
NLRP3 Upregulation in Retinal Pigment Epithelium in Age-Related Macular Degeneration

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results and Discussion
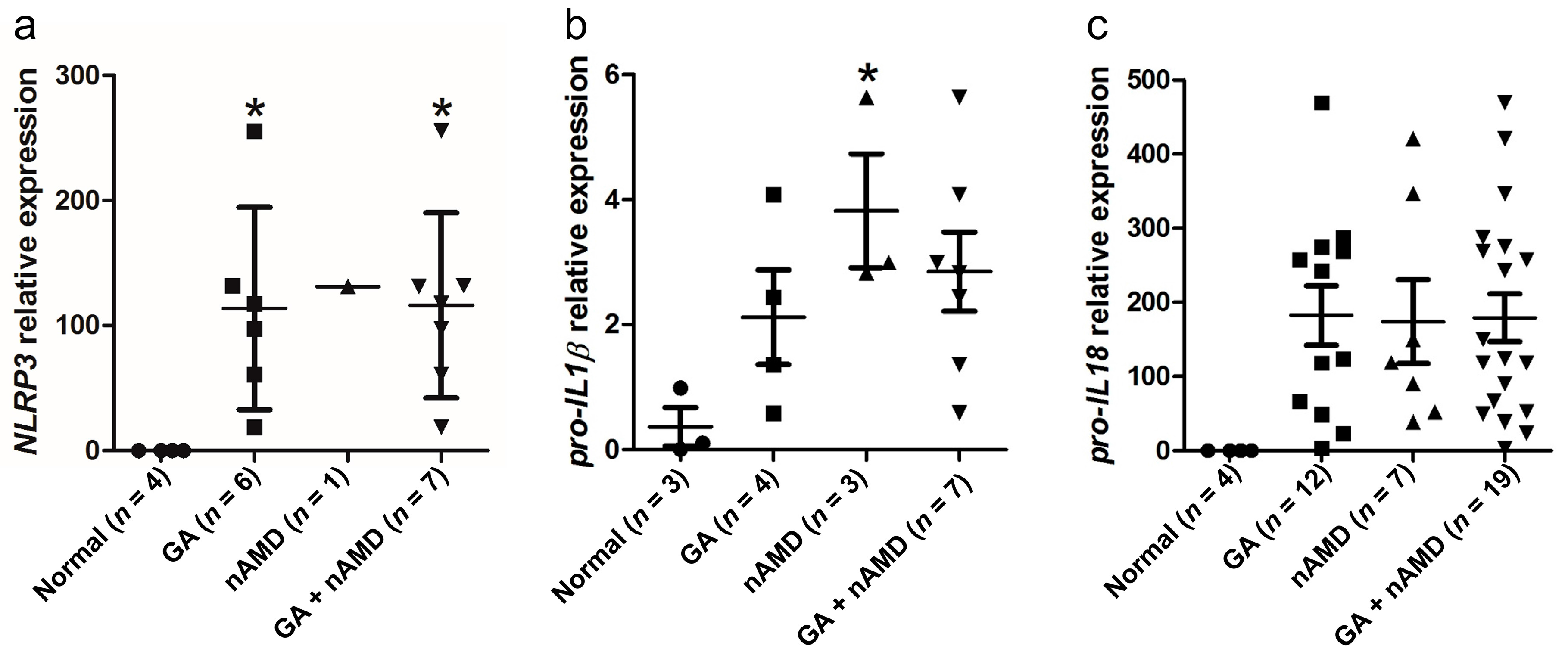
2.1. Upregulation of NLRP3 Inflammasome in the Maculae of GA and nAMD

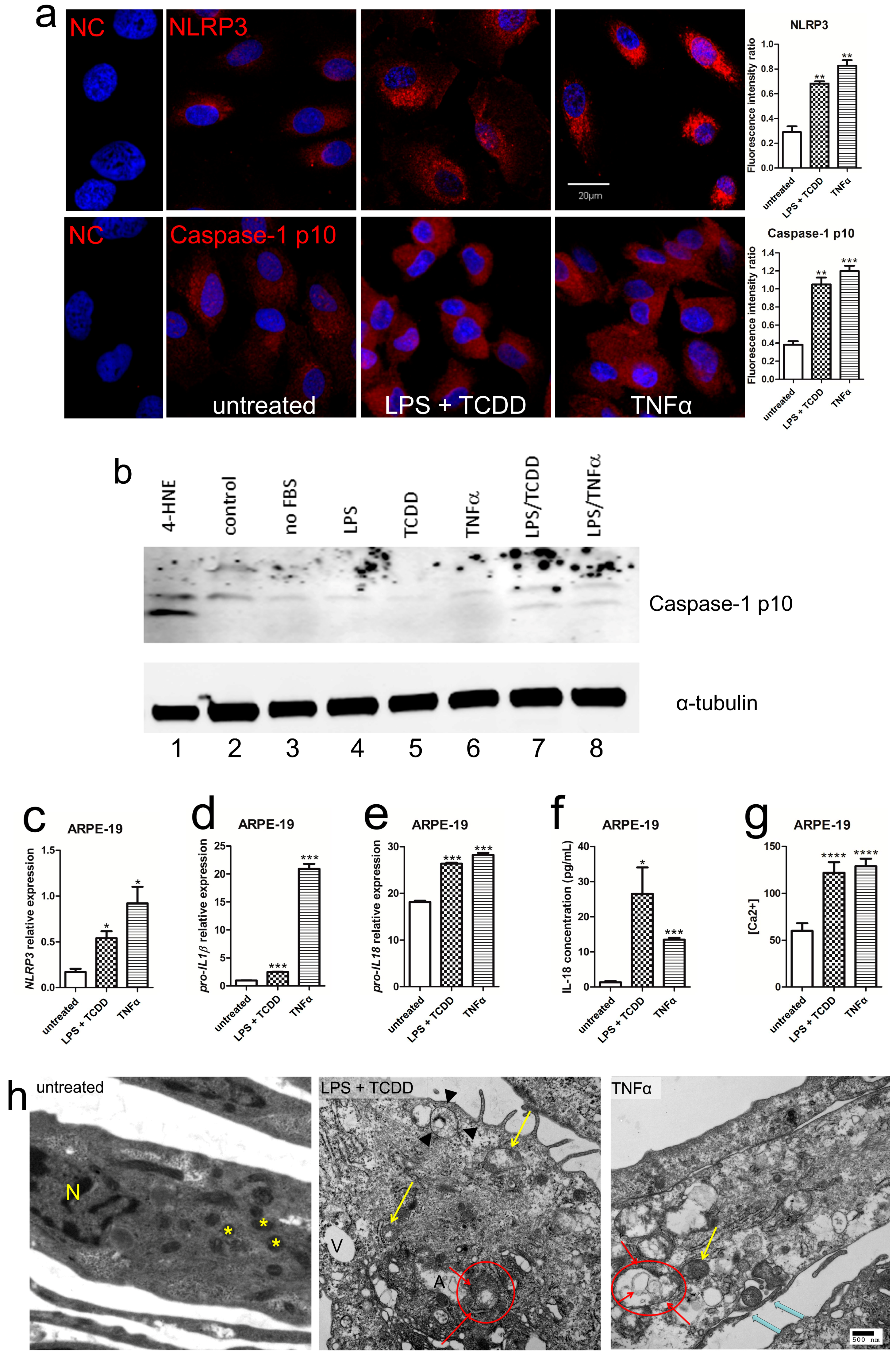
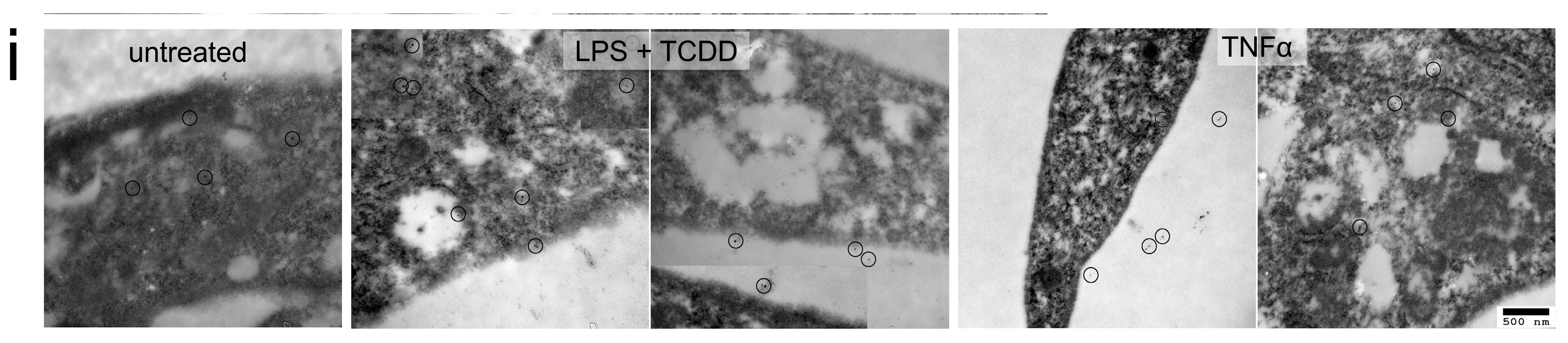
2.2. Activation of NLRP3 Inflammasome in Human RPE under Inflammation and Oxidative Stress


2.3. Silencing of NLRP3 Inhibits Inflammasome Activation in Human RPE Cells under Inflammation and Oxidative Stress

2.4. Upregulation of NLRP3 Inflammasome in Ccl2/Cx3cr1 double knockout on C57BL/6N (Crb1 rd8) (DKO rd8) Mouse Retina

2.5. Discussion
3. Experimental Section
3.1. Human Tissue
3.2. Cells and Stimulations
3.3. Mice
3.4. NLRP3 siRNA Transfection
3.5. Immunohistochemistry
3.6. qRT-PCR
3.7. Enzyme-Linked Immunosorbent Assay (ELISA)
3.8. Western Blot
3.9. Transmission Electron Microscopy (TEM)
3.10. Ca2+ Imaging
3.11. Statistical Analysis
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- De Jong, P.T. Age-related macular degeneration. N. Engl. J. Med. 2006, 355, 1474–1485. [Google Scholar] [CrossRef] [PubMed]
- Coleman, H.R.; Chan, C.C.; Ferris, F.L., 3rd; Chew, E.Y. Age-related macular degeneration. Lancet 2008, 372, 1835–1845. [Google Scholar] [CrossRef]
- Klein, R.; Chou, C.F.; Klein, B.E.; Zhang, X.; Meuer, S.M.; Saaddine, J.B. Prevalence of age-related macular degeneration in the US population. Arch. Ophthalmol. 2011, 129, 75–80. [Google Scholar] [CrossRef] [PubMed]
- Lim, L.S.; Mitchell, P.; Seddon, J.M.; Holz, F.G.; Wong, T.Y. Age-related macular degeneration. Lancet 2012, 379, 1728–1738. [Google Scholar] [CrossRef]
- Ambati, J.; Fowler, B.J. Mechanisms of age-related macular degeneration. Neuron 2012, 75, 26–39. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; Chen, M.; Forrester, J.V. Para-inflammation in the aging retina. Prog. Retin. Eye Res. 2009, 28, 348–368. [Google Scholar] [CrossRef] [PubMed]
- Rutar, M.; Natoli, R.; Kozulin, P.; Valter, K.; Gatenby, P.; Provis, J.M. Analysis of complement expression in light-induced retinal degeneration: Synthesis and deposition of C3 by microglia/macrophages is associated with focal photoreceptor degeneration. Investig. Ophthalmol. Vis. Sci. 2011, 52, 5347–5358. [Google Scholar] [CrossRef] [PubMed]
- Colak, E.; Majkic-Singh, N.; Zoric, L.; Radosavljevic, A.; Kosanovic-Jakovic, N. The impact of inflammation to the antioxidant defense parameters in AMD patients. Aging Clin. Exp. Res. 2012, 24, 588–594. [Google Scholar] [PubMed]
- Kauppinen, A.; Niskanen, H.; Suuronen, T.; Kinnunen, K.; Salminen, A.; Kaarniranta, K. Oxidative stress activates NLRP3 inflammasomes in ARPE-19 cells—Implications for age-related macular degeneration (AMD). Immunol. Lett. 2012, 147, 29–33. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Wang, V.M.; Chan, C.C. The role of anti-inflammatory agents in age-related macular degeneration (AMD) treatment. Eye 2011, 25, 127–139. [Google Scholar] [CrossRef] [PubMed]
- Tarallo, V.; Hirano, Y.; Gelfand, B.D.; Dridi, S.; Kerur, N.; Kim, Y.; Cho, W.G.; Kaneko, H.; Fowler, B.J.; Bogdanovich, S.; et al. DICER1 loss and Alu RNA induce age-related macular degeneration via the NLRP3 inflammasome and MyD88. Cell 2012, 149, 847–859. [Google Scholar] [CrossRef] [PubMed]
- Ardeljan, D.; Chan, C.C. Aging is not a disease: Distinguishing age-related macular degeneration from aging. Prog. Retin. Eye Res. 2013, 37, 68–89. [Google Scholar] [CrossRef] [PubMed]
- Fujihara, M.; Nagai, N.; Sussan, T.E.; Biswal, S.; Handa, J.T. Chronic cigarette smoke causes oxidative damage and apoptosis to retinal pigmented epithelial cells in mice. PLoS ONE 2008, 3, e3119. [Google Scholar] [CrossRef] [PubMed]
- Ding, X.; Patel, M.; Shen, D.; Herzlich, A.A.; Cao, X.; Villasmil, R.; Klupsch, K.; Tuo, J.; Downward, J.; Chan, C.C. Enhanced HtrA2/Omi expression in oxidative injury to retinal pigment epithelial cells and murine models of neurodegeneration. Investig. Ophthalmol. Vis. Sci. 2009, 50, 4957–4966. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Shen, D.; Wang, V.M.; Yu, C.R.; Wang, R.X.; Tuo, J.; Chan, C.C. Enhanced apoptosis in retinal pigment epithelium under inflammatory stimuli and oxidative stress. Apoptosis 2012, 17, 1144–1155. [Google Scholar] [CrossRef] [PubMed]
- Medzhitov, R. Origin and physiological roles of inflammation. Nature 2008, 454, 428–435. [Google Scholar] [CrossRef] [PubMed]
- Doyle, S.L.; Campbell, M.; Ozaki, E.; Salomon, R.G.; Mori, A.; Kenna, P.F.; Farrar, G.J.; Kiang, A.S.; Humphries, M.M.; Lavelle, E.C.; et al. NLRP3 has a protective role in age-related macular degeneration through the induction of IL-18 by drusen components. Nat. Med. 2012, 18, 791–798. [Google Scholar] [CrossRef] [PubMed]
- Tseng, W.A.; Thein, T.; Kinnunen, K.; Lashkari, K.; Gregory, M.S.; D’Amore, P.A.; Ksander, B.R. NLRP3 inflammasome activation in retinal pigment epithelial cells by lysosomal destabilization: Implications for age-related macular degeneration. Investig. Ophthalmol. Vis. Sci. 2013, 54, 110–120. [Google Scholar] [CrossRef] [PubMed]
- Kerur, N.; Hirano, Y.; Tarallo, V.; Fowler, B.J.; Bastos-Carvalho, A.; Yasuma, T.; Yasuma, R.; Kim, Y.; Hinton, D.R.; Kirschning, C.J.; et al. TLR-independent and P2X7-dependent signaling mediate Alu RNA-induced NLRP3 inflammasome activation in geographic atrophy. Investig. Ophthalmol. Vis. Sci. 2013, 54, 7395–7401. [Google Scholar] [CrossRef] [PubMed]
- Marneros, A.G. NLRP3 inflammasome blockade inhibits VEGF-A-induced age-related macular degeneration. Cell Rep. 2013, 4, 945–958. [Google Scholar] [CrossRef] [PubMed]
- Strowig, T.; Henao-Mejia, J.; Elinav, E.; Flavell, R. Inflammasomes in health and disease. Nature 2012, 481, 278–286. [Google Scholar] [CrossRef] [PubMed]
- Fowler, B.J.; Gelfand, B.D.; Kim, Y.; Kerur, N.; Tarallo, V.; Hirano, Y.; Amarnath, S.; Fowler, D.H.; Radwan, M.; Young, M.T.; et al. Nucleoside reverse transcriptase inhibitors possess intrinsic anti-inflammatory activity. Science 2014, 346, 1000–1003. [Google Scholar] [CrossRef] [PubMed]
- Von Ahlfen, S.; Missel, A.; Bendrat, K.; Schlumpberger, M. Determinants of RNA quality from FFPE samples. PLoS ONE 2007, 2, e1261. [Google Scholar] [CrossRef] [PubMed]
- Chung, J.Y.; Braunschweig, T.; Williams, R.; Guerrero, N.; Hoffmann, K.M.; Kwon, M.; Song, Y.K.; Libutti, S.K.; Hewitt, S.M. Factors in tissue handling and processing that impact RNA obtained from formalin-fixed, paraffin-embedded tissue. J. Histochem. Cytochem. 2008, 56, 1033–1042. [Google Scholar] [CrossRef] [PubMed]
- Klettner, A.; Kauppinen, A.; Blasiak, J.; Roider, J.; Salminen, A.; Kaarniranta, K. Cellular and molecular mechanisms of age-related macular degeneration: From impaired autophagy to neovascularization. Int. J. Biochem. Cell Biol. 2013, 45, 1457–1467. [Google Scholar] [CrossRef] [PubMed]
- Tuo, J.; Bojanowski, C.M.; Zhou, M.; Shen, D.; Ross, R.J.; Rosenberg, K.I.; Cameron, D.J.; Yin, C.; Kowalak, J.A.; Zhuang, Z.; et al. Murine Ccl2/Cx3cr1 deficiency results in retinal lesions mimicking human age-related macular degeneration. Investig. Ophthalmol. Vis. Sci. 2007, 48, 3827–3836. [Google Scholar] [CrossRef] [PubMed]
- Mattapallil, M.J.; Wawrousek, E.F.; Chan, C.C.; Zhao, H.; Roychoudhury, J.; Ferguson, T.A.; Caspi, R.R. The Rd8 mutation of the Crb1 gene is present in vendor lines of C57BL/6N mice and embryonic stem cells, and confounds ocular induced mutant phenotypes. Investig. Ophthalmol. Vis. Sci. 2012, 53, 2921–2927. [Google Scholar] [CrossRef] [PubMed]
- Chu, X.K.; Wang, Y.; Ardeljan, D.; Tuo, J.; Chan, C.C. Controversial view of a genetically altered mouse model of focal retinal degeneration. Bioengineered 2012, 4, 130–135. [Google Scholar] [CrossRef] [PubMed]
- Popp, N.; Chu, X.K.; Shen, D.; Tuo, J.; Chan, C.C. Evaluating potential therapies in a mouse model of focal retinal degeneration with age-related macular degeneration (AMD)-like lesions. J. Clin. Exp. Ophthalmol. 2013, 4. [Google Scholar] [CrossRef]
- Chen, M.; Hombrebueno, J.R.; Luo, C.; Penalva, R.; Zhao, J.; Colhoun, L.; Pandi, S.P.; Forrester, J.V.; Xu, H. Age- and light-dependent development of localised retinal atrophy in CCL2−/−CX3CR1GFP/GFP mice. PLoS ONE 2013, 8, e61381. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cao, X.; Liu, M.; Tuo, J.; Shen, D.; Chan, C.C. The effects of quercetin in cultured human RPE cells under oxidative stress and in Ccl2/Cx3cr1 double deficient mice. Exp. Eye Res. 2010, 91, 15–25. [Google Scholar] [CrossRef] [PubMed]
- Tuo, J.; Cao, X.; Shen, D.; Wang, Y.; Zhang, J.; Oh, J.Y.; Prockop, D.J.; Chan, C.C. Anti-inflammatory recombinant TSG-6 stabilizes the progression of focal retinal degeneration in a murine model. J. Neuroinflamm. 2012, 9. [Google Scholar] [CrossRef] [PubMed]
- Shen, D.; Cao, X.; Zhao, L.; Tuo, J.; Wong, W.T.; Chan, C.C. Naloxone ameliorates retinal lesions in Ccl2/Cx3cr1 double-deficient mice via modulation of microglia. Investig. Ophthalmol. Vis. Sci. 2011, 52, 2897–2904. [Google Scholar] [CrossRef] [PubMed]
- Ardeljan, D.; Wang, Y.; Park, S.; Shen, D.; Chu, X.K.; Yu, C.R.; Abu-Asab, M.; Tuo, J.; Eberhart, C.G.; Olsen, T.W.; et al. Interleukin-17 retinotoxicity is prevented by gene transfer of a soluble interleukin-17 receptor acting as a cytokine blocker: Implications for age-related macular degeneration. PLoS ONE 2014, 9, e95900. [Google Scholar] [CrossRef] [PubMed]
- Ijima, R.; Kaneko, H.; Ye, F.; Nagasaka, Y.; Takayama, K.; Kataoka, K.; Kachi, S.; Iwase, T.; Terasaki, H. Interleukin-18 induces retinal pigment epithelium degeneration in mice. Investig. Ophthalmol. Vis. Sci. 2014, 18, 6673–6678. [Google Scholar] [CrossRef] [PubMed]
- Doyle, S.L.; Ozaki, E.; Brennan, K.; Humphries, M.M.; Mulfaul, K.; Keaney, J.; Kenna, P.F.; Maminishkis, A.; Kiang, A.S.; Saunders, S.P.; et al. IL-18 attenuates experimental choroidal neovascularization as a potential therapy for wet age-related macular degeneration. Sci. Transl. Med. 2014, 6. [Google Scholar] [CrossRef] [PubMed]
- Oh, H.; Takagi, H.; Takagi, C.; Suzuma, K.; Otani, A.; Ishida, K.; Matsumura, M.; Ogura, Y.; Honda, Y. The potential angiogenic role of macrophages in the formation of choroidal neovascular membranes. Investig. Ophthalmol. Vis. Sci. 1999, 40, 1891–1898. [Google Scholar]
- Shen, J.; Choy, D.F.; Yoshida, T.; Iwase, T.; Hafiz, G.; Xie, B.; Hackett, S.F.; Arron, J.R.; Campochiaro, P.A. Interleukin-18 has antipermeablity and antiangiogenic activities in the eye: Reciprocal suppression with VEGF. J. Cell. Physiol. 2014, 229, 947–983. [Google Scholar] [CrossRef] [PubMed]
- Parekh, A.B. Mitochondrial regulation of store-operated CRAC channels. Cell Calcium 2008, 44, 6–13. [Google Scholar] [CrossRef] [PubMed]
- Murakami, T.; Ockinger, J.; Yu, J.; Byles, V.; McColl, A.; Hofer, A.M.; Horng, T. Critical role for calcium mobilization in activation of the NLRP3 inflammasome. Proc. Natl. Acad. Sci. USA 2012, 109, 11282–11287. [Google Scholar] [CrossRef] [PubMed]
- Lemasters, J.J.; Theruvath, T.P.; Zhong, Z.; Nieminen, A.L. Mitochondrial calcium and the permeability transition in cell death. Biochim. Biophys. Acta 2009, 1787, 1395–1401. [Google Scholar] [CrossRef] [PubMed]
- Dong, Z.; Saikumar, P.; Weinberg, J.M.; Venkatachalam, M.A. Calcium in cell injury and death. Annu. Rev. Pathol. 2006, 1, 405–434. [Google Scholar] [CrossRef] [PubMed]
- Behrends, C.; Sowa, M.E.; Gygi, S.P.; Harper, J.W. Network organization of the human autophagy system. Nature 2010, 466, 68–76. [Google Scholar] [CrossRef] [PubMed]
- Zhou, R.; Yazdi, A.S.; Menu, P.; Tschopp, J. A role for mitochondria in NLRP3 inflammasome activation. Nature 2011, 469, 221–225. [Google Scholar] [CrossRef] [PubMed]
- Shi, C.S.; Shenderov, K.; Huang, N.N.; Kabat, J.; Abu-Asab, M.; Fitzgerald, K.A.; Sher, A.; Kehrl, J.H. Activation of autophagy by inflammatory signals limits IL-1β production by targeting ubiquitinated inflammasomes for destruction. Nat. Immunol. 2012, 13, 255–263. [Google Scholar] [CrossRef] [PubMed]
- Ramkumar, H.L.; Tuo, J.; de Shen, F.; Zhang, J.; Cao, X.; Chew, E.Y.; Chan, C.C. Nutrient supplementation with n3 polyunsaturated fatty acids, lutein, and zeaxanthin decrease A2E accumulation and VEGF expression in the retinas of Ccl2/Cx3cr1-deficient mice on Crb1rd8 background. J. Nutr. 2013, 143, 1129–1135. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Subramanian, P.; Shen, D.; Tuo, J.; Becerra, S.P.; Chan, C.C. Pigment epithelium-derived factor reduces apoptosis and pro-inflammatory cytokine gene expression in a murine model of focal retinal degeneration. ASN Neuro 2013, 5. [Google Scholar] [CrossRef] [PubMed]
- Tuo, J.; Pang, J.J.; Cao, X.; Shen, D.; Zhang, J.; Scaria, A.; Wadsworth, S.C.; Pechan, P.; Boye, S.L.; Hauswirth, W.W.; et al. AAV5-mediated sFLT01 gene therapy arrests retinal lesions in Ccl2−/−/Cx3cr1−/− mice. Neurobiol. Aging 2012, 33, 433.e1–433.e10. [Google Scholar] [CrossRef] [PubMed]
- Ebrahimi, K.B.; Handa, J.T. Lipids, lipoproteins, and age-related macular degeneration. J. Lipids 2011, 2011. [Google Scholar] [CrossRef] [PubMed]
- Kishan, A.U.; Modjtahedi, B.S.; Martins, E.N.; Modjtahedi, S.P.; Morse, L.S. Lipids and age-related macular degeneration. Surv. Ophthalmol. 2011, 56, 195–213. [Google Scholar] [CrossRef] [PubMed]
- Nagineni, C.N.; Detrick, B.; Hooks, J.J. Synergistic effects of gamma interferon on inflammatory mediators that induce interleukin-6 gene expression and secretion by human retinal pigment epithelial cells. Clin. Diagn. Lab. Immunol. 1994, 1, 569–577. [Google Scholar] [PubMed]
- Ogilvy, A.J.; Shen, D.; Wang, Y.; Chan, C.C.; Abu-Asab, M.S. Implications of DNA leakage in eyes of mutant mice. Ultrastruct. Pathol. 2014, 38, 335–343. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Popp, N.; Chan, C.C. Animal models of age-related macular degeneration and their translatability into the clinic. Exp. Rev. Ophthalmol. 2014, 9, 285–295. [Google Scholar] [CrossRef]
- Gee, K.R.; Brown, K.A.; Chen, W.N.; Bishop-Stewart, J.; Gray, D.; Johnson, I. Chemical and physiological characterization of fluo-4 Ca2+-indicator dyes. Cell Calcium 2000, 27, 97–106. [Google Scholar] [CrossRef] [PubMed]
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, Y.; Hanus, J.W.; Abu-Asab, M.S.; Shen, D.; Ogilvy, A.; Ou, J.; Chu, X.K.; Shi, G.; Li, W.; Wang, S.; et al. NLRP3 Upregulation in Retinal Pigment Epithelium in Age-Related Macular Degeneration. Int. J. Mol. Sci. 2016, 17, 73. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms17010073
Wang Y, Hanus JW, Abu-Asab MS, Shen D, Ogilvy A, Ou J, Chu XK, Shi G, Li W, Wang S, et al. NLRP3 Upregulation in Retinal Pigment Epithelium in Age-Related Macular Degeneration. International Journal of Molecular Sciences. 2016; 17(1):73. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms17010073
Chicago/Turabian StyleWang, Yujuan, Jakub W. Hanus, Mones S. Abu-Asab, Defen Shen, Alexander Ogilvy, Jingxing Ou, Xi K. Chu, Guangpu Shi, Wei Li, Shusheng Wang, and et al. 2016. "NLRP3 Upregulation in Retinal Pigment Epithelium in Age-Related Macular Degeneration" International Journal of Molecular Sciences 17, no. 1: 73. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms17010073