
Role of Serum Uric Acid and Ferritin in the Development and Progression of NAFLD

Abstract
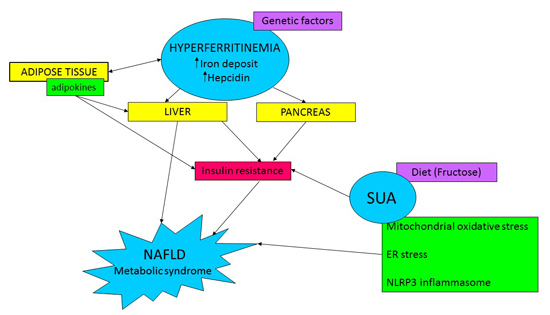
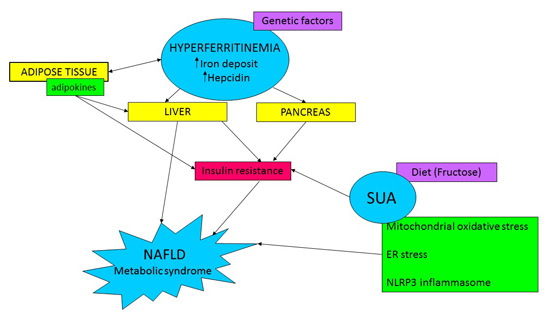
:
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Uric Acid
3. Serum Uric Acid and Metabolic Syndrome Clinical Manifestations
4. Serum Uric Acid and Nonalcoholic Fatty Liver Disease (NAFLD)
5. Relationship between Uric Acid and NAFLD/Metabolic Syndrome: Possible Mechanisms
5.1. Interaction between Uric Acid and Insulin
5.2. Uric Acid and Lipid Metabolism
5.3. Mitochondrial and Endoplasmic Reticulum (ER) Oxidative Stress
5.4. The Ucleotide-Binding Oligomerization Domain-Like (NOD-Like) Receptor Family Pyrin Domain Containing 3 (NLRP3) Inflammasome
6. Ferritin
7. Ferritin and Metabolic Syndrome Clinical Manifestations
8. Ferritin and NAFLD
9. Relationship between Ferritin and NAFLD/Metabolic Syndrome: Possible Mechanisms
9.1. Pathogenesis of DIOS (Dysmetabolic Iron Overload Syndrome)
9.2. Hyperferritinemia and Insulin-Resistance
9.3. Hyperferritinemia and Adipose Tissue
10. Hyperferritinemia and Severity of Liver Damage in NAFLD
11. Does Hyperferritinemia Reflect Iron Overload?
12. Iron Depletion in Patients with Hyperferritinemia, Metabolic Alterations and NAFLD
13. Conclusions
Author Contributions
Conflicts of Interest
References
- Williams, C.D.; Stengel, J.; Asike, M.I.; Torres, D.M.; Shaw, J.; Contreras, M.; Landt, C.L.; Harrison, S.A. Prevalence of nonalcoholic fatty liver disease and nonalcoholic steatohepatitis among a largely middle-aged population utilizing ultrasound and liver biopsy: A prospective study. Gastroenterology 2011, 140, 124–131. [Google Scholar] [CrossRef] [PubMed]
- Lonardo, A.; Ballestri, S.; Marchesini, G.; Angulo, P.; Loria, P. Nonalcoholic fatty liver disease: A precursor of the metabolic syndrome. Dig. Liver Dis. 2015, 47, 181–190. [Google Scholar] [CrossRef] [PubMed]
- Anstee, Q.M.; Targher, G.; Day, C.P. Progression of NAFLD to diabetes mellitus, cardiovascular disease or cirrhosis. Nat. Rev. Gastroenterol. Hepatol. 2013, 10, 330–344. [Google Scholar] [CrossRef] [PubMed]
- Serfaty, L.; Lemoine, M. Definition and natural history of metabolic steatosis: Clinical aspects of NAFLD, NASH and cirrhosis. Diabetes Metab. 2008, 34, 634–637. [Google Scholar] [CrossRef]
- Buzzetti, E.; Lombardi, R.; de Luca, L.; Tsochatzis, E.A. Noninvasive assessment of fibrosis in patients with nonalcoholic fatty liver disease. Int. J. Endocrinol. 2015, 2015, 343828. [Google Scholar] [CrossRef] [PubMed]
- Tsochatzis, E.A.; Gurusamy, K.S.; Ntaoula, S.; Cholongitas, E.; Davidson, B.R.; Burroughs, A.K. Elastography for the diagnosis of severity of fibrosis in chronic liver disease: A meta-analysis of diagnostic accuracy. J. Hepatol. 2011, 54, 650–659. [Google Scholar] [CrossRef] [PubMed]
- Mount, D.B.; Kwon, C.Y.; Zandi-Nejad, K. Renal urate transport. Rheum. Dis. Clin. N. Am. 2006, 32, 313–331. [Google Scholar] [CrossRef] [PubMed]
- Richette, P.; Bardin, T. Gout. Lancet 2010, 375, 318–328. [Google Scholar] [CrossRef]
- Zhang, M.L.; Gao, Y.X.; Wang, X.; Chang, H.; Huang, G.W. Serum uric acid and appropriate cutoff value for prediction of metabolic syndrome among Chinese adults. J. Clin. Biochem. Nutr. 2013, 52, 38–42. [Google Scholar] [CrossRef] [PubMed]
- Choi, H.K.; Ford, E.S. Prevalence of the metabolic syndrome in individuals with hyperuricemia. Am. J. Med. 2007, 120, 442–447. [Google Scholar] [CrossRef] [PubMed]
- Kodama, S.; Saito, K.; Yachi, Y.; Asumi, M.; Sugawara, A.; Totsuka, K.; Saito, A.; Sone, H. Association between serum uric acid and development of type 2 diabetes. Diabetes Care 2009, 32, 1737–1742. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, S.D.; Tsai, D.H.; Hsu, S.R. Association between serum uric acid level and components of the metabolic syndrome. J. Chin. Med. Assoc. 2006, 69, 512–516. [Google Scholar] [CrossRef]
- Lv, Q.; Meng, X.F.; He, F.F.; Chen, S.; Su, H.; Xiong, J.; Gao, P.; Tian, X.J.; Liu, J.S.; Zhu, Z.H.; et al. High serum uric acid and increased risk of type 2 diabetes: A systemic review and meta-analysis of prospective cohort studies. PLoS ONE 2013, 8, e56864. [Google Scholar]
- Lonardo, A.; Loria, P.; Leonardi, F.; Borsatti, A.; Neri, P.; Pulvirenti, M.; Verrone, A.M.; Bagni, A.; Bertolotti, M.; Ganazzi, D.; et al. Fasting insulin and uric acid levels but not indices of iron metabolism are independent predictors of non-alcoholic fatty liver disease. A case-control study. Dig. Liver Dis. 2002, 34, 204–211. [Google Scholar] [CrossRef]
- Li, Y.; Xu, C.; Yu, C.; Xu, L.; Miao, M. Association of serum uric acid level with non-alcoholic fatty liver disease: A cross-sectional study. J. Hepatol. 2009, 50, 1029–1034. [Google Scholar] [PubMed]
- Ryu, S.; Chang, Y.; Kim, S.G.; Cho, J.; Guallar, E. Serum uric acid levels predict incident nonalcoholic fatty liver disease in healthy Korean men. Metabolism 2011, 60, 860–866. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Que, S.; Zhou, L.; Zheng, S. Dose-response relationship of serum uric acid with metabolic syndrome and non-alcoholic fatty liver disease incidence: A meta-analysis of prospective studies. Sci. Rep. 2015, 5, 14325. [Google Scholar] [CrossRef] [PubMed]
- Yuan, H.; Yu, C.; Li, X.; Sun, L.; Zhu, X.; Zhao, C.; Zhang, Z.; Yang, Z. Serum uric acid levels and risk of metabolic syndrome: A dose-response meta-analysis of prospective studies. J. Clin. Endocrinol. Metab. 2015, 100, 4198–4207. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.H.; Chuang, S.Y.; Chen, H.J.; Yeh, W.T.; Pan, W.H. Serum uric acid level as an independent risk factor for all-cause, cardiovascular, and ischemic stroke mortality: A Chinese cohort study. Arthritis Rheum. 2009, 61, 225–232. [Google Scholar] [CrossRef] [PubMed]
- Xu, C.; Wan, X.; Xu, L.; Weng, H.; Yan, M.; Miao, M.; Sun, Y.; Xu, G.; Dooley, S.; Li, Y.; et al. Xanthine oxidase in non-alcoholic fatty liver disease and hyperuricemia: One stone hits two birds. J. Hepatol. 2015, 62, 1412–1419. [Google Scholar] [CrossRef] [PubMed]
- Sirota, J.C.; McFann, K.; Targher, G.; Johnson, R.J.; Chonchol, M.; Jalal, D.I. Elevated serum uric acid levels are associated with non-alcoholic fatty liver disease independently of metabolic syndrome features in the United States: Liver ultrasound data from the National Health and Nutrition Examination Survey. Metabolism 2013, 62, 392–399. [Google Scholar] [CrossRef] [PubMed]
- Petta, S.; Camma, C.; Cabibi, D.; Di Marco, V.; Craxi, A. Hyperuricemia is associated with histological liver damage in patients with non-alcoholic fatty liver disease. Aliment. Pharmacol. Ther. 2011, 34, 757–766. [Google Scholar] [PubMed]
- Afzali, A.; Weiss, N.S.; Boyko, E.J.; Ioannou, G.N. Association between serum uric acid level and chronic liver disease in the United States. Hepatology 2010, 52, 578–589. [Google Scholar] [CrossRef] [PubMed]
- Abreu, E.; Fonseca, M.J.; Santos, A.C. Association between hyperuricemia and insulin resistance. Acta Med. Port. 2011, 24, 565–574. [Google Scholar] [PubMed]
- Tan, Y.; Liu, X.; Zhou, K.; He, X.; Lu, C.; He, B.; Niu, X.; Xiao, C.; Xu, G.; Bian, Z.; et al. The potential biomarkers to identify the development of steatosis in hyperuricemia. PLoS ONE 2016, 11, e0149043. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Hsieh, M.C.; Chang, S.J. Metabolic syndrome, diabetes, and hyperuricemia. Curr. Opin. Rheumatol. 2013, 25, 210–216. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Hu, Y.; Huang, T.; Zhang, Y.; Li, Z.; Luo, C.; Luo, Y.; Yuan, H.; Hisatome, I.; Yamamoto, T.; et al. High uric acid directly inhibits insulin signalling and induces insulin resistance. Biochem. Biophys. Res. Commun. 2014, 447, 707–714. [Google Scholar] [CrossRef] [PubMed]
- Vandanmagsar, B.; Youm, Y.H.; Ravussin, A.; Galgani, J.E.; Stadler, K.; Mynatt, R.L.; Ravussin, E.; Stephens, J.M.; Dixit, V.D. The NLRP3 inflammasome instigates obesity-induced inflammation and insulin resistance. Nat. Med. 2011, 17, 179–188. [Google Scholar] [CrossRef] [PubMed]
- Lanaspa, M.A.; Sanchez-Lozada, L.G.; Choi, Y.J.; Cicerchi, C.; Kanbay, M.; Roncal-Jimenez, C.A.; Ishimoto, T.; Li, N.; Marek, G.; Duranay, M.; et al. Uric acid induces hepatic steatosis by generation of mitochondrial oxidative stress: potential role in fructose-dependent and -independent fatty liver. J. Biol. Chem. 2012, 287, 40732–40744. [Google Scholar] [CrossRef] [PubMed]
- Choi, Y.J.; Shin, H.S.; Choi, H.S.; Park, J.W.; Jo, I.; Oh, E.S.; Lee, K.Y.; Lee, B.H.; Johnson, R.J.; Kang, D.H. Uric acid induces fat accumulation via generation of endoplasmic reticulum stress and SREBP-1c activation in hepatocytes. Lab. Investig. 2014, 94, 1114–1125. [Google Scholar] [CrossRef] [PubMed]
- Wan, X.; Xu, C.; Lin, Y.; Lu, C.; Li, D.; Sang, J.; He, H.; Liu, X.; Li, Y.; Yu, C. Uric acid regulates hepatic steatosis and insulin resistance through the NLRP3 inflammasome-dependent mechanism. J. Hepatol. 2015. [Google Scholar] [CrossRef]
- Ackerman, Z.; Oron-Herman, M.; Grozovski, M.; Rosenthal, T.; Pappo, O.; Link, G.; Sela, B.A. Fructose-induced fatty liver disease: hepatic effects of blood pressure and plasma triglyceride reduction. Hypertension 2005, 45, 1012–1018. [Google Scholar] [CrossRef] [PubMed]
- Lanaspa, M.A.; Sanchez-Lozada, L.G.; Cicerchi, C.; Li, N.; Roncal-Jimenez, C.A.; Ishimoto, T.; Le, M.; Garcia, G.E.; Thomas, J.B.; Rivard, C.J.; et al. Uric acid stimulates fructokinase and accelerates fructose metabolism in the development of fatty liver. PLoS ONE 2012, 7, e47948. [Google Scholar] [CrossRef] [PubMed]
- Pagliassotti, M.J. Endoplasmic reticulum stress in nonalcoholic fatty liver disease. Annu. Rev. Nutr. 2012, 32, 17–33. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Chen, X.; Zhu, R.M.; Zhang, Y.; Yu, T.; Wang, H.; Zhao, H.; Zhao, M.; Ji, Y.L.; Chen, Y.H.; et al. Endoplasmic reticulum stress is involved in hepatic SREBP-1c activation and lipid accumulation in fructose-fed mice. Toxicol. Lett. 2012, 212, 229–240. [Google Scholar] [CrossRef] [PubMed]
- De Oliveira, E.P.; Burini, R.C. High plasma uric acid concentration: causes and consequences. Diabetol. Metab. Syndr. 2012, 4, 12. [Google Scholar] [CrossRef] [PubMed]
- Martinon, F.; Petrilli, V.; Mayor, A.; Tardivel, A.; Tschopp, J. Gout-associated uric acid crystals activate the NALP3 inflammasome. Nature 2006, 440, 237–241. [Google Scholar] [CrossRef] [PubMed]
- Bugianesi, E.; Manzini, P.; D’Antico, S.; Vanni, E.; Longo, F.; Leone, N.; Massarenti, P.; Piga, A.; Marchesini, G.; Rizzetto, M. Relative contribution of iron burden, HFE mutations, and insulin resistance to fibrosis in nonalcoholic fatty liver. Hepatology 2004, 39, 179–187. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Real, J.M.; Ricart-Engel, W.; Arroyo, E.; Balanca, R.; Casamitjana-Abella, R.; Cabrero, D.; Fernandez-Castaner, M.; Soler, J. Serum ferritin as a component of the insulin resistance syndrome. Diabetes Care 1998, 21, 62–68. [Google Scholar] [CrossRef] [PubMed]
- Ford, E.S.; Cogswell, M.E. Diabetes and serum ferritin concentration among U.S. adults. Diabetes Care 1999, 22, 1978–1983. [Google Scholar] [CrossRef] [PubMed]
- Salonen, J.T.; Tuomainen, T.P.; Nyyssonen, K.; Lakka, H.M.; Punnonen, K. Relation between iron stores and non-insulin dependent diabetes in men: Case-control study. BMJ 1998, 317, 727. [Google Scholar] [CrossRef] [PubMed]
- Kim, C.W.; Chang, Y.; Sung, E.; Shin, H.; Ryu, S. Serum ferritin levels predict incident non-alcoholic fatty liver disease in healthy Korean men. Metabolism 2012, 61, 1182–1188. [Google Scholar] [CrossRef] [PubMed]
- Gillum, R.F. Association of serum ferritin and indices of body fat distribution and obesity in Mexican American men—The third national health and nutrition examination survey. Int. J. Obes. Relat. Metab. Disord. 2001, 25, 639–645. [Google Scholar] [CrossRef] [PubMed]
- Piperno, A.; Trombini, P.; Gelosa, M.; Mauri, V.; Pecci, V.; Vergani, A.; Salvioni, A.; Mariani, R.; Mancia, G. Increased serum ferritin is common in men with essential hypertension. J. Hypertens. 2002, 20, 1513–1518. [Google Scholar] [CrossRef] [PubMed]
- Williams, M.J.; Poulton, R.; Williams, S. Relationship of serum ferritin with cardiovascular risk factors and inflammation in young men and women. Atherosclerosis 2002, 165, 179–184. [Google Scholar] [CrossRef]
- Iwasaki, T.; Nakajima, A.; Yoneda, M.; Yamada, Y.; Mukasa, K.; Fujita, K.; Fujisawa, N.; Wada, K.; Terauchi, Y. Serum ferritin is associated with visceral fat area and subcutaneous fat area. Diabetes Care 2005, 28, 2486–2491. [Google Scholar] [CrossRef] [PubMed]
- Alam, F.; Memon, A.S.; Fatima, S.S. Increased Body Mass Index may lead to Hyperferritinemia Irrespective of Body Iron Stores. Pak. J. Med. Sci. 2015, 31, 1521–1526. [Google Scholar] [PubMed]
- Ellervik, C.; Marott, J.L.; Tybjaerg-Hansen, A.; Schnohr, P.; Nordestgaard, B.G. Total and cause-specific mortality by moderately and markedly increased ferritin concentrations: General population study and metaanalysis. Clin. Chem. 2014, 60, 1419–1428. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Real, J.M.; Lopez-Bermejo, A.; Ricart, W. Cross-talk between iron metabolism and diabetes. Diabetes 2002, 51, 2348–2354. [Google Scholar] [CrossRef] [PubMed]
- Zelber-Sagi, S.; Nitzan-Kaluski, D.; Halpern, Z.; Oren, R. NAFLD and hyperinsulinemia are major deter4.minants of serum ferritin levels. J. Hepatol. 2007, 46, 700–707. [Google Scholar] [CrossRef] [PubMed]
- Moirand, R.; Mortaji, A.M.; Loreal, O.; Paillard, F.; Brissot, P.; Deugnier, Y. A new syndrome of liver iron overload with normal transferrin saturation. Lancet 1997, 349, 95–97. [Google Scholar] [CrossRef]
- Corradini, E.; Meynard, D.; Wu, Q.; Chen, S.; Ventura, P.; Pietrangelo, A.; Babitt, J.L. Serum and liver iron differently regulate the bone morphogenetic protein 6 (BMP6)-SMAD signaling pathway in mice. Hepatology 2011, 54, 273–284. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vecchi, C.; Montosi, G.; Zhang, K.; Lamberti, I.; Duncan, S.A.; Kaufman, R.J.; Pietrangelo, A. ER stress controls iron metabolism through induction of hepcidin. Science 2009, 325, 877–880. [Google Scholar] [CrossRef] [PubMed]
- Fargion, S.; Valenti, L.; Fracanzani, A.L. Beyond hereditary hemochromatosis: New insights into the relationship between iron overload and chronic liver diseases. Dig. Liver Dis. 2011, 43, 89–95. [Google Scholar] [CrossRef] [PubMed]
- Murotomi, K.; Arai, S.; Uchida, S.; Endo, S.; Mitsuzumi, H.; Tabei, Y.; Yoshida, Y.; Nakajima, Y. Involvement of splenic iron accumulation in the development of nonalcoholic steatohepatitis in Tsumura Suzuki Obese Diabetes mice. Sci. Rep. 2016, 6, 22476. [Google Scholar] [CrossRef] [PubMed]
- Corradini, E.; Pietrangelo, A. Iron and steatohepatitis. J. Gastroenterol. Hepatol. 2012, 27, 42–46. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dongiovanni, P.; Fracanzani, A.L.; Fargion, S.; Valenti, L. Iron in fatty liver and in the metabolic syndrome: a promising therapeutic target. J. Hepatol. 2011, 55, 920–932. [Google Scholar] [CrossRef] [PubMed]
- Boga, S.; Alkim, H.; Alkim, C.; Koksal, A.R.; Bayram, M.; Yilmaz Ozguven, M.B.; Tekin Neijmann, S. The relationship of serum hemojuvelin and hepcidin levels with iron overload in nonalcoholic fatty liver disease. J. Gastrointest. Liver Dis. 2015, 24, 293–300. [Google Scholar]
- Senates, E.; Yilmaz, Y.; Colak, Y.; Ozturk, O.; Altunoz, M.E.; Kurt, R.; Ozkara, S.; Aksaray, S.; Tuncer, I.; Ovunc, A.O. Serum levels of hepcidin in patients with biopsy-proven nonalcoholic fatty liver disease. Metab. Syndr. Relat. Disord. 2011, 9, 287–290. [Google Scholar] [CrossRef] [PubMed]
- Demircioglu, F.; Gorunmez, G.; Dagistan, E.; Goksugur, S.B.; Bekdas, M.; Tosun, M.; Kizildag, B.; Kismet, E. Serum hepcidin levels and iron metabolism in obese children with and without fatty liver: Case-control study. Eur. J. Pediatr. 2014, 173, 947–951. [Google Scholar] [CrossRef] [PubMed]
- Neri, S.; Pulvirenti, D.; Signorelli, S.; Ignaccolo, L.; Tsami, A.; Mauceri, B.; Misseri, M.; Interlandi, D.; Cutuli, N.; Castellino, P. The HFE gene heterozygosis H63D: A cofactor for liver damage in patients with steatohepatitis? Epidemiological and clinical considerations. Intern. Med. J. 2008, 38, 254–258. [Google Scholar] [CrossRef] [PubMed]
- Valenti, L.; Dongiovanni, P.; Fracanzani, A.L.; Fargion, S. HFE mutations in nonalcoholic fatty liver disease. Hepatology 2008, 47, 1794–1795. [Google Scholar] [CrossRef] [PubMed]
- McClain, D.A.; Abraham, D.; Rogers, J.; Brady, R.; Gault, P.; Ajioka, R.; Kushner, J.P. High prevalence of abnormal glucose homeostasis secondary to decreased insulin secretion in individuals with hereditary haemochromatosis. Diabetologia 2006, 49, 1661–1669. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.S.; Koh, I.U.; Lee, H.J.; Kim, W.H.; Song, J. Effects of excess dietary iron and fat on glucose and lipid metabolism. J. Nutr. Biochem. 2013, 24, 1634–1644. [Google Scholar] [CrossRef] [PubMed]
- Dongiovanni, P.; Ruscica, M.; Rametta, R.; Recalcati, S.; Steffani, L.; Gatti, S.; Girelli, D.; Cairo, G.; Magni, P.; Fargion, S.; et al. Dietary iron overload induces visceral adipose tissue insulin resistance. Am. J. Pathol. 2013, 182, 2254–2263. [Google Scholar] [CrossRef] [PubMed]
- Vecchi, C.; Montosi, G.; Garuti, C.; Corradini, E.; Sabelli, M.; Canali, S.; Pietrangelo, A. Gluconeogenic signals regulate iron homeostasis via hepcidin in mice. Gastroenterology 2014, 146, 1060–1069. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marra, F.; Bertolani, C. Adipokines in liver diseases. Hepatology 2009, 50, 957–969. [Google Scholar] [CrossRef] [PubMed]
- Bekri, S.; Gual, P.; Anty, R.; Luciani, N.; Dahman, M.; Ramesh, B.; Iannelli, A.; Staccini-Myx, A.; Casanova, D.; Ben Amor, I.; et al. Increased adipose tissue expression of hepcidin in severe obesity is independent from diabetes and NASH. Gastroenterology 2006, 131, 788–796. [Google Scholar] [CrossRef] [PubMed]
- Green, A.; Basile, R.; Rumberger, J.M. Transferrin and iron induce insulin resistance of glucose transport in adipocytes. Metabolism 2006, 55, 1042–1045. [Google Scholar] [CrossRef] [PubMed]
- Item, F.; Konrad, D. Visceral fat and metabolic inflammation: the portal theory revisited. Obes. Rev. 2012, 13, 30–39. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fargion, S.; Mattioli, M.; Fracanzani, A.L.; Sampietro, M.; Tavazzi, D.; Fociani, P.; Taioli, E.; Valenti, L.; Fiorelli, G. Hyperferritinemia, iron overload, and multiple metabolic alterations identify patients at risk for nonalcoholic steatohepatitis. Am. J. Gastroenterol. 2001, 96, 2448–2455. [Google Scholar] [CrossRef] [PubMed]
- Manousou, P.; Kalambokis, G.; Grillo, F.; Watkins, J.; Xirouchakis, E.; Pleguezuelo, M.; Leandro, G.; Arvaniti, V.; Germani, G.; Patch, D.; et al. Serum ferritin is a discriminant marker for both fibrosis and inflammation in histologically proven non-alcoholic fatty liver disease patients. Liver Int. 2011, 31, 730–739. [Google Scholar] [CrossRef] [PubMed]
- Kowdley, K.V.; Belt, P.; Wilson, L.A.; Yeh, M.M.; Neuschwander-Tetri, B.A.; Chalasani, N.; Sanyal, A.J.; Nelson, J.E.; Network, N.C.R. Serum ferritin is an independent predictor of histologic severity and advanced fibrosis in patients with nonalcoholic fatty liver disease. Hepatology 2012, 55, 77–85. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.S.; Jung, E.S.; Hur, W.; Bae, S.H.; Choi, J.Y.; Song, M.J.; Kim, C.W.; Jo, S.H.; Lee, C.D.; Lee, Y.S.; et al. Noninvasive predictors of nonalcoholic steatohepatitis in Korean patients with histologically proven nonalcoholic fatty liver disease. Clin. Mol. Hepatol. 2013, 19, 120–130. [Google Scholar] [CrossRef] [PubMed]
- Angulo, P.; George, J.; Day, C.P.; Vanni, E.; Russell, L.; de la Cruz, A.C.; Liaquat, H.; Mezzabotta, L.; Lee, E.; Bugianesi, E. Serum ferritin levels lack diagnostic accuracy for liver fibrosis in patients with nonalcoholic fatty liver disease. Clin. Gastroenterol. Hepatol. 2014, 12, 1163–1169. [Google Scholar] [CrossRef] [PubMed]
- Yoneda, M.; Thomas, E.; Sumida, Y.; Imajo, K.; Eguchi, Y.; Hyogo, H.; Fujii, H.; Ono, M.; Kawaguchi, T.; Schiff, E.R. Clinical usage of serum ferritin to assess liver fibrosis in patients with non-alcoholic fatty liver disease: Proceed with caution. Hepatol. Res. 2014, 44, E499–E502. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, A.; Yoneda, M.; Sumida, Y.; Eguchi, Y.; Fujii, H.; Hyogo, H.; Ono, M.; Suzuki, Y.; Kawaguchi, T.; Aoki, N.; et al. Modification of a simple clinical scoring system as a diagnostic screening tool for non-alcoholic steatohepatitis in Japanese patients with non-alcoholic fatty liver disease. J. Diabetes Investig. 2013, 4, 651–658. [Google Scholar] [CrossRef] [PubMed]
- Cales, P.; Boursier, J.; Oberti, F.; Hubert, I.; Gallois, Y.; Rousselet, M.C.; Dib, N.; Moal, V.; Macchi, L.; Chevailler, A.; et al. FibroMeters: A family of blood tests for liver fibrosis. Gastroenterol. Clin. Biol. 2008, 32, 40–51. [Google Scholar] [CrossRef] [Green Version]
- Valenti, L.; Canavesi, E.; Galmozzi, E.; Dongiovanni, P.; Rametta, R.; Maggioni, P.; Maggioni, M.; Fracanzani, A.L.; Fargion, S. β-Globin mutations are associated with parenchymal siderosis and fibrosis in patients with non-alcoholic fatty liver disease. J. Hepatol. 2010, 53, 927–933. [Google Scholar] [CrossRef] [PubMed]
- Facchini, F.S.; Hua, N.W.; Stoohs, R.A. Effect of iron depletion in carbohydrate-intolerant patients with clinical evidence of nonalcoholic fatty liver disease. Gastroenterology 2002, 122, 931–939. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Real, J.M.; Penarroja, G.; Castro, A.; Garcia-Bragado, F.; Hernandez-Aguado, I.; Ricart, W. Blood letting in high-ferritin type 2 diabetes: effects on insulin sensitivity and β-cell function. Diabetes 2002, 51, 1000–1004. [Google Scholar] [CrossRef] [PubMed]
- Valenti, L.; Fracanzani, A.L.; Dongiovanni, P.; Bugianesi, E.; Marchesini, G.; Manzini, P.; Vanni, E.; Fargion, S. Iron depletion by phlebotomy improves insulin resistance in patients with nonalcoholic fatty liver disease and hyperferritinemia: evidence from a case-control study. Am. J. Gastroenterol. 2007, 102, 1251–1258. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.C.; Huang, Y.F.; Wang, J.D. Hyperferritinemia and hyperuricemia may be associated with liver function abnormality in obese adolescents. PLoS ONE 2012, 7, e48645. [Google Scholar] [CrossRef] [PubMed]
- Ghio, A.J.; Ford, E.S.; Kennedy, T.P.; Hoidal, J.R. The association between serum ferritin and uric acid in humans. Free Radic. Res. 2005, 39, 337–342. [Google Scholar] [CrossRef] [PubMed]
- Mainous, A.G., 3rd; Knoll, M.E.; Everett, C.J.; Matheson, E.M.; Hulihan, M.M.; Grant, A.M. Uric acid as a potential cue to screen for iron overload. J. Am. Board Fam. Med. 2011, 24, 415–421. [Google Scholar] [CrossRef] [PubMed]


© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lombardi, R.; Pisano, G.; Fargion, S. Role of Serum Uric Acid and Ferritin in the Development and Progression of NAFLD. Int. J. Mol. Sci. 2016, 17, 548. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms17040548
Lombardi R, Pisano G, Fargion S. Role of Serum Uric Acid and Ferritin in the Development and Progression of NAFLD. International Journal of Molecular Sciences. 2016; 17(4):548. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms17040548
Chicago/Turabian StyleLombardi, Rosa, Giuseppina Pisano, and Silvia Fargion. 2016. "Role of Serum Uric Acid and Ferritin in the Development and Progression of NAFLD" International Journal of Molecular Sciences 17, no. 4: 548. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms17040548