan, M.; Callaway, L.; Williams, G.; Najman, J.; Lawlor, D.A. Associations of gestational weight gain with offspring body mass index and blood pressure at 21 years of age: Evidence from a birth cohort study. Circulation 2009, 119, 1720–1727. [Google Scholar] [CrossRef] [PubMed]
- Hochner, H.; Friedlander, Y.; Calderon-Margalit, R.; Meiner, V.; Sagy, Y.; Avgil-Tsadok, M.; Burger, A.; Savitsky, B.; Siscovick, D.S.; Manor, O. Associations of maternal pre-pregnancy body mass index and gestational weight gain with adult offspring cardiometabolic risk factors: The jerusalem perinatal family follow-up study. Circulation 2012, 125, 1381–1389. [Google Scholar] [CrossRef] [PubMed]
- Stein, A.D.; Zybert, P.A.; van der Pal-de Bruin, K.; Lumey, L.H. Exposure to famine during gestation, size at birth, and blood pressure at age 59 y: Evidence from the Dutch famine. Eur. J. Epidemiol. 2006, 21, 759–765. [Google Scholar] [CrossRef] [PubMed]
- Van Abeelen, A.F.; Veenendaal, M.V.; Painter, R.C.; de Rooij, S.R.; Thangaratinam, S.; van der Post, J.A.; Bossuyt, P.M.; Elias, S.G.; Uiterwaal, C.S.; Grobbee, D.E.; et al. The fetal origins of hypertension: A systematic review and meta-analysis of the evidence from animal experiments of maternal undernutrition. J. Hypertens. 2012, 30, 2255–2267. [Google Scholar] [CrossRef] [PubMed]
- Samuelsson, A.M.; Morris, A.; Igosheva, N.; Kirk, S.L.; Pombo, J.M.; Coen, C.W.; Poston, L.; Taylor, P.D. Evidence for sympathetic origins of hypertension in juvenile offspringof obese rats. Hypertension 2010, 55, 76–82. [Google Scholar] [CrossRef] [PubMed]
- MohanKumar, S.M.; King, A.; Shin, A.C.; Sirivelu, M.P.; MohanKumar, P.S.; Fink, G.D. Developmental programming of cardiovascular disorders: Focus on hypertension. Rev. Endocr. Metab. Disord. 2007, 8, 115–125. [Google Scholar] [CrossRef] [PubMed]
- Santos, M.S.; Joles, J.A. Early determinants of cardiovascular disease. Best Pract. Res. Clin. Endocrinol. Metab. 2012, 26, 581–597. [Google Scholar] [CrossRef] [PubMed]
- Tain, Y.L.; Huang, L.T. Restoration of asymmetric dimethylarginine-nitric oxide balance to prevent the development of hypertension. Int. J. Mol. Sci. 2014, 15, 11773–11782. [Google Scholar] [CrossRef] [PubMed]
- Tain, Y.L.; Huang, L.T.; Chan, J.Y. Transcriptional regulation of programmed hypertension by melatonin: An epigenetic perspective. Int. J. Mol. Sci. 2014, 15, 18484–18495. [Google Scholar] [CrossRef] [PubMed]
- Hussain, N. Epigenetic influences that modulate infant growth, development, and disease. Antioxid. Redox Signal. 2012, 17, 224–236. [Google Scholar] [CrossRef] [PubMed]
- Ji, Y.; Wu, Z.; Dai, Z.; Sun, K.; Wang, J.; Wu, G. Nutritional epigenetics with a focus on amino acids: Implications for the development and treatment of metabolic syndrome. J. Nutr. Biochem. 2015. [Google Scholar] [CrossRef] [PubMed]
- Franco Mdo, C.; Akamine, E.H.; Aparecida de Oliveira, M.; Fortes, Z.B.; Tostes, R.C.; Carvalho, M.H.; Nigro, D. Vitamins C and E improve endothelial dysfunction in intrauterine undernourished rats by decreasing vascular superoxide anion concentration. J. Cardiovasc. Pharmacol. 2003, 42, 211–217. [Google Scholar] [CrossRef] [PubMed]
- Franco Mdo, C.; Ponzio, B.F.; Gomes, G.N.; Gil, F.Z.; Tostes, R.; Carvalho, M.H.; Fortes, Z.B. Micronutrient prenatal supplementation prevents the development of hypertension and vascular endothelial damage induced by intrauterine malnutrition. Life Sci. 2009, 85, 327–333. [Google Scholar] [CrossRef] [PubMed]
- Racasan, S.; Braam, B.; van der Giezen, D.M.; Goldschmeding, R.; Boer, P.; Koomans, H.A.; Joles, J.A. Perinatal l-arginine and antioxidant supplements reduce adult blood pressure in spontaneously hypertensive rats. Hypertension 2004, 44, 83–88. [Google Scholar] [CrossRef] [PubMed]
- Koeners, M.P.; Racasan, S.; Koomans, H.A.; Joles, J.A.; Braam, B. Nitric oxide, superoxide and renal blood flow autoregulation in SHR after perinatal l-arginine and antioxidants. Acta Physiol. 2007, 190, 329–338. [Google Scholar] [CrossRef] [PubMed]
- Koeners, M.P.; Braam, B.; van der Giezen, D.M.; Goldschmeding, R.; Joles, J.A. Perinatal micronutrient supplements ameliorate hypertension and proteinuria in adult fawn-hooded hypertensive rats. Am. J. Hypertens. 2010, 23, 802–808. [Google Scholar] [CrossRef] [PubMed]
- Tain, Y.L.; Hsieh, C.S.; Lin, I.C.; Chen, C.C.; Sheen, J.M.; Huang, L.T. Effects of maternal l-citrulline supplementation on renal function and blood pressure in offspring exposed to maternal caloric restriction: The impact of nitric oxide pathway. Nitric. Oxide 2010, 23, 34–41. [Google Scholar] [CrossRef] [PubMed]
- Koeners, M.P.; van Faassen, E.E.; Wesseling, S.; Sain-van der Velden, M.; Koomans, H.A.; Braam, B.; Joles, J.A. Maternal supplementation with citrulline increases renal nitric oxide in young spontaneously hypertensive rats and has long-term antihypertensive effects. Hypertension 2007, 50, 1077–1084. [Google Scholar] [CrossRef] [PubMed]
- Tain, Y.L.; Lee, W.C.; Hsu, C.N.; Lee, W.C.; Huang, L.T.; Lee, C.T.; Lin, C.Y. Asymmetric dimethylarginine is associated with developmental programming of adult kidney disease and hypertension in offspring of streptozotocin-treated mothers. PLoS ONE 2013, 8, e55420. [Google Scholar] [CrossRef] [PubMed]
- Tain, Y.L.; Sheen, J.M.; Chen, C.C.; Yu, H.R.; Tiao, M.M.; Kuo, H.C.; Huang, L.T. Maternal citrulline supplementation prevents prenatal dexamethasone-induced programmed hypertension. Free Radic. Res. 2014, 48, 580–586. [Google Scholar] [CrossRef] [PubMed]
- Tain, Y.L.; Lee, C.T.; Huang, L.T. Long-term effects of maternal citrulline supplementation on renal transcriptome prevention of nitric oxide depletion-related programmed hypertension: The impact of gene-nutrient interactions. Int. J. Mol. Sci. 2014, 15, 23255–23268. [Google Scholar] [CrossRef] [PubMed]
- Tain, Y.L.; Huang, L.T.; Lee, C.T.; Chan, J.Y.; Hsu, C.N. Maternal citrulline supplementation prevents prenatal NG-nitro-l-arginine-methyl ester(L-NAME)-induced programmed hypertension in rats. Biol. Reprod. 2015, 92, 7. [Google Scholar] [CrossRef] [PubMed]
- Koeners, M.P.; Braam, B.; van der Giezen, D.M.; Goldschmeding, R.; Joles, J.A. A perinatal nitric oxide donor increases renal vascular resistance and ameliorates hypertension and glomerular injury in adult fawn-hooded hypertensive rats. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2008, 294, R1847–R1855. [Google Scholar] [CrossRef] [PubMed]
- Racasan, S.; Braam, B.; Koomans, H.A.; Joles, J.A. Programming blood pressure in adult SHR by shifting perinatal balance of NO and reactive oxygen species toward NO: The inverted barker phenomenon. Am. J. Physiol. Ren. Physiol. 2005, 288, F626–F636. [Google Scholar] [CrossRef] [PubMed]
- Wesseling, S.; Essers, P.B.; Koeners, M.P.; Pereboom, T.C.; Braam, B.; van Faassen, E.E.; Macinnes, A.W.; Joles, J.A. Perinatal exogenous nitric oxide in fawn-hooded hypertensive rats reduces renal ribosomal biogenesis in early life. Front. Genet. 2011, 2, 52. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.; Siuda, D.; Xia, N.; Reifenberg, G.; Daiber, A.; Münzel, T.; Förstermann, U.; Li, H. Maternal treatment of spontaneously hypertensive rats with pentaerythritoltetranitrate reduces blood pressure in female offspring. Hypertension 2015, 65, 232–237. [Google Scholar] [CrossRef] [PubMed]
- Tain, Y.L.; Chen, C.C.; Sheen, J.M.; Yu, H.R.; Tiao, M.M.; Kuo, H.C.; Huang, L.T. Melatonin attenuates prenatal dexamethasone-induced blood pressure increase in a rat model. J. Am. Soc. Hypertens. 2014, 8, 216–226. [Google Scholar] [CrossRef] [PubMed]
- Wu, T.H.; Kuo, H.C.; Lin, I.C.; Chien, S.J.; Huang, L.T.; Tain, Y.L. Melatonin prevents neonatal dexamethasone induced programmed hypertension: Histone deacetylase inhibition. J. Steroid Biochem. Mol. Biol. 2014, 144, 253–259. [Google Scholar] [CrossRef] [PubMed]
- Tain, Y.L.; Leu, S.; Wu, K.L.; Lee, W.C.; Chan, J.Y. Melatonin prevents maternal fructose intake-induced programmed hypertension in the offspring: Roles of nitric oxide and arachidonic acid metabolites. J. Pineal Res. 2014, 57, 80–89. [Google Scholar] [CrossRef] [PubMed]
- Cambonie, G.; Comte, B.; Yzydorczyk, C.; Ntimbane, T.; Germain, N.; Lê, N.L.; Pladys, P.; Gauthier, C.; Lahaie, I.; Abran, D.; et al. Antenatal antioxidant prevents adult hypertension, vascular dysfunction, and microvascular rarefaction associated with in utero exposure to a low-protein diet. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2007, 292, R1236–R1245. [Google Scholar] [CrossRef] [PubMed]
- Koeners, M.P.; Braam, B.; Joles, J.A. Perinatal inhibition of NF-κB has long-term antihypertensive effects in spontaneouslyhypertensive rats. J. Hypertens. 2011, 29, 1160–1166. [Google Scholar] [CrossRef] [PubMed]
- Koeners, M.P.; Wesseling, S.; Sánchez, M.; Braam, B.; Joles, J.A. Perinatal inhibition of NF-kappaB has long-term antihypertensive and renoprotective effects infawn-hooded hypertensive rats. Am. J. Hypertens. 2016, 29, 123–131. [Google Scholar] [CrossRef] [PubMed]
- Chan, S.H.; Chan, J.Y. Brain stem NOS and ROS in neural mechanisms of hypertension. Antioxid. Redox Signal. 2014, 20, 146–163. [Google Scholar] [CrossRef] [PubMed]
- Cowley, A.W., Jr.; Abe, M.; Mori, T.; O’Connor, P.M.; Ohsaki, Y.; Zheleznova, N.N. Reactive oxygen species as important determinants of medullary flow, sodium excretion, and hypertension. Am. J. Physiol. Ren. Physiol. 2015, 308, F179–F197. [Google Scholar] [CrossRef] [PubMed]
- Brown, D.I.; Griendling, K.K. Regulation of signal transduction by reactive oxygen species in the cardiovascular system. Circ. Res. 2015, 116, 531–549. [Google Scholar] [CrossRef] [PubMed]
- Chang, S.Y.; Chen, Y.W.; Zhao, X.P.; Chenier, I.; Tran, S.; Sauvé, A.; Ingelfinger, J.R.; Zhang, S.L. Catalase prevents maternal diabetes-induced perinatal programming via the Nrf2-HO-1 defense system. Diabetes 2012, 61, 2565–2574. [Google Scholar] [CrossRef] [PubMed]
- Konior, A.; Schramm, A.; Czesnikiewicz-Guzik, M.; Guzik, T.J. NADPH oxidases in vascular pathology. Antioxid. Redox Signal. 2014, 20, 2794–2814. [Google Scholar] [CrossRef] [PubMed]
- Panieri, E.; Santoro, M.M. ROS signaling and redox biology in endothelial cells. Cell. Mol. Life Sci. 2015, 72, 3281–3303. [Google Scholar] [CrossRef] [PubMed]
- Racasan, S.; Hahnel, B.; van der Giezen, D.M.; Blezer, E.L.; Goldschmeding, R.; Braam, B.; Kriz, W.; Koomans, H.A.; Joles, J.A. Temporary losartan or captopril in young SHR induces malignant hypertension despite initial normotension. Kidney Int. 2004, 65, 575–581. [Google Scholar] [CrossRef] [PubMed]
- Gwathmey, T.M.; Alzayadneh, E.M.; Pendergrass, K.D.; Chappell, M.C. Novel roles of nuclear angiotensin receptors and signaling mechanisms. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2012, 302, R518–R530. [Google Scholar] [CrossRef] [PubMed]
- Harrap, S.B.; van der Merwe, W.M.; Griffin, S.A.; Macpherson, F.; Lever, A.F. Brief angiotensin converting enzyme inhibitor treatment in young spontaneously hypertensive rats reduces blood pressure long-term. Hypertension 1990, 16, 603–614. [Google Scholar] [CrossRef] [PubMed]
- Tain, Y.L.; Hsu, C.N.; Lin, C.Y.; Huang, L.T.; Lau, Y.T. Aliskiren prevents hypertension and reduces asymmetric dimethylarginine in young spontaneously hypertensive rats. Eur. J. Pharmacol. 2011, 670, 561–565. [Google Scholar] [CrossRef] [PubMed]
- Smallegange, C.; Hale, T.M.; Bushfield, T.L.; Adams, M.A. Persistent lowering of pressure by transplanting kidneys from adult spontaneously hypertensive ratstreated with brief antihypertensive therapy. Hypertension 2004, 44, 89–94. [Google Scholar] [CrossRef] [PubMed]
- Sherman, R.C.; Langley-Evans, S.C. Early administration of angiotensin-converting enzyme inhibitor captopril, prevents the development of hypertension programmed by intrauterine exposure to a maternal low-protein diet in the rat. Clin. Sci. 1998, 94, 373–381. [Google Scholar] [CrossRef] [PubMed]
- Sherman, R.C.; Langley-Evans, S.C. Antihypertensive treatment in early postnatal life modulates prenatal dietary influences upon blood pressure in the rat. Clin. Sci. 2000, 98, 269–275. [Google Scholar] [CrossRef] [PubMed]
- Manning, J.; Vehaskari, V.M. Postnatal modulation of prenatally programmed hypertension by dietary Na and ACE inhibition. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2005, 288, R80–R84. [Google Scholar] [CrossRef] [PubMed]
- Hsu, C.N.; Lee, C.T.; Huang, L.T.; Tain, Y.L. Aliskiren in early postnatal life prevents hypertension and reduces asymmetric dimethylarginine in offspring exposed to maternal caloric restriction. J. Renin Angiotensin Aldosterone Syst. 2015, 16, 506–513. [Google Scholar] [CrossRef] [PubMed]
- Satterfield, M.C.; Bazer, F.W.; Spencer, T.E.; Wu, G. Sildenafil citrate treatment enhances amino acid availability in the conceptus and fetal growth in an ovine model of intrauterine growth restriction. J. Nutr. 2010, 140, 251–258. [Google Scholar] [CrossRef] [PubMed]
- Tain, Y.L.; Hsu, C.N.; Chan, J.Y.; Huang, L.T. Renal transcriptome analysis of programmed hypertension induced by maternal nutritional insults. Int. J. Mol. Sci. 2015, 16, 17826–17837. [Google Scholar] [CrossRef] [PubMed]
- Sheen, J.M.; Yu, H.R.; Tiao, M.M.; Chen, C.C.; Huang, L.T.; Chang, H.Y.; Tain, Y.L. Prenatal dexamethasone-induced programmed hypertension and renal programming. Life Sci. 2015, 132, 41–48. [Google Scholar] [CrossRef] [PubMed]
- Tain, Y.L.; Wu, K.L.; Lee, W.C.; Leu, S.; Chan, J.Y. Maternal fructose-intake-induced renal programming in adult male offspring. J. Nutr. Biochem. 2015, 26, 642–650. [Google Scholar] [CrossRef] [PubMed]
- Tain, Y.L.; Huang, L.T.; Chan, J.Y.; Lee, C.T. Transcriptome analysis in rat kidneys: Importance of genes involved in programmed hypertension. Int. J. Mol. Sci. 2015, 16, 4744–4758. [Google Scholar] [CrossRef] [PubMed]
- Wesseling, S.; Koeners, M.P.; Kantouh, F.; Joles, J.A.; Braam, B. Consequences of perinatal treatment with l-arginine and antioxidants for the renal transcriptome in spontaneously hypertensive rats. Pflugers. Arch. 2009, 458, 513–524. [Google Scholar] [CrossRef] [PubMed]
- Costantini, F. GDNF/Ret signaling and renal branching morphogenesis: From mesenchymal signals to epithelial cell behaviors. Organogenesis 2010, 6, 252–262. [Google Scholar] [CrossRef] [PubMed]
- Benz, K.; Campean, V.; Cordasic, N.; Karpe, B.; Neuhuber, W.; Mall, G.; Hartner, A.; Hilgers, K.F.; Amann, K. Early glomerular alterations in genetically determined low nephron number. Am. J. Physiol. Ren. Physiol. 2011, 300, F521–F530. [Google Scholar] [CrossRef] [PubMed]
- Dagan, A.; Gattineni, J.; Habib, S.; Baum, M. Effect of prenatal dexamethasone on postnatal serum and urinary angiotensin II levels. Am. J. Hypertens. 2010, 23, 420–424. [Google Scholar] [CrossRef] [PubMed]
- Habib, S.; Gattineni, J.; Twombley, K.; Baum, M. Evidence that prenatal programming of hypertension by dietary protein deprivation is mediated by fetal glucocorticoid exposure. Am. J. Hypertens. 2011, 24, 96–101. [Google Scholar] [CrossRef] [PubMed]
- Rexhaj, E.; Pireva, A.; Paoloni-Giacobino, A.; Allemann, Y.; Cerny, D.; Dessen, P.; Sartori, C.; Scherrer, U.; Rimoldi, S.F. Prevention of vascular dysfunction and arterial hypertension in mice generated by assisted reproductive technologies by addition of melatonin to culture media. Am. J. Physiol. Heart Circ. Physiol. 2015, 309, H1151–H1156. [Google Scholar] [CrossRef] [PubMed]
- Pechanova, O.; Paulis, L.; Simko, F. Peripheral and central effects of melatonin on blood pressure regulation. Int. J. Mol. Sci. 2014, 15, 17920–17937. [Google Scholar] [CrossRef] [PubMed]
- Koeners, M.P.; Wesseling, S.; Ulu, A.; Sepúlveda, R.L.; Morisseau, C.; Braam, B.; Hammock, B.D.; Joles, J.A. Soluble epoxide hydrolase in the generation and maintenance of high blood pressure in spontaneously hypertensive rats. Am. J. Physiol. Endocrinol. Metab. 2011, 300, E691–E698. [Google Scholar] [CrossRef] [PubMed]
- Morisseau, C.; Hammock, B.D. Epoxide hydrolases: Mechanisms, inhibitor designs, and biological roles. Annu. Rev. Pharmacol. Toxicol. 2005, 45, 311–333. [Google Scholar] [CrossRef] [PubMed]
- Imig, J.D. Epoxide hydrolase and epoxygenase metabolites as therapeutic targets for renal diseases. Am. J. Physiol. Ren. Physiol. 2005, 289, F496–F503. [Google Scholar] [CrossRef] [PubMed]
- Sinal, C.J.; Miyata, M.; Tohkin, M.; Nagata, K.; Bend, J.R.; Gonzalez, F.J. Targeted disruption of soluble epoxide hydrolase reveals a role in blood pressure regulation. J. Biol. Chem. 2000, 275, 40504–40510. [Google Scholar] [CrossRef] [PubMed]
- Manhiani, M.; Quigley, J.E.; Knight, S.F.; Tasoobshirazi, S.; Moore, T.; Brands, M.W.; Hammock, B.D.; Imig, J.D. Soluble epoxide hydrolase gene deletion attenuates renal injury and inflammation with DOCA-salt hypertension. Am. J. Physiol. Ren. Physiol. 2009, 297, F740–F748. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Li, N.; Pang, W.; Zhang, X.; Hammock, B.D.; Ai, D.; Zhu, Y. Opposite effects of gene deficiency and pharmacological inhibition of soluble epoxide hydrolase on cardiac fibrosis. PLoS ONE 2014, 9, e94092. [Google Scholar] [CrossRef] [PubMed]
- Ortiz, L.A.; Quan, A.; Zarzar, F.; Weinberg, A.; Baum, M. Prenatal dexamethasone programs hypertension and renal injury in the rat. Hypertension 2003, 41, 328–334. [Google Scholar] [CrossRef] [PubMed]
- De Vries, W.B.; van den Borne, P.; Goldschmeding, R.; de Weger, R.A.; Bal, M.P.; van Bel, F.; van Oosterhout, M.F. Neonatal dexamethasone treatment in the rat leads to kidneydamage in adulthood. Pediatr. Res. 2010, 67, 72–76. [Google Scholar] [CrossRef] [PubMed]
- Oparil, S.; Schmieder, R.E. New approaches in the treatment of hypertension. Circ. Res. 2015, 116, 1074–1095. [Google Scholar] [CrossRef] [PubMed]
- Von Dadelszen, P.; Dwinnell, S.; Magee, L.A.; Carleton, B.C.; Gruslin, A.; Lee, B.; Lim, K.I.; Liston, R.M.; Miller, S.P.; Rurak, D.; et al. Research into advanced fetal diagnosis and therapy (RAFT) group. Sildenafil citrate therapy for severe early-onset intrauterine growth restriction. BJOG 2011, 118, 624–628. [Google Scholar] [CrossRef] [PubMed]
- Ganzevoort, W.; Alfirevic, Z.; von Dadelszen, P.; Kenny, L.; Papageorghiou, A.; van Wassenaer-Leemhuis, A.; Gluud, C.; Mol, B.W.; Baker, P.N. STRIDER: Sildenafil therapy in dismal prognosis early-onset intrauterine growth restriction—A protocol for a systematic review with individual participant data and aggregate data meta-analysis and trial sequential analysis. Syst. Rev. 2014, 3, 23. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tain, Y.-L.; Joles, J.A. Reprogramming: A Preventive Strategy in Hypertension Focusing on the Kidney. Int. J. Mol. Sci. 2016, 17, 23. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms17010023
Tain Y-L, Joles JA. Reprogramming: A Preventive Strategy in Hypertension Focusing on the Kidney. International Journal of Molecular Sciences. 2016; 17(1):23. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms17010023
Chicago/Turabian StyleTain, You-Lin, and Jaap A. Joles. 2016. "Reprogramming: A Preventive Strategy in Hypertension Focusing on the Kidney" International Journal of Molecular Sciences 17, no. 1: 23. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms17010023