
Roles of RNA-Binding Proteins in DNA Damage Response

{kind=link}
Abstract
:1. Introduction
2. Control of DNA Damage Response (DDR)-Gene Expression by RNA-Binding Protein (RBP)
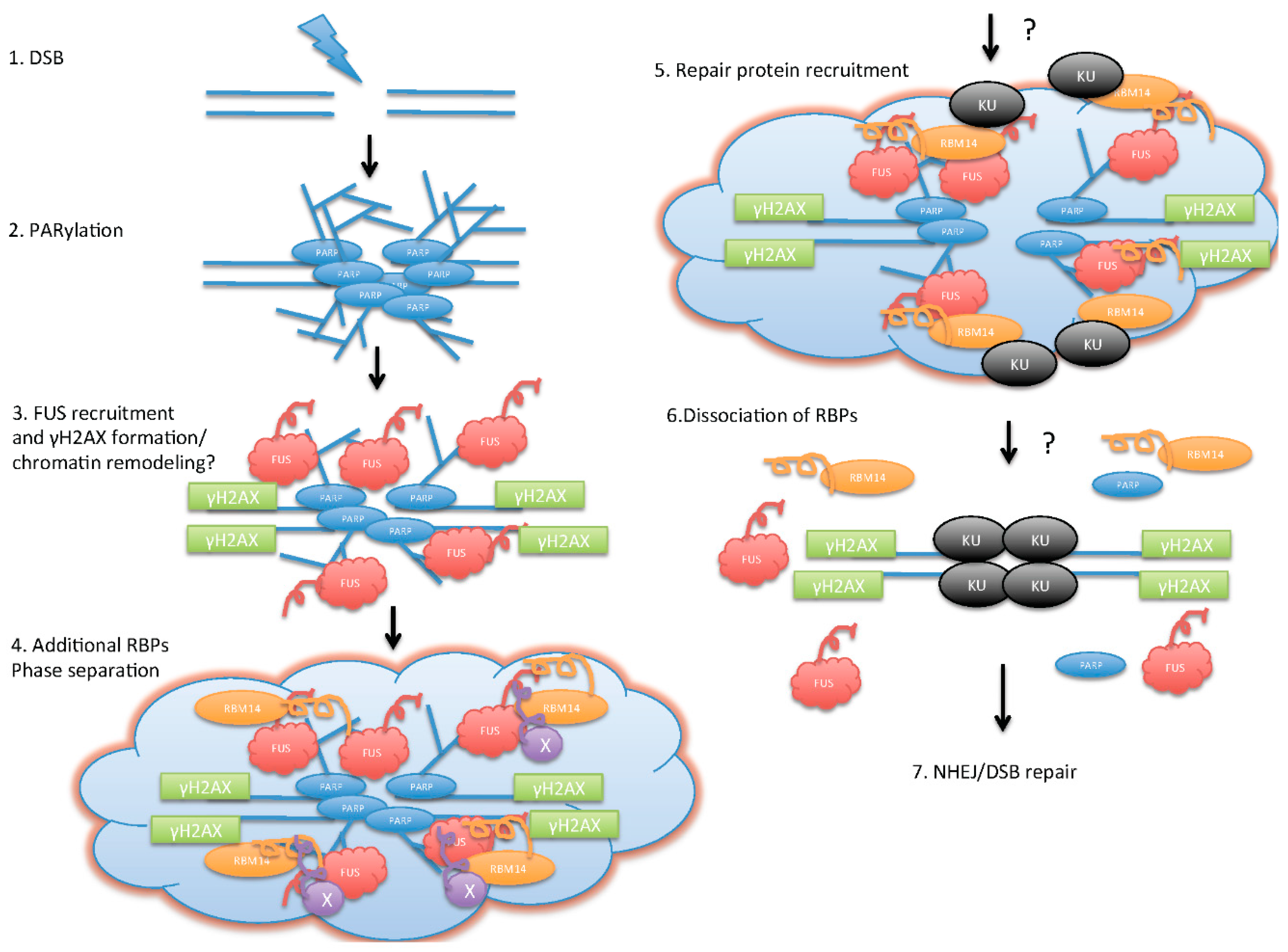
3. Direct Roles of RBPs in DDR
4. Conclusions
Conflicts of Interest
References
- Matsuoka, S.; Ballif, B.A.; Smogorzewska, A.; McDonald, E.R., III; Hurov, K.E.; Luo, J.; Bakalarski, C.E.; Zhao, Z.; Solimini, N.; Lerenthal, Y.; et al. ATM and ATR substrate analysis reveals extensive protein networks responsive to DNA damage. Science 2007, 316, 1160–1166. [Google Scholar] [CrossRef] [PubMed]
- Paulsen, R.D.; Soni, D.V.; Wollman, R.; Hahn, A.T.; Yee, M.C.; Guan, A.; Hesley, J.A.; Miller, S.C.; Cromwell, E.F.; Solow-Cordero, D.E.; et al. A genome-wide siRNA screen reveals diverse cellular processes and pathways that mediate genome stability. Mol. Cell 2009, 35, 228–239. [Google Scholar] [CrossRef] [PubMed]
- Lackner, D.H.; Durocher, D.; Karlseder, J. A siRNA-based screen for genes involved in chromosome end protection. PLoS ONE 2011, 6, e21407. [Google Scholar] [CrossRef] [PubMed]
- Paronetto, M.P.; Minana, B.; Valcarcel, J. The ewing sarcoma protein regulates DNA damage-induced alternative splicing. Mol. Cell 2011, 43, 353–368. [Google Scholar] [CrossRef] [PubMed]
- Savage, K.I.; Gorski, J.J.; Barros, E.M.; Irwin, G.W.; Manti, L.; Powell, A.J.; Pellagatti, A.; Lukashchuk, N.; McCance, D.J.; McCluggage, W.G.; et al. Identification of a BRCA1-mRNA splicing complex required for efficient DNA repair and maintenance of genomic stability. Mol. Cell 2014, 54, 445–459. [Google Scholar] [CrossRef] [PubMed]
- Kastan, M.B.; Onyekwere, O.; Sidransky, D.; Vogelstein, B.; Craig, R.W. Participation of p53 protein in the cellular response to DNA damage. Cancer Res. 1991, 51, 6304–6311. [Google Scholar] [PubMed]
- Giaccia, A.J.; Kastan, M.B. The complexity of p53 modulation: Emerging patterns from divergent signals. Genes Dev. 1998, 12, 2973–2983. [Google Scholar] [CrossRef] [PubMed]
- Mazan-Mamczarz, K.; Galban, S.; Lopez de Silanes, I.; Martindale, J.L.; Atasoy, U.; Keene, J.D.; Gorospe, M. RNA-binding protein HuR enhances p53 translation in response to ultraviolet light irradiation. Proc. Natl. Acad. Sci. USA 2003, 100, 8354–8359. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Furneaux, H.; Cheng, H.; Caldwell, M.C.; Hutter, D.; Liu, Y.; Holbrook, N.; Gorospe, M. HuR regulates p21 mRNA stabilization by UV light. Mol. Cell. Biol. 2000, 20, 760–769. [Google Scholar] [CrossRef] [PubMed]
- Masuda, K.; Abdelmohsen, K.; Kim, M.M.; Srikantan, S.; Lee, E.K.; Tominaga, K.; Selimyan, R.; Martindale, J.L.; Yang, X.; Lehrmann, E.; et al. Global dissociation of HuR-mRNA complexes promotes cell survival after ionizing radiation. EMBO J. 2011, 30, 1040–1053. [Google Scholar] [CrossRef] [PubMed]
- Mazan-Mamczarz, K.; Hagner, P.R.; Zhang, Y.; Dai, B.; Lehrmann, E.; Becker, K.G.; Keene, J.D.; Gorospe, M.; Liu, Z.; Gartenhaus, R.B. ATM regulates a DNA damage response posttranscriptional RNA operon in lymphocytes. Blood 2011, 117, 2441–2450. [Google Scholar] [CrossRef] [PubMed]
- Abdelmohsen, K.; Pullmann, R., Jr.; Lal, A.; Kim, H.H.; Galban, S.; Yang, X.; Blethrow, J.D.; Walker, M.; Shubert, J.; Gillespie, D.A.; et al. Phosphorylation of HuR by Chk2 regulates SIRT1 expression. Mol. Cell 2007, 25, 543–557. [Google Scholar] [CrossRef] [PubMed]
- Adamson, B.; Smogorzewska, A.; Sigoillot, F.D.; King, R.W.; Elledge, S.J. A genome-wide homologous recombination screen identifies the RNA-binding protein RBMX as a component of the DNA-damage response. Nat. Cell Biol. 2012, 14, 318–328. [Google Scholar] [CrossRef] [PubMed]
- Heinrich, B.; Zhang, Z.; Raitskin, O.; Hiller, M.; Benderska, N.; Hartmann, A.M.; Bracco, L.; Elliott, D.; Ben-Ari, S.; Soreq, H.; et al. Heterogeneous nuclear ribonucleoprotein G regulates splice site selection by binding to CC(A/C)-rich regions in pre-mRNA. J. Biol. Chem. 2009, 284, 14303–14315. [Google Scholar] [CrossRef] [PubMed]
- Chan, S.P.; Cheng, S.C. The PRP19-associated complex is required for specifying interactions of U5 and U6 with pre-mRNA during spliceosome activation. J. Biol. Chem. 2005, 280, 31190–31199. [Google Scholar] [CrossRef] [PubMed]
- Chan, S.P.; Kao, D.I.; Tsai, W.Y.; Cheng, S.C. The PRP19p-associated complex in spliceosome activation. Science 2003, 302, 279–282. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.H.; Kao, D.I.; Chan, S.P.; Kao, T.C.; Lin, J.Y.; Cheng, S.C. Functional links between the PRP19-associated complex, U4/U6 biogenesis, and spliceosome recycling. RNA 2006, 12, 765–774. [Google Scholar] [CrossRef] [PubMed]
- Song, E.J.; Werner, S.L.; Neubauer, J.; Stegmeier, F.; Aspden, J.; Rio, D.; Harper, J.W.; Elledge, S.J.; Kirschner, M.W.; Rape, M. The PRP19 complex and the Usp4Sart3 deubiquitinating enzyme control reversible ubiquitination at the spliceosome. Genes Dev. 2010, 24, 1434–1447. [Google Scholar] [CrossRef] [PubMed]
- Chanarat, S.; Seizl, M.; Strasser, K. The PRP19 complex is a novel transcription elongation factor required for trex occupancy at transcribed genes. Genes Dev. 2011, 25, 1147–1158. [Google Scholar] [CrossRef] [PubMed]
- David, C.J.; Boyne, A.R.; Millhouse, S.R.; Manley, J.L. The RNA polymerase II C-terminal domain promotes splicing activation through recruitment of a U2AF65-PRP19 complex. Genes Dev. 2011, 25, 972–983. [Google Scholar] [CrossRef] [PubMed]
- Cheng, S.C.; Tarn, W.Y.; Tsao, T.Y.; Abelson, J. PRP19: A novel spliceosomal component. Mol. Cell. Biol. 1993, 13, 1876–1882. [Google Scholar] [CrossRef] [PubMed]
- Tarn, W.Y.; Lee, K.R.; Cheng, S.C. The yeast PRP19 protein is not tightly associated with small nuclear RNAs, but appears to associate with the spliceosome after binding of U2 to the pre-mRNA and prior to formation of the functional spliceosome. Mol. Cell. Biol. 1993, 13, 1883–1891. [Google Scholar] [CrossRef] [PubMed]
- Grey, M.; Dusterhoft, A.; Henriques, J.A.; Brendel, M. Allelism of PSO4 and PRP19 links pre-mRNA processing with recombination and error-prone DNA repair in saccharomyces cerevisiae. Nucleic Acids Res. 1996, 24, 4009–4014. [Google Scholar] [CrossRef] [PubMed]
- Mahajan, K.N.; Mitchell, B.S. Role of human PSO4 in mammalian DNA repair and association with terminal deoxynucleotidyl transferase. Proc. Natl. Acad. Sci. USA 2003, 100, 10746–10751. [Google Scholar] [CrossRef] [PubMed]
- Zhang, N.; Kaur, R.; Lu, X.; Shen, X.; Li, L.; Legerski, R.J. The PSO4 mRNA splicing and DNA repair complex interacts with WRN for processing of DNA interstrand cross-links. J. Biol. Chem. 2005, 280, 40559–40567. [Google Scholar] [CrossRef] [PubMed]
- Zhang, N.; Kaur, R.; Akhter, S.; Legerski, R.J. CDC5L interacts with atr and is required for the S-phase cell-cycle checkpoint. EMBO Rep. 2009, 10, 1029–1035. [Google Scholar] [CrossRef] [PubMed]
- Marechal, A.; Li, J.M.; Ji, X.Y.; Wu, C.S.; Yazinski, S.A.; Nguyen, H.D.; Liu, S.; Jimenez, A.E.; Jin, J.; Zou, L. PRP19 transforms into a sensor of RPA-ssDNA after DNA damage and drives ATR activation via a ubiquitin-mediated circuitry. Mol. Cell 2014, 53, 235–246. [Google Scholar] [CrossRef] [PubMed]
- Wan, L.; Huang, J. The pso4 protein complex associates with replication protein a (RPA) and modulates the activation of ataxia telangiectasia-mutated and RAD3-related (ATR). J. Biol. Chem. 2014, 289, 6619–6626. [Google Scholar] [CrossRef] [PubMed]
- Polo, S.E.; Blackford, A.N.; Chapman, J.R.; Baskcomb, L.; Gravel, S.; Rusch, A.; Thomas, A.; Blundred, R.; Smith, P.; Kzhyshkowska, J.; et al. Regulation of DNA-end resection by hnRNPU-like proteins promotes DNA double-strand break signaling and repair. Mol. Cell 2012, 45, 505–516. [Google Scholar] [CrossRef] [PubMed]
- Hong, Z.; Jiang, J.; Ma, J.; Dai, S.; Xu, T.; Li, H.; Yasui, A. The role of hnRPUL1 involved in DNA damage response is related to PARP1. PLoS ONE 2013, 8, e60208. [Google Scholar] [CrossRef] [PubMed]
- Yang, D.; Zhang, Z.J.; Oldenburg, M.; Ayala, M.; Zhang, S.C. Human embryonic stem cell-derived dopaminergic neurons reverse functional deficit in parkinsonian rats. Stem Cells 2008, 26, 55–63. [Google Scholar] [CrossRef] [PubMed]
- Auboeuf, D.; Dowhan, D.H.; Li, X.; Larkin, K.; Ko, L.; Berget, S.M.; O’Malley, B.W. CoAA, a nuclear receptor coactivator protein at the interface of transcriptional coactivation and RNA splicing. Mol. Cell. Biol. 2004, 24, 442–453. [Google Scholar] [CrossRef] [PubMed]
- Law, W.J.; Cann, K.L.; Hicks, G.G. TLS, EWS and TAF15: A model for transcriptional integration of gene expression. Brief. Funct. Genom. Proteom. 2006, 5, 8–14. [Google Scholar] [CrossRef] [PubMed]
- MacKenzie, L.; McCall, P.; Hatziieremia, S.; Catlow, J.; Adams, C.; McArdle, P.; Seywright, M.; Tanahill, C.; Paul, A.; Underwood, M.; et al. Nuclear factor κB predicts poor outcome in patients with hormone-naive prostate cancer with high nuclear androgen receptor. Hum. Pathol. 2012, 43, 1491–1500. [Google Scholar] [CrossRef] [PubMed]
- Sui, Y.; Yang, Z.; Xiong, S.; Zhang, L.; Blanchard, K.L.; Peiper, S.C.; Dynan, W.S.; Tuan, D.; Ko, L. Gene amplification and associated loss of 5′ regulatory sequences of CoAA in human cancers. Oncogene 2007, 26, 822–835. [Google Scholar] [CrossRef] [PubMed]
- Perani, M.; Antonson, P.; Hamoudi, R.; Ingram, C.J.; Cooper, C.S.; Garrett, M.D.; Goodwin, G.H. The proto-oncoprotein SYT interacts with SYT-interacting protein/co-activator activator (SIP/CoAA), a human nuclear receptor co-activator with similarity to ews and TLS/FUS family of proteins. J. Biol. Chem. 2005, 280, 42863–42876. [Google Scholar] [CrossRef] [PubMed]
- Iwasaki, T.; Chin, W.W.; Ko, L. Identification and characterization of RRM-containing coactivator activator (CoAA) as TRBP-interacting protein, and its splice variant as a coactivator modulator (CoAM). J. Biol. Chem. 2001, 276, 33375–33383. [Google Scholar] [CrossRef] [PubMed]
- Verreman, K.; Baert, J.L.; Verger, A.; Drobecq, H.; Ferreira, E.; de Launoit, Y.; Monte, D. The coactivator activator CoAA regulates PEA3 group member transcriptional activity. Biochem. J. 2011, 439, 469–477. [Google Scholar] [CrossRef] [PubMed]
- Iwasaki, T.; Koibuchi, N.; Chin, W.W. Synovial sarcoma translocation (SYT) encodes a nuclear receptor coactivator. Endocrinology 2005, 146, 3892–3899. [Google Scholar] [CrossRef] [PubMed]
- Brooks, Y.S.; Wang, G.; Yang, Z.; Smith, K.K.; Bieberich, E.; Ko, L. Functional pre-mRNA trans-splicing of coactivator coaa and corepressor RBM4 during stem/progenitor cell differentiation. J. Biol. Chem. 2009, 284, 18033–18046. [Google Scholar] [CrossRef] [PubMed]
- Yuan, M.; Eberhart, C.G.; Kai, M. RNA binding protein RBM14 promotes radio-resistance in glioblastoma by regulating DNA repair and cell differentiation. Oncotarget 2014, 5, 2820–2826. [Google Scholar] [CrossRef] [PubMed]
- Hirose, T.; Virnicchi, G.; Tanigawa, A.; Naganuma, T.; Li, R.; Kimura, H.; Yokoi, T.; Nakagawa, S.; Benard, M.; Fox, A.H.; et al. NEAT1 long noncoding RNA regulates transcription via protein sequestration within subnuclear bodies. Mol. Biol. Cell 2014, 25, 169–183. [Google Scholar] [CrossRef] [PubMed]
- Hennig, S.; Kong, G.; Mannen, T.; Sadowska, A.; Kobelke, S.; Blythe, A.; Knott, G.J.; Iyer, K.S.; Ho, D.; Newcombe, E.A.; et al. Prion-like domains in RNA binding proteins are essential for building subnuclear paraspeckles. J. Cell Biol. 2015, 210, 529–539. [Google Scholar] [CrossRef] [PubMed]
- Krietsch, J.; Caron, M.C.; Gagne, J.P.; Ethier, C.; Vignard, J.; Vincent, M.; Rouleau, M.; Hendzel, M.J.; Poirier, G.G.; Masson, J.Y. Parp activation regulates the RNA-binding protein NONO in the DNA damage response to DNA double-strand breaks. Nucleic Acids Res. 2012, 40, 10287–10301. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, W.Y.; Pan, L.; Su, S.C.; Quinn, E.J.; Sasaki, M.; Jimenez, J.C.; Mackenzie, I.R.; Huang, E.J.; Tsai, L.H. Interaction of FUS and HDAC1 regulates DNA damage response and repair in neurons. Nat. Neurosci. 2013, 16, 1383–1391. [Google Scholar] [CrossRef] [PubMed]
- Mastrocola, A.S.; Kim, S.H.; Trinh, A.T.; Rodenkirch, L.A.; Tibbetts, R.S. The RNA-binding protein fused in sarcoma (FUS) functions downstream of poly(ADP-ribose) polymerase (PARP) in response to DNA damage. J. Biol. Chem. 2013, 288, 24731–24741. [Google Scholar] [CrossRef] [PubMed]
- Baechtold, H.; Kuroda, M.; Sok, J.; Ron, D.; Lopez, B.S.; Akhmedov, A.T. Human 75-kDa DNA-pairing protein is identical to the pro-oncoprotein TLS/FUS and is able to promote D-loop formation. J. Biol. Chem. 1999, 274, 34337–34342. [Google Scholar] [CrossRef] [PubMed]
- Bertrand, P.; Akhmedov, A.T.; Delacote, F.; Durrbach, A.; Lopez, B.S. Human POMp75 is identified as the pro-oncoprotein TLS/FUS: Both POMp75 and POMp100 DNA homologous pairing activities are associated to cell proliferation. Oncogene 1999, 18, 4515–4521. [Google Scholar] [CrossRef] [PubMed]
- Rulten, S.L.; Rotheray, A.; Green, R.L.; Grundy, G.J.; Moore, D.A.; Gomez-Herreros, F.; Hafezparast, M.; Caldecott, K.W. PARP-1 dependent recruitment of the amyotrophic lateral sclerosis-associated protein FUS/TLS to sites of oxidative DNA damage. Nucleic Acids Res. 2014, 42, 307–314. [Google Scholar] [CrossRef] [PubMed]
- Altmeyer, M.; Neelsen, K.J.; Teloni, F.; Pozdnyakova, I.; Pellegrino, S.; Grofte, M.; Rask, M.B.; Streicher, W.; Jungmichel, S.; Nielsen, M.L.; et al. Liquid demixing of intrinsically disordered proteins is seeded by poly(ADP-ribose). Nat Commun. 2015, 6. [Google Scholar] [CrossRef] [PubMed]
- Patel, A.; Lee, H.O.; Jawerth, L.; Maharana, S.; Jahnel, M.; Hein, M.Y.; Stoynov, S.; Mahamid, J.; Saha, S.; Franzmann, T.M.; et al. A liquid-to-solid phase transition of the als protein FUS accelerated by disease mutation. Cell 2015, 162, 1066–1077. [Google Scholar] [CrossRef] [PubMed]
- Britton, S.; Dernoncourt, E.; Delteil, C.; Froment, C.; Schiltz, O.; Salles, B.; Frit, P.; Calsou, P. DNA damage triggers SAF-A and RNA biogenesis factors exclusion from chromatin coupled to R-loops removal. Nucleic Acids Res. 2014, 42, 9047–9062. [Google Scholar] [CrossRef] [PubMed]
- D’Amours, D.; Desnoyers, S.; D’Silva, I.; Poirier, G.G. Poly(ADP-ribosyl)ation reactions in the regulation of nuclear functions. Biochem. J. 1999, 342 Pt 2, 249–268. [Google Scholar] [CrossRef] [PubMed]
- Pears, C.J.; Couto, C.A.; Wang, H.Y.; Borer, C.; Kiely, R.; Lakin, N.D. The role of ADP-ribosylation in regulating DNA double-strand break repair. Cell Cycle 2012, 11, 48–56. [Google Scholar] [CrossRef] [PubMed]
- Haince, J.F.; McDonald, D.; Rodrigue, A.; Dery, U.; Masson, J.Y.; Hendzel, M.J.; Poirier, G.G. PARP1-dependent kinetics of recruitment of MRE11 and NBS1 proteins to multiple DNA damage sites. J. Biol. Chem. 2008, 283, 1197–1208. [Google Scholar] [CrossRef] [PubMed]
- Spagnolo, L.; Barbeau, J.; Curtin, N.J.; Morris, E.P.; Pearl, L.H. Visualization of a DNA-PK/PARP1 complex. Nucleic Acids Res. 2012, 40, 4168–4177. [Google Scholar] [CrossRef] [PubMed]
- Mandraju, R.; Chekuri, A.; Bhaskar, C.; Duning, K.; Kremerskothen, J.; Kondapi, A.K. Topoisomerase iibeta associates with Ku70 and PARP-1 during double strand break repair of DNA in neurons. Arch. Biochem. Biophys. 2011, 516, 128–137. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.G.; Cortes, U.; Patnaik, S.; Jasin, M.; Wang, Z.Q. Ablation of PARP-1 does not interfere with the repair of DNA double-strand breaks, but compromises the reactivation of stalled replication forks. Oncogene 2004, 23, 3872–3882. [Google Scholar] [CrossRef] [PubMed]
- Audebert, M.; Salles, B.; Calsou, P. Involvement of poly(ADP-ribose) polymerase-1 and XRCC1/DNA ligase III in an alternative route for DNA double-strand breaks rejoining. J. Biol. Chem. 2004, 279, 55117–55126. [Google Scholar] [CrossRef] [PubMed]
- Audebert, M.; Salles, B.; Weinfeld, M.; Calsou, P. Involvement of polynucleotide kinase in a poly(ADP-ribose) polymerase-1-dependent DNA double-strand breaks rejoining pathway. J. Mol. Biol. 2006, 356, 257–265. [Google Scholar] [CrossRef] [PubMed]
- Paddock, M.N.; Bauman, A.T.; Higdon, R.; Kolker, E.; Takeda, S.; Scharenberg, A.M. Competition between PARP-1 and Ku70 control the decision between high-fidelity and mutagenic DNA repair. DNA Repair 2011, 10, 338–343. [Google Scholar] [CrossRef] [PubMed]
- Schultz, N.; Lopez, E.; Saleh-Gohari, N.; Helleday, T. Poly(ADP-ribose) polymerase (PARP-1) has a controlling role in homologous recombination. Nucleic Acids Res. 2003, 31, 4959–4964. [Google Scholar] [CrossRef] [PubMed]
- Hochegger, H.; Dejsuphong, D.; Fukushima, T.; Morrison, C.; Sonoda, E.; Schreiber, V.; Zhao, G.Y.; Saberi, A.; Masutani, M.; Adachi, N.; et al. PARP-1 protects homologous recombination from interference by Ku and ligase IV in vertebrate cells. EMBO J. 2006, 25, 1305–1314. [Google Scholar] [CrossRef] [PubMed]
- Berti, M.; Ray Chaudhuri, A.; Thangavel, S.; Gomathinayagam, S.; Kenig, S.; Vujanovic, M.; Odreman, F.; Glatter, T.; Graziano, S.; Mendoza-Maldonado, R.; et al. Human RECQ1 promotes restart of replication forks reversed by DNA topoisomerase I inhibition. Nat. Struct. Mol. Biol. 2013, 20, 347–354. [Google Scholar] [CrossRef] [PubMed]
- Sugimura, K.; Takebayashi, S.; Taguchi, H.; Takeda, S.; Okumura, K. PARP-1 ensures regulation of replication fork progression by homologous recombination on damaged DNA. J. Cell Biol. 2008, 183, 1203–1212. [Google Scholar] [CrossRef] [PubMed]
- Bryant, H.E.; Petermann, E.; Schultz, N.; Jemth, A.S.; Loseva, O.; Issaeva, N.; Johansson, F.; Fernandez, S.; McGlynn, P.; Helleday, T. PARP is activated at stalled forks to mediate MRE11-dependent replication restart and recombination. EMBO J. 2009, 28, 2601–2615. [Google Scholar] [CrossRef] [PubMed]

© 2016 by the author; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kai, M. Roles of RNA-Binding Proteins in DNA Damage Response. Int. J. Mol. Sci. 2016, 17, 310. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms17030310
Kai M. Roles of RNA-Binding Proteins in DNA Damage Response. International Journal of Molecular Sciences. 2016; 17(3):310. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms17030310
Chicago/Turabian StyleKai, Mihoko. 2016. "Roles of RNA-Binding Proteins in DNA Damage Response" International Journal of Molecular Sciences 17, no. 3: 310. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms17030310