
Unraveling Molecular Differences of Gastric Cancer by Label-Free Quantitative Proteomics Analysis


 and
and
Abstract
:
1. Introduction
2. Results
2.1. Overall Protein Changes Identified by Label-Free Quantitative Strategy


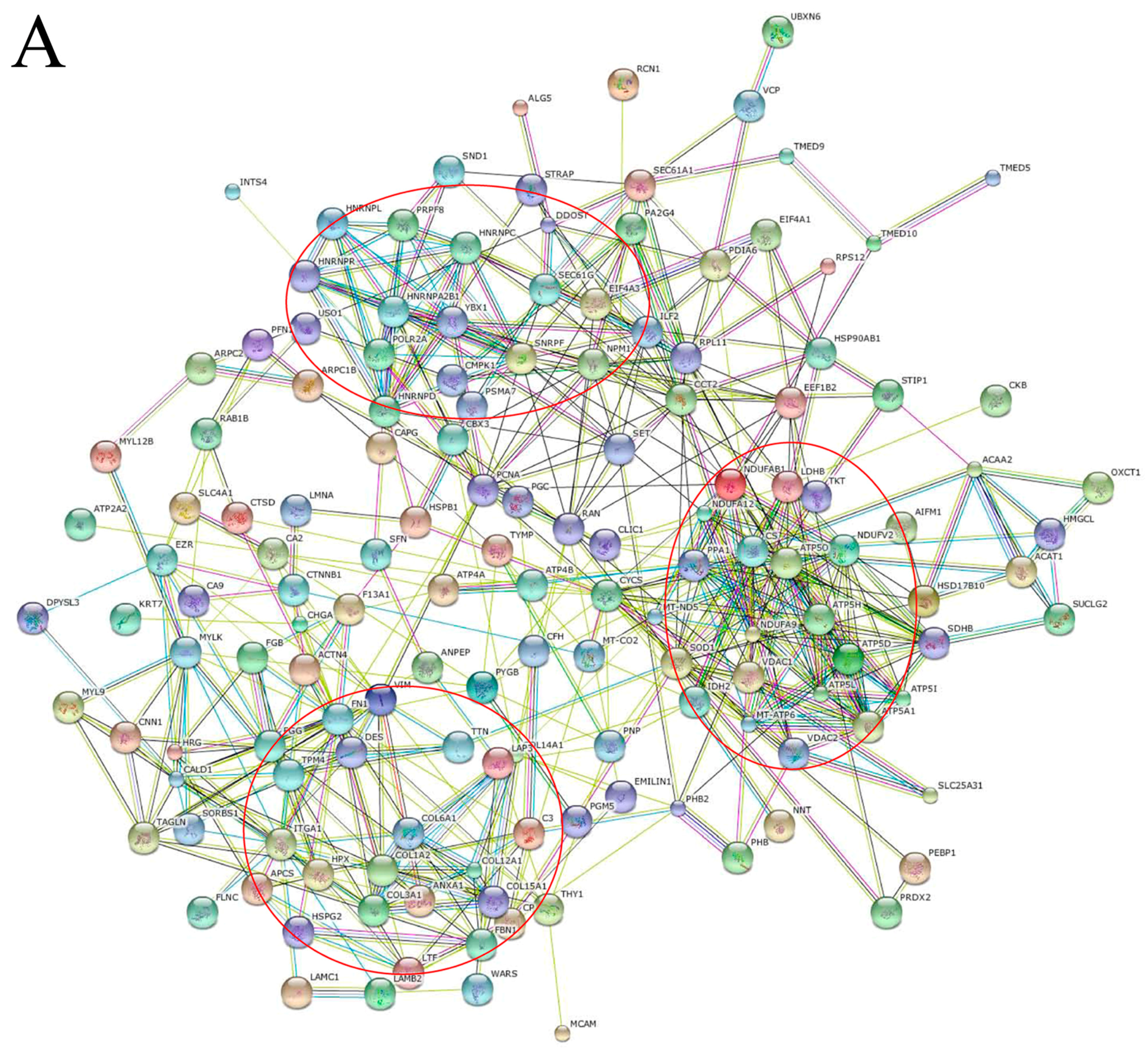
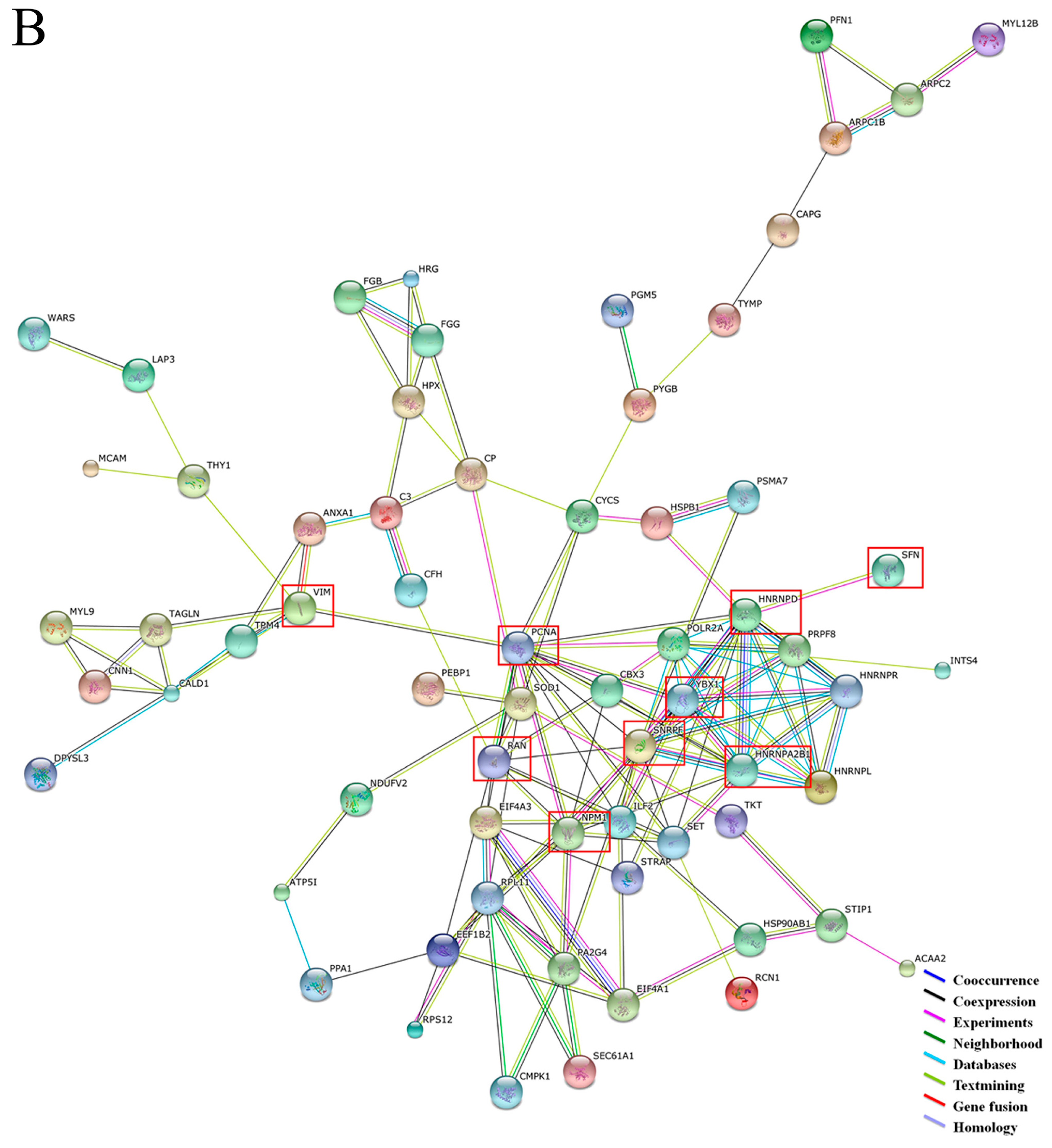
2.2. Functional Annotation of Proteins between GC and Adjacent Tissues

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Disease and Disorder | No. of Molecules | p-Value | Protein Names |
|---|---|---|---|
| Cancer | 117 | 2.62 × 10−11–2.63 × 10−3 | ANPEP, ANXA1, ATP2A2, ATP4A, CBX3, HNRNPA2B1, HNRNPC, HNRNPL, HSP90AB1, ILF2, NPM1, RAN, SNRPF, VIM, YBX1, … |
| Gastrointestinal Disease | 99 | 2.62 × 10−11–2.98 × 10−3 | ANPEP, ANXA1, FN1, HNRNPA2B1, HNRNPC, HNRNPL, HPX, NPM1, RAN, SFN, SNRPF, TAGLN, VIM, WARS, YBX1, … |
| Function | No. of Molecules | p-Value | Protein Names |
|---|---|---|---|
| Cellular Growth and Proliferation | 78 | 4.95 × 10−13–2.42 × 10−3 | ACAT1, HNRNPA2B1, HNRNPC, HNRNPD, HNRNPL, HNRNPR, HPX, HRG, HSP90AB1, HSPB1, LF2, NPM1, RAN, VIM, YBX1, … |
| Nucleic Acid Metabolism | 25 | 7.36 × 10−12-1.83 × 10−3 | ACAA2, ATP2A2, ATP4A, ATP4B, CS, CYCS, EIF4A3, HMGCL, PNP, PPA1, SET,SOD1, TYMP, VCP, VDAC1, … |
| Small Molecule Biochemistry | 36 | 7.36 × 10−12–3.02 × 10−3 | ANXA1, ATP2A2, ATP4A, ATP4B, CMPK1, CYCS, EIF4A3, MT-ATP6, PNP, PPA1, SET, SOD1, TYMP, VCP, VDAC1, … |
| Cell Death and Survival | 74 | 1.98 × 10−10–2.53 × 10−3 | ACAT1, CCT2, CFH, CP, CTNNB1, CYCS, DPYSL3, EZR, F13A1, FGG, FN1, HNRNPC, NPM1, VIM, YBX1, … |
| Cellular Movement | 52 | 5.38 × 10−9–2.97 × 10−3 | ACTN4, ANXA1, CNN1, CTNNB1, DPYSL3, FN1, HNRNPA2B1, HNRNPL, HRG, HSP90AB1, NPM1, SFN, VIM, WARS, YBX1, … |



2.3. Validation of Dysregulated hnRNPA2B1, hnRNPD, hnRNPL and YBX-1 by Nano-LC-MS/MS, qRT-PCR and Western Blot

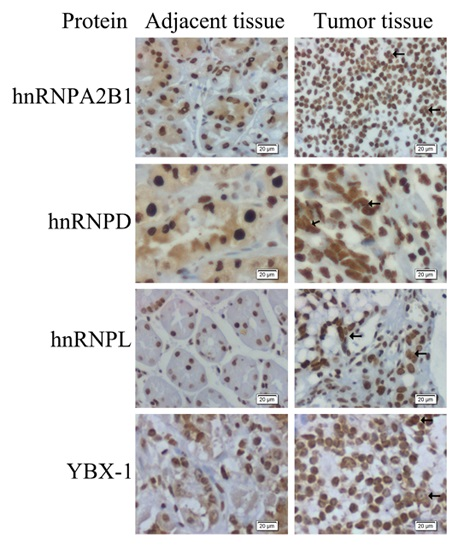
2.4. Expression and Distribution Detection of hnRNPA2B1, hnRNPD, hnRNPL and YBX-1 by Immunohistochemistry

3. Discussion
4. Experimental Section
4.1. Clinical Tissue Samples
4.2. Protein Preparation
4.3. In-Solution Tryptic Digestion
4.4. SDS-PAGE and in-Gel Tryptic Digestion
4.5. Nano-LC-MS/MS Analysis
4.6. Label-Free Quantification
4.7. Bioinformatics Analysis
4.8. Validation of Dysregulated Proteins by qRT-PCR, Western Blot and Immunohistochemistry
4.9. Data Analysis
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Ferlay, J.; Soerjomataram, I.; Dikshit, R.; Eser, S.; Mathers, C.; Rebelo, M.; Parkin, D.M.; Forman, D.; Bray, F. Cancer incidence and mortality worldwide: Sources, methods and major patterns in GLOBOCAN 2012. Int. J. Cancer 2015, 136, 359–386. [Google Scholar] [CrossRef] [PubMed]
- Torre, L.A.; Bray, F.; Siegel, R.L.; Ferlay, J.; Lortet-Tieulent, J.; Jemal, A. Global cancer statistics, 2012. CA Cancer J. Clin. 2015, 65, 87–108. [Google Scholar] [CrossRef] [PubMed]
- Kinugasa, H.; Nouso, K.; Tanaka, T.; Miyahara, K.; Morimoto, Y.; Dohi, C.; Matsubara, T.; Okada, H.; Yamamoto, K. Droplet digital PCR measurement of HER2 in patients with gastric cancer. Br. J. Cancer 2015, 112, 1652–1655. [Google Scholar] [CrossRef] [PubMed]
- Karimi, P.; Islami, F.; Anandasabapathy, S.; Freedman, N.D.; Kamangar, F. Gastric cancer: Descriptive epidemiology, risk factors, screening, and prevention. Cancer Epidemiol. Biomark. Prev. 2014, 23, 700–713. [Google Scholar] [CrossRef] [PubMed]
- Wadhwa, R.; Song, S.; Lee, J.S.; Yao, Y.; Wei, Q.; Ajani, J.A. Gastric cancer-molecular and clinical dimensions. Nat. Rev. Clin. Oncol. 2013, 10, 643–655. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.; Raufi, A.; Klempner, S.J. Targeted therapy for gastric cancer: Molecular pathways and ongoing investigations. Biochim. Biophys. Acta 2014, 1846, 232–237. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.H.; Lin, W.C.; Tsai, K.W. Advances in molecular biomarkers for gastric cancer: miRNAs as emerging novel cancer markers. Expert Rev. Mol. Med. 2014. [Google Scholar] [CrossRef] [PubMed]
- Li, P.; Zhang, D.; Guo, C. Serum biomarker screening for the diagnosis of early gastric cancer using SELDI-TOF-MS. Mol. Med. Rep. 2012, 5, 1531–1535. [Google Scholar] [PubMed]
- Piazuelo, M.B.; Correa, P. Gastric cancer: Overview. Colomb. Med 2013, 44, 192–201. [Google Scholar] [PubMed]
- Uppal, D.S.; Powell, S.M. Genetics/genomics/proteomics of gastric adenocarcinoma. Gastroenterol. Clin. N. Am. 2013, 42, 241–260. [Google Scholar] [CrossRef] [PubMed]
- Deyati, A.; Younesi, E.; Hofmann-Apitius, M.; Novac, N. Challenges and opportunities for oncology biomarker discovery. Drug Discov. Today 2013, 18, 614–624. [Google Scholar] [CrossRef] [PubMed]
- Cho, W.C. Proteomics technologies and challenges. Genom. Proteom. Bioinform. 2007, 5, 77–85. [Google Scholar] [CrossRef]
- Uhlen, M.; Fagerberg, L.; Hallstrom, B.M.; Lindskog, C.; Oksvold, P.; Mardinoglu, A.; Sivertsson, A.; Kampf, C.; Sjostedt, E.; Asplund, A.; et al. Proteomics. Tissue-based map of the human proteome. Science 2015, 347, 1260419. [Google Scholar] [CrossRef] [PubMed]
- Sallam, R.M. Proteomics in cancer biomarkers discovery: Challenges and applications. Dis. Markers 2015, 2015, 321370. [Google Scholar] [CrossRef] [PubMed]
- Wong, J.W.; Cagney, G. An overview of label-free quantitation methods in proteomics by mass spectrometry. Methods Mol. Biol. 2010, 604, 273–283. [Google Scholar] [PubMed]
- Megger, D.A.; Bracht, T.; Meyer, H.E.; Sitek, B. Label-free quantification in clinical proteomics. Biochim. Biophys. Acta 2013, 1834, 1581–1590. [Google Scholar] [CrossRef] [PubMed]
- Marcus, K.; Sitek, B.; Waldera-Lupa, D.M.; Poschmann, G.; Meyer, H.E.; Stühler, K. Application of Label-Free Proteomics for Differential Analysis of Lung Carcinoma Cell Line A549. In Quantitative Methods in Proteomics; Marcus, K., Ed.; Humana Press: New York, NY, USA, 2012; pp. 241–248. [Google Scholar]
- Atrih, A.; Mudaliar, M.A.; Zakikhani, P.; Lamont, D.J.; Huang, J.T.; Bray, S.E.; Barton, G.; Fleming, S.; Nabi, G. Quantitative proteomics in resected renal cancer tissue for biomarker discovery and profiling. Br. J. Cancer 2014, 110, 1622–1633. [Google Scholar] [CrossRef] [PubMed]
- Wu, W.; Chung, M.C. The gastric fluid proteome as a potential source of gastric cancer biomarkers. J. Proteom. 2013, 90, 3–13. [Google Scholar] [CrossRef] [PubMed]
- Lin, L.L.; Huang, H.C.; Juan, H.F. Discovery of biomarkers for gastric cancer: A proteomics approach. J. Proteom. 2012, 75, 3081–3097. [Google Scholar] [CrossRef] [PubMed]
- Hu, C.W.; Tseng, C.W.; Chien, C.W.; Huang, H.C.; Ku, W.C.; Lee, S.J.; Chen, Y.J.; Juan, H.F. Quantitative proteomics reveals diverse roles of miR-148a from gastric cancer progression to neurological development. J. Proteome Res. 2013, 12, 3993–4004. [Google Scholar] [CrossRef] [PubMed]
- Mi, H.; Muruganujan, A.; Thomas, P.D. PANTHER in 2013: Modeling the evolution of gene function, and other gene attributes, in the context of phylogenetic trees. Nucleic Acids Res. 2013, 41, D377–D386. [Google Scholar] [CrossRef] [PubMed]
- Huang, D.W.; Sherman, B.T.; Lempicki, R.A. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 2009, 4, 44–57. [Google Scholar] [CrossRef] [PubMed]
- Szklarczyk, D.; Franceschini, A.; Wyder, S.; Forslund, K.; Heller, D.; Huerta-Cepas, J.; Simonovic, M.; Roth, A.; Santos, A.; Tsafou, K.P.; et al. STRING v10: Protein-protein interaction networks, integrated over the tree of life. Nucleic Acids Res. 2015, 43, D447–D452. [Google Scholar] [CrossRef] [PubMed]
- Croft, D.; Mundo, A.F.; Haw, R.; Milacic, M.; Weiser, J.; Wu, G.; Caudy, M.; Garapati, P.; Gillespie, M.; Kamdar, M.R.; et al. The Reactome pathway knowledgebase. Nucleic Acids Res. 2014, 42, D472–D477. [Google Scholar] [CrossRef] [PubMed]
- Iuga, C.; Seicean, A.; Iancu, C.; Buiga, R.; Sappa, P.K.; Volker, U.; Hammer, E. Proteomic identification of potential prognostic biomarkers in resectable pancreatic ductal adenocarcinoma. Proteomics 2014, 14, 945–955. [Google Scholar] [CrossRef] [PubMed]
- Lundgren, D.H.; Hwang, S.I.; Wu, L.; Han, D.K. Role of spectral counting in quantitative proteomics. Expert Rev. Proteom. 2010, 7, 39–53. [Google Scholar] [CrossRef] [PubMed]
- Aquino, P.F.; Fischer, J.S.; Neves-Ferreira, A.G.; Perales, J.; Domont, G.B.; Araujo, G.D.; Barbosa, V.C.; Viana, J.; Chalub, S.R.; Lima, D.S.A.; et al. Are gastric cancer resection margin proteomic profiles more similar to those from controls or tumors? J. Proteome Res. 2012, 11, 5836–5842. [Google Scholar] [CrossRef] [PubMed]
- Uen, Y.H.; Lin, K.Y.; Sun, D.P.; Liao, C.C.; Hsieh, M.S.; Huang, Y.K.; Chen, Y.W.; Huang, P.H.; Chen, W.J.; Tai, C.C.; et al. Comparative proteomics, network analysis and post-translational modification identification reveal differential profiles of plasma Con A-bound glycoprotein biomarkers in gastric cancer. J. Proteom. 2013, 83, 197–213. [Google Scholar] [CrossRef] [PubMed]
- Qiao, J.; Cui, S.J.; Xu, L.L.; Chen, S.J.; Yao, J.; Jiang, Y.H.; Peng, G.; Fang, C.Y.; Yang, P.Y.; Liu, F. Filamin C, a dysregulated protein in cancer revealed by label-free quantitative proteomic analyses of human gastric cancer cells. Oncotarget 2015, 6, 1171–1189. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.D.; Wang, C.S.; Huang, Y.H.; Chien, K.Y.; Liang, Y.; Chen, W.J.; Lin, K.H. Overexpression of CLIC1 in human gastric carcinoma and its clinicopathological significance. Proteomics 2007, 7, 155–167. [Google Scholar] [CrossRef] [PubMed]
- Muhlmann, G.; Ofner, D.; Zitt, M.; Muller, H.M.; Maier, H.; Moser, P.; Schmid, K.W.; Zitt, M.; Amberger, A. 14–3-3 sigma and p53 expression in gastric cancer and its clinical applications. Dis. Markers 2010, 29, 21–29. [Google Scholar] [CrossRef] [PubMed]
- Kuramitsu, Y.; Baron, B.; Yoshino, S.; Zhang, X.; Tanaka, T.; Yashiro, M.; Hirakawa, K.; Oka, M.; Nakamura, K. Proteomic differential display analysis shows up-regulation of 14-3-3 sigma protein in human scirrhous-type gastric carcinoma cells. Anticancer Res. 2010, 30, 4459–4465. [Google Scholar] [PubMed]
- Kocevar, N.; Odreman, F.; Vindigni, A.; Grazio, S.F.; Komel, R. Proteomic analysis of gastric cancer and immunoblot validation of potential biomarkers. World J. Gastroenterol. 2012, 18, 1216–1228. [Google Scholar] [CrossRef] [PubMed]
- Bai, Z.; Ye, Y.; Liang, B.; Xu, F.; Zhang, H.; Zhang, Y.; Peng, J.; Shen, D.; Cui, Z.; Zhang, Z.; et al. Proteomics-based identification of a group of apoptosis-related proteins and biomarkers in gastric cancer. Int. J. Oncol. 2011, 38, 375–383. [Google Scholar] [PubMed]
- Liu, Z.; Chen, L.; Zhang, X.; Xu, X.; Xing, H.; Zhang, Y.; Li, W.; Yu, H.; Zeng, J.; Jia, J. RUNX3 regulates vimentin expression via miR-30a during epithelial-mesenchymal transition in gastric cancer cells. J. Cell Mol. Med. 2014, 18, 610–623. [Google Scholar] [CrossRef] [PubMed]
- Han, N.; Li, W.; Zhang, M. The function of the RNA-binding protein hnRNP in cancer metastasis. J. Cancer Res. Ther. 2013, 9, S129–S134. [Google Scholar] [PubMed]
- Carpenter, B.; MacKay, C.; Alnabulsi, A.; MacKay, M.; Telfer, C.; Melvin, W.T.; Murray, G.I. The roles of heterogeneous nuclear ribonucleoproteins in tumour development and progression. Biochim. Biophys. Acta 2006, 1765, 85–100. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.H.; Lum, J.H.; Cheung, B.P.; Wong, M.S.; Butt, Y.K.; Tam, M.F.; Chan, W.Y.; Chow, C.; Hui, P.K.; Kwok, F.S.; et al. Identification of the heterogeneous nuclear ribonucleoprotein A2/B1 as the antigen for the gastrointestinal cancer specific monoclonal antibody MG7. Proteomics 2005, 5, 1160–1166. [Google Scholar] [CrossRef] [PubMed]
- Jing, G.J.; Xu, D.H.; Shi, S.L.; Li, Q.F.; Wang, S.Y.; Wu, F.Y.; Kong, H.Y. Aberrant expression and localization of hnRNP-A2/B1 is a common event in human gastric adenocarcinoma. J. Gastroenterol. Hepatol. 2011, 26, 108–115. [Google Scholar] [CrossRef] [PubMed]
- Siveke, J.T. The increasing diversity of KRAS signaling in pancreatic cancer. Gastroenterology 2014, 147, 736–739. [Google Scholar] [CrossRef] [PubMed]
- Barcelo, C.; Etchin, J.; Mansour, M.R.; Sanda, T.; Ginesta, M.M.; Sanchez-Arevalo, L.V.; Real, F.X.; Capella, G.; Estanyol, J.M.; Jaumot, M.; et al. Ribonucleoprotein HNRNPA2B1 interacts with and regulates oncogenic KRAS in pancreatic ductal adenocarcinoma cells. Gastroenterology 2014, 147, 882–892. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gouble, A.; Grazide, S.; Meggetto, F.; Mercier, P.; Delsol, G.; Morello, D. A new player in oncogenesis: AUF1/hnRNPD overexpression leads to tumorigenesis in transgenic mice. Cancer Res. 2002, 62, 1489–1495. [Google Scholar] [PubMed]
- Trojanowicz, B.; Brodauf, L.; Sekulla, C.; Lorenz, K.; Finke, R.; Dralle, H.; Hoang-Vu, C. The role of AUF1 in thyroid carcinoma progression. Endocr. Relat. Cancer 2009, 16, 857–871. [Google Scholar] [CrossRef] [PubMed]
- Kosnopfel, C.; Sinnberg, T.; Schittek, B. Y-box binding protein 1—A prognostic marker and target in tumour therapy. Eur. J. Cell Biol. 2014, 93, 61–70. [Google Scholar] [CrossRef] [PubMed]
- Lasham, A.; Print, C.G.; Woolley, A.G.; Dunn, S.E.; Braithwaite, A.W. YB-1: Oncoprotein, prognostic marker and therapeutic target? Biochem. J. 2013, 449, 11–23. [Google Scholar] [CrossRef] [PubMed]
- Guo, T.; Yu, Y.; Yip, G.W.; Baeg, G.H.; Thike, A.A.; Lim, T.K.; Tan, P.H.; Matsumoto, K.; Bay, B.H. Y-box binding protein 1 is correlated with lymph node metastasis in intestinal-type gastric cancer. Histopathology 2015, 66, 491–499. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Wang, K.Y.; Li, Z.; Liu, Y.P.; Izumi, H.; Yamada, S.; Uramoto, H.; Nakayama, Y.; Ito, K.; Kohno, K. Y-box binding protein 1 expression in gastric cancer subtypes and association with cancer neovasculature. Clin. Transl. Oncol. 2015, 17, 152–159. [Google Scholar] [CrossRef] [PubMed]
- Shibata, T.; Kan, H.; Murakami, Y.; Ureshino, H.; Watari, K.; Kawahara, A.; Kage, M.; Hattori, S.; Ono, M.; Kuwano, M. Y-box binding protein-1 contributes to both HER2/ErbB2 expression and lapatinib sensitivity in human gastric cancer cells. Mol. Cancer Ther. 2013, 12, 737–746. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Z.; Wu, F.; Ding, S.; Sun, L.; Liu, Z.; Ding, K.; Lu, J. Label-free quantitative proteomic analysis reveals potential biomarkers and pathways in renal cell carcinoma. Tumour Biol. 2015, 36, 939–951. [Google Scholar] [CrossRef] [PubMed]
- UniProtKB. Available online: http://www.uniprot.org/ (accessed on 10 November 2015).
- Theron, L.; Gueugneau, M.; Coudy, C.; Viala, D.; Bijlsma, A.; Butler-Browne, G.; Maier, A.; Bechet, D.; Chambon, C. Label-free quantitative protein profiling of vastus lateralis muscle during human aging. Mol. Cell. Proteom. 2014, 13, 283–294. [Google Scholar] [CrossRef] [PubMed]
- PANTHER 9.0. Available online: http://www.pantherdb.org (accessed on 10 November 2015).
- Ingenuity Pathway Analysis (IPA). Available online: http://www.ingenuity.com/ (accessed on 10 November 2015).
- STRING (version 9.1). Available online: http://www.string-db.org/ (accessed on 10 November 2015).
- Reactome. Available online: http://www.reactome.org/ (accessed on 10 November 2015).
- Database for Annotation, Visualization and Integrated Discovery (DAVID, Bioinformatics Resources 6.7). Available online: http://david.abcc.ncifcrf.gov/ (accessed on 10 November 2015).
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dai, P.; Wang, Q.; Wang, W.; Jing, R.; Wang, W.; Wang, F.; Azadzoi, K.M.; Yang, J.-H.; Yan, Z. Unraveling Molecular Differences of Gastric Cancer by Label-Free Quantitative Proteomics Analysis. Int. J. Mol. Sci. 2016, 17, 69. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms17010069
Dai P, Wang Q, Wang W, Jing R, Wang W, Wang F, Azadzoi KM, Yang J-H, Yan Z. Unraveling Molecular Differences of Gastric Cancer by Label-Free Quantitative Proteomics Analysis. International Journal of Molecular Sciences. 2016; 17(1):69. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms17010069
Chicago/Turabian StyleDai, Peng, Qin Wang, Weihua Wang, Ruirui Jing, Wei Wang, Fengqin Wang, Kazem M. Azadzoi, Jing-Hua Yang, and Zhen Yan. 2016. "Unraveling Molecular Differences of Gastric Cancer by Label-Free Quantitative Proteomics Analysis" International Journal of Molecular Sciences 17, no. 1: 69. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms17010069