
Confocal Spectroscopy to Study Dimerization, Oligomerization and Aggregation of Proteins: A Practical Guide

Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
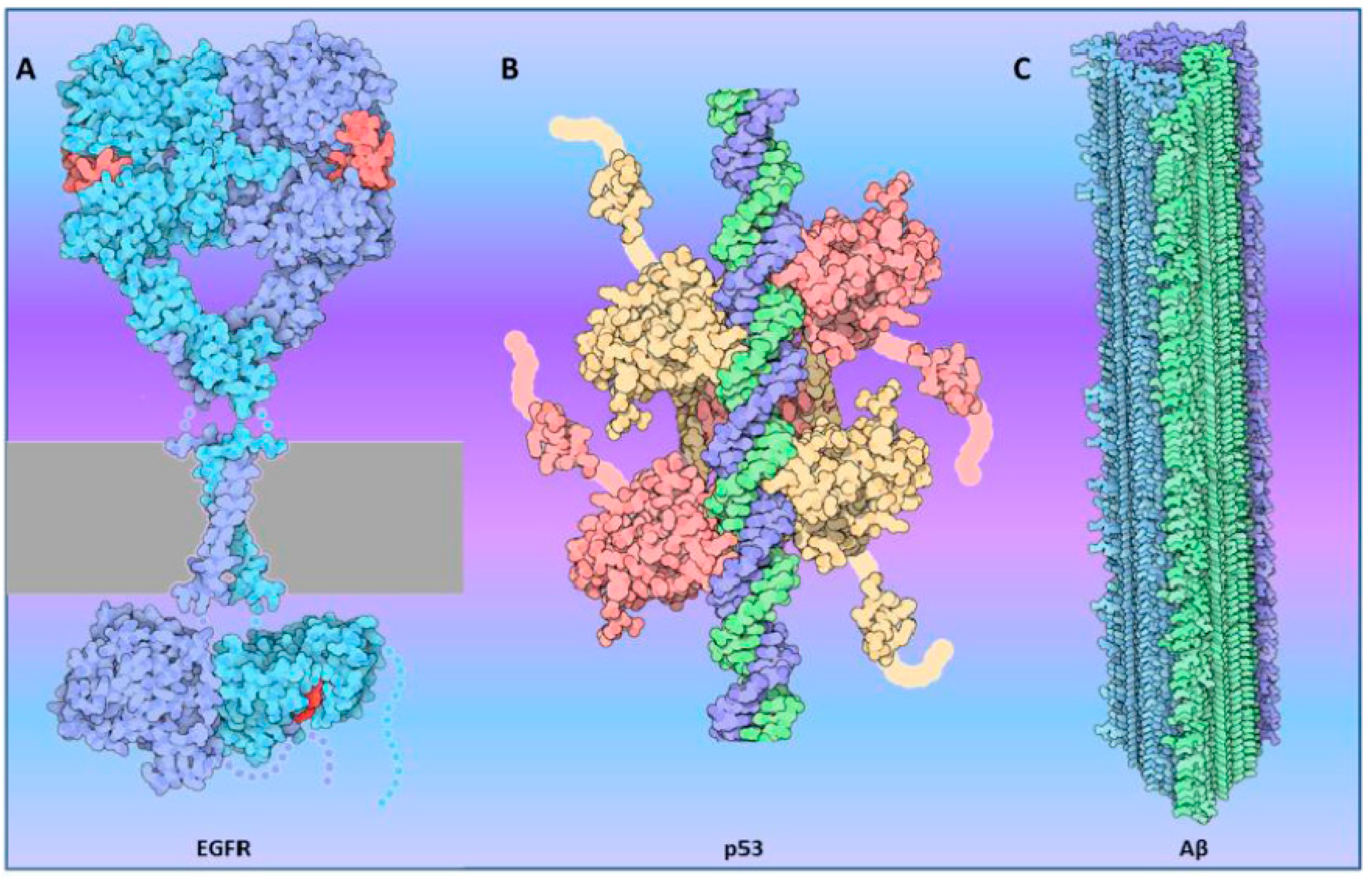
1.1. Oligomerization: Function and Dysfunction
1.2. Single Molecule Detection and Confocal Spectroscopy for the Study of Protein Self-Assembly
2. Characterization of the Brightness Parameter in Confocal Spectroscopy
2.1. Theory
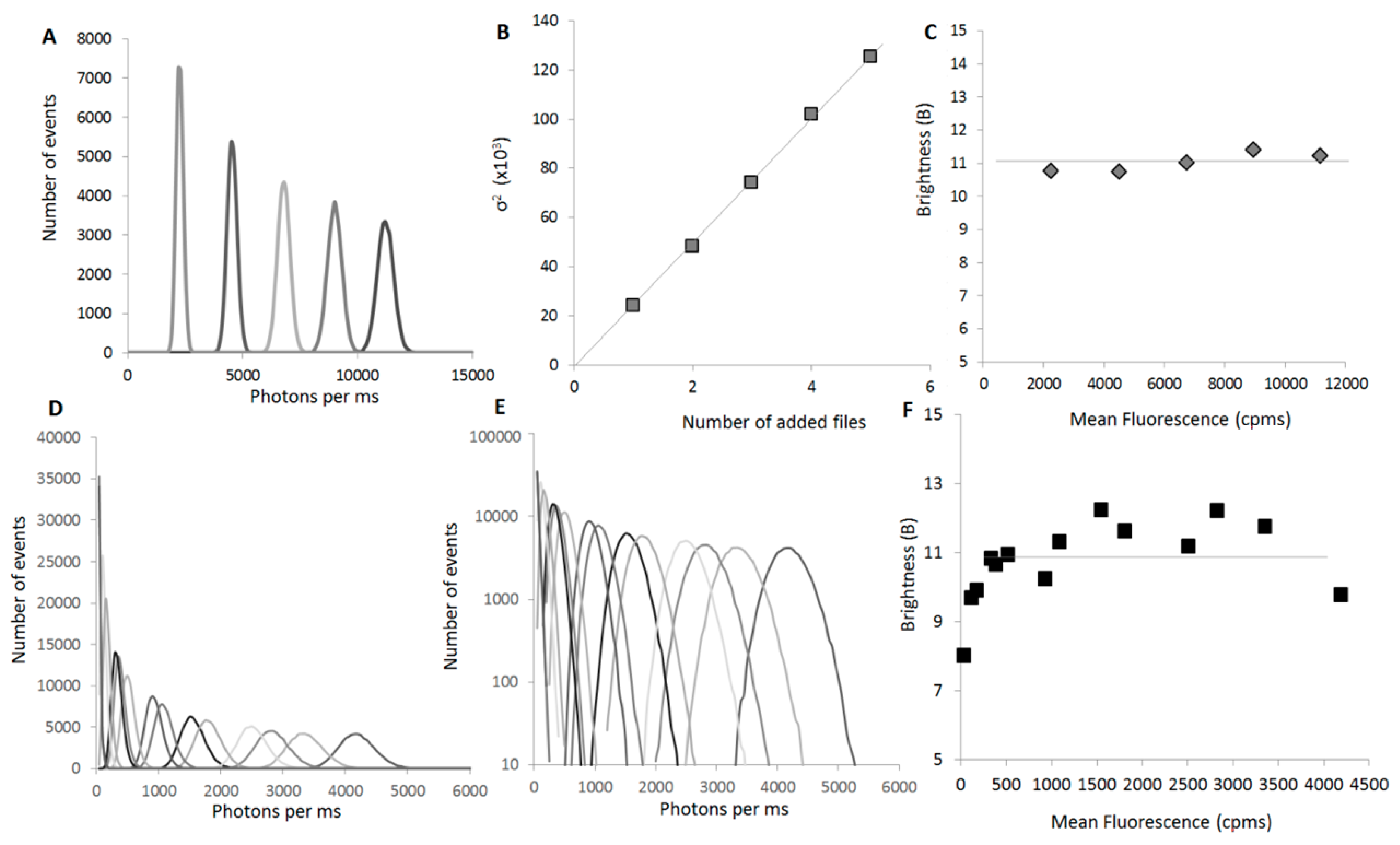
2.2. Experimental Measure of the Size of Oligomers
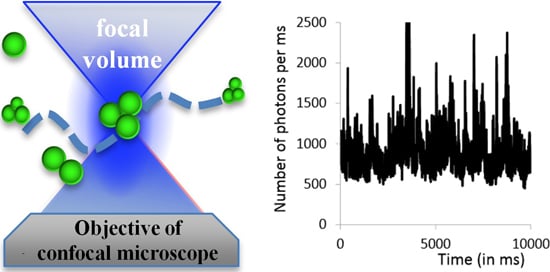
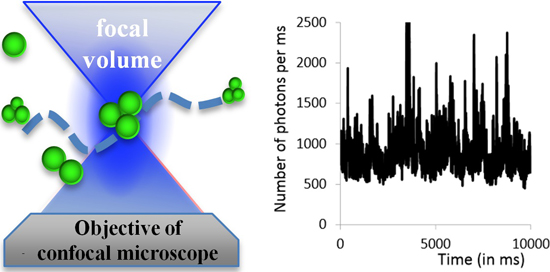
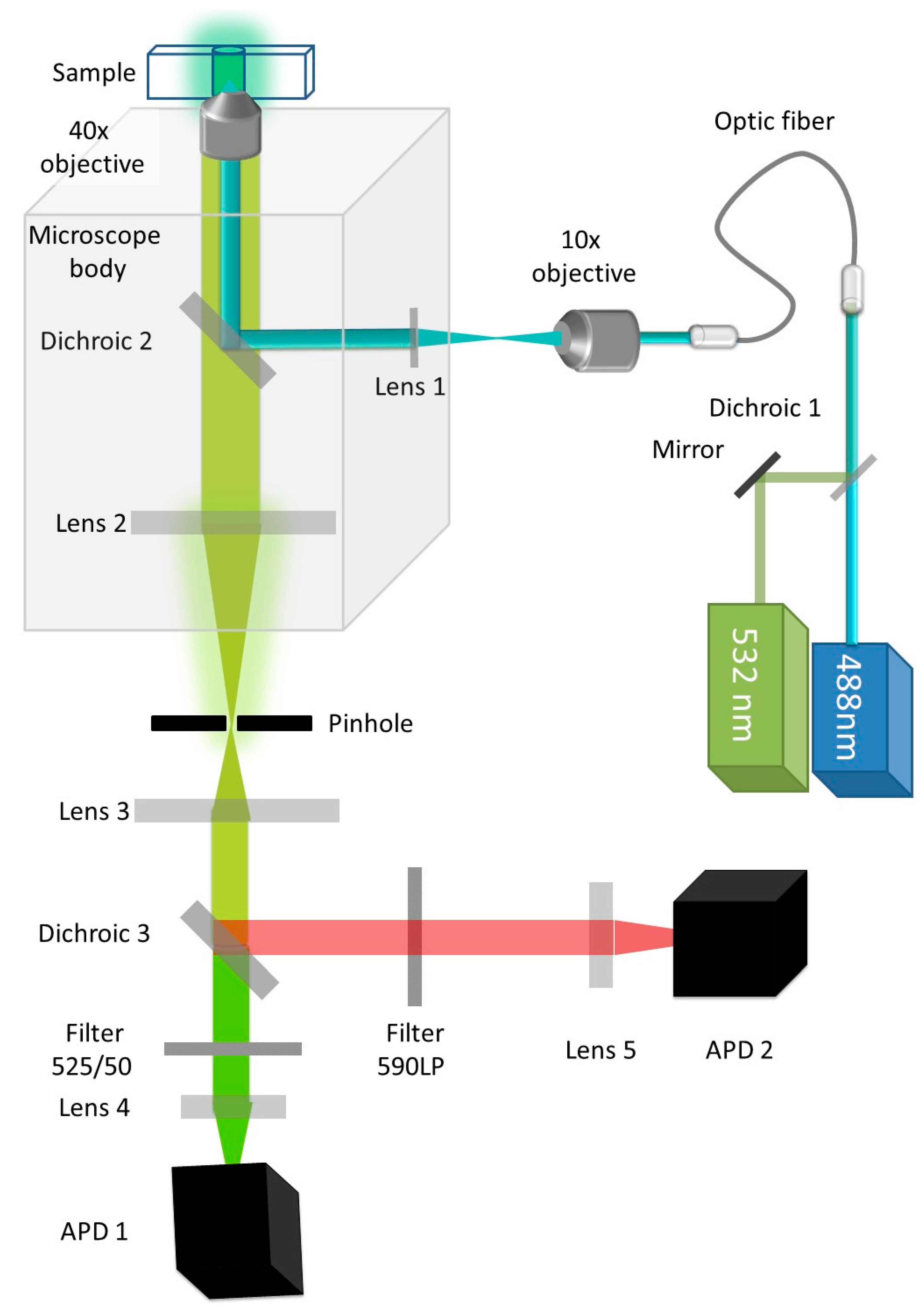
2.2.1. Experimental Setup
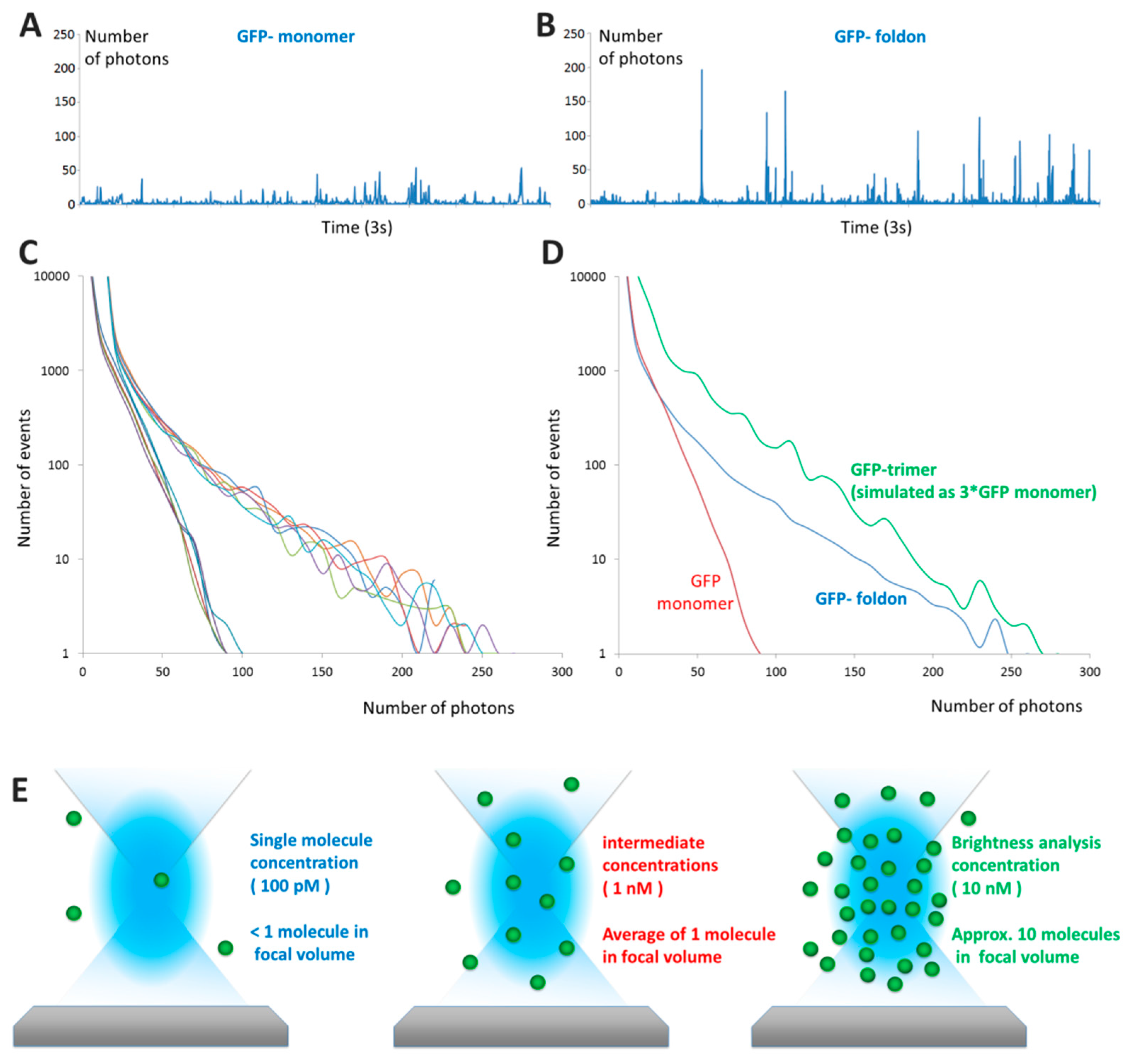
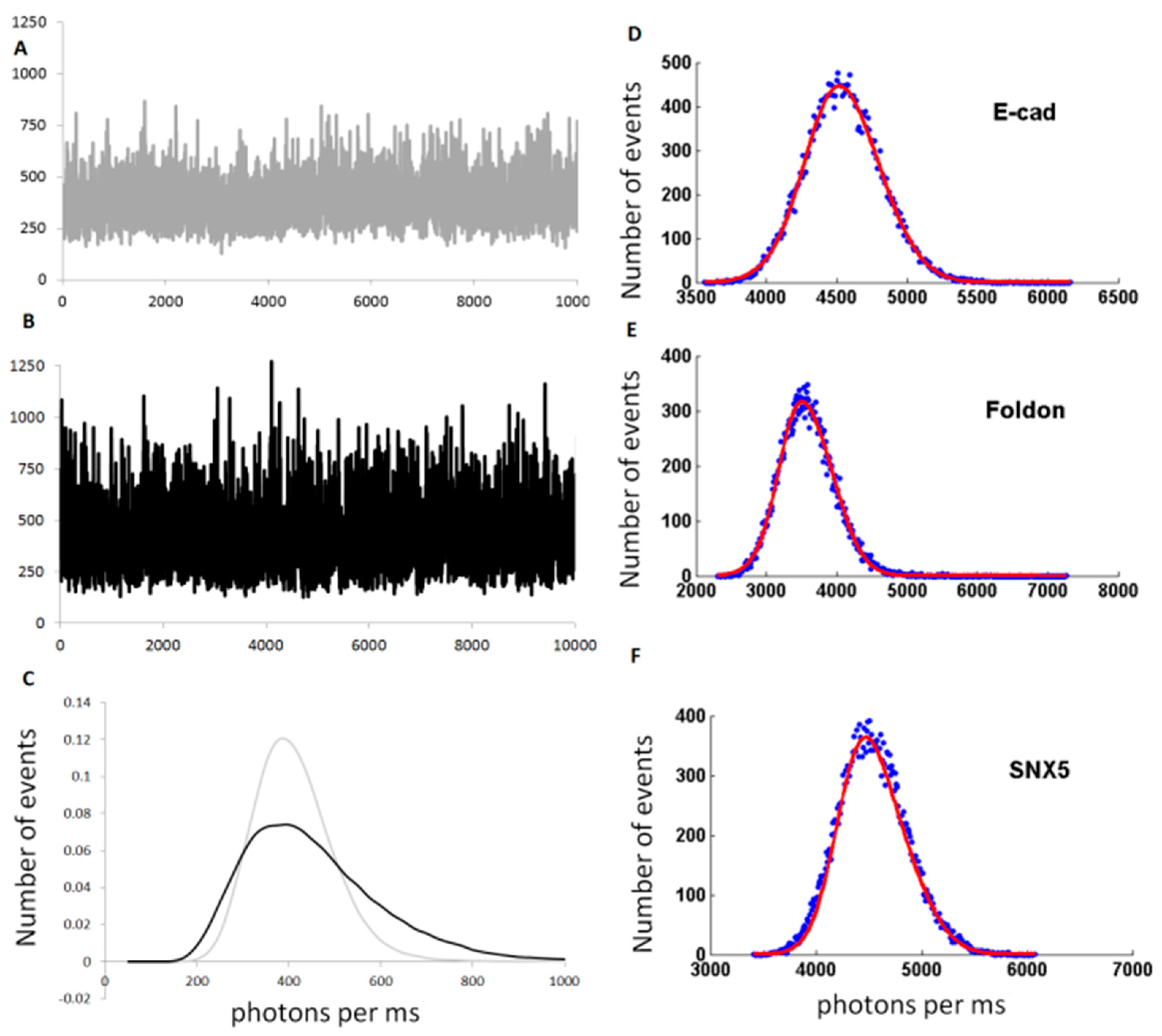
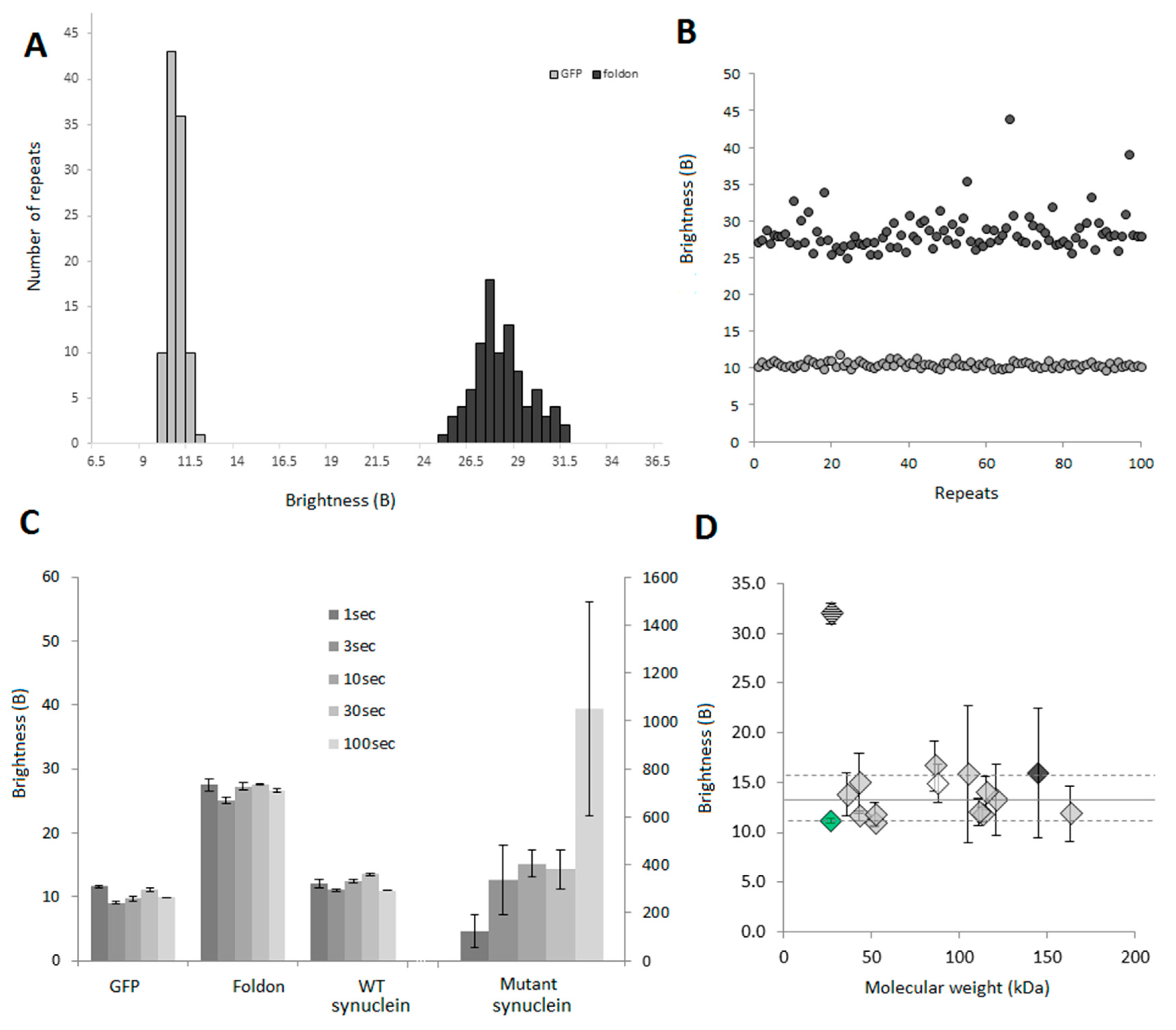
2.2.2. Examples: Monomers, Dimers and Trimers
2.3. A High-Throughput Screening Tool
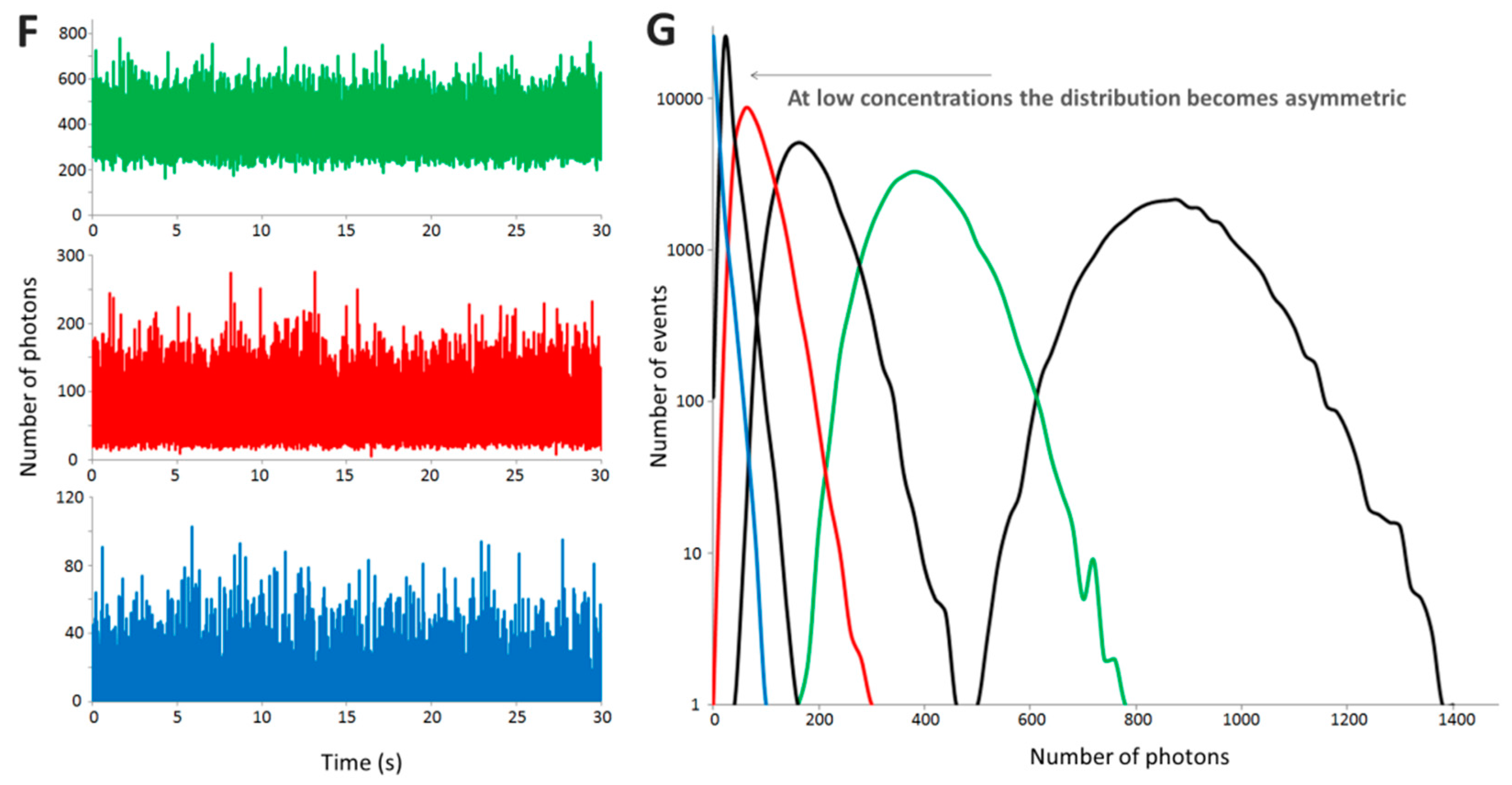
2.3.1. B Is a Concentration-Independent Parameter for a Wide Range of Concentrations
2.3.2. B Is a Highly Reproducible Parameter
2.3.3. B Can Be Acquired Rapidly
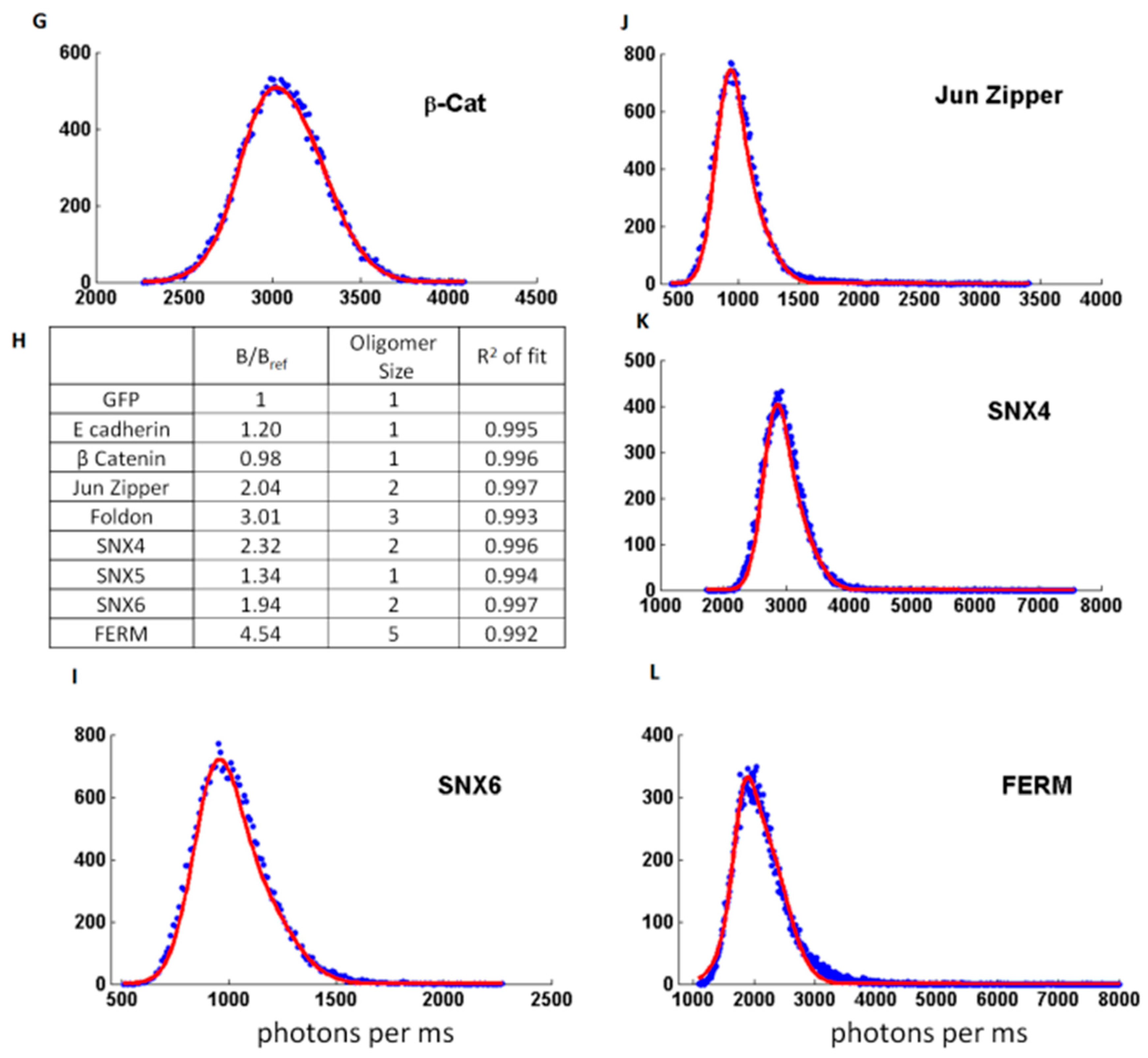
2.3.4. B Is Mostly Unaffected by the Size of the Protein
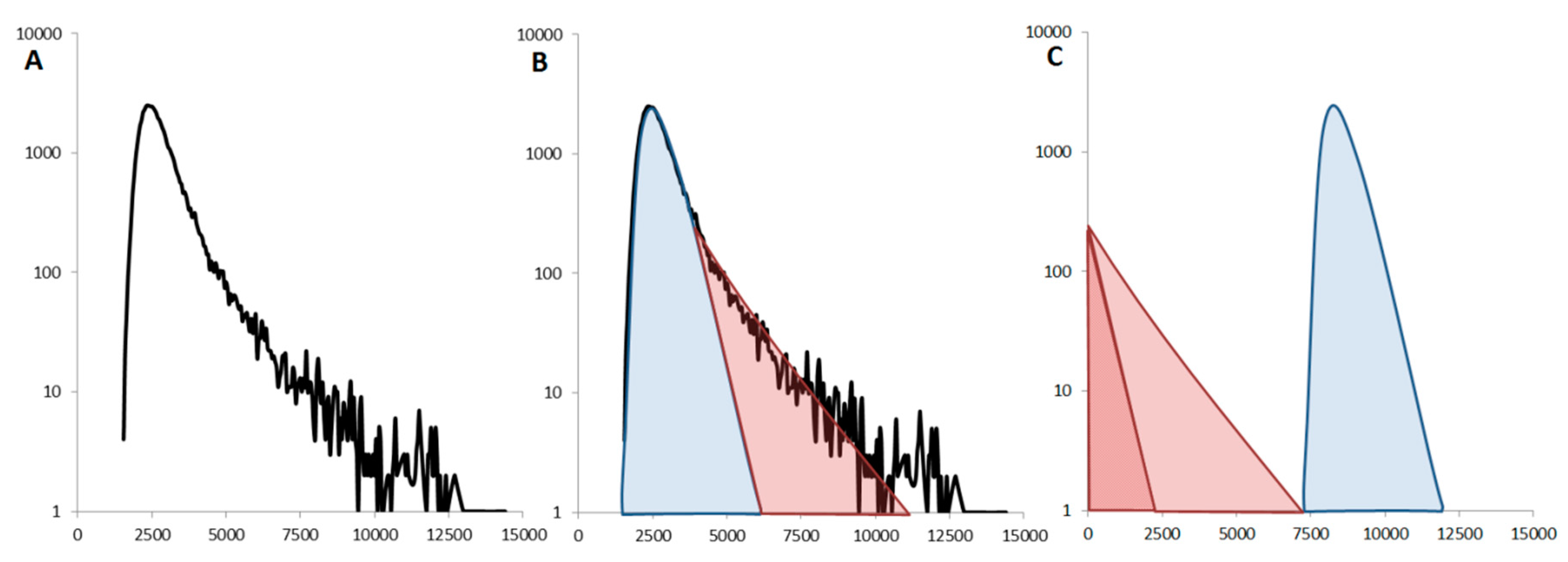
2.3.5. Oligomers or Aggregates?
3. Examples of Aggregation Assays
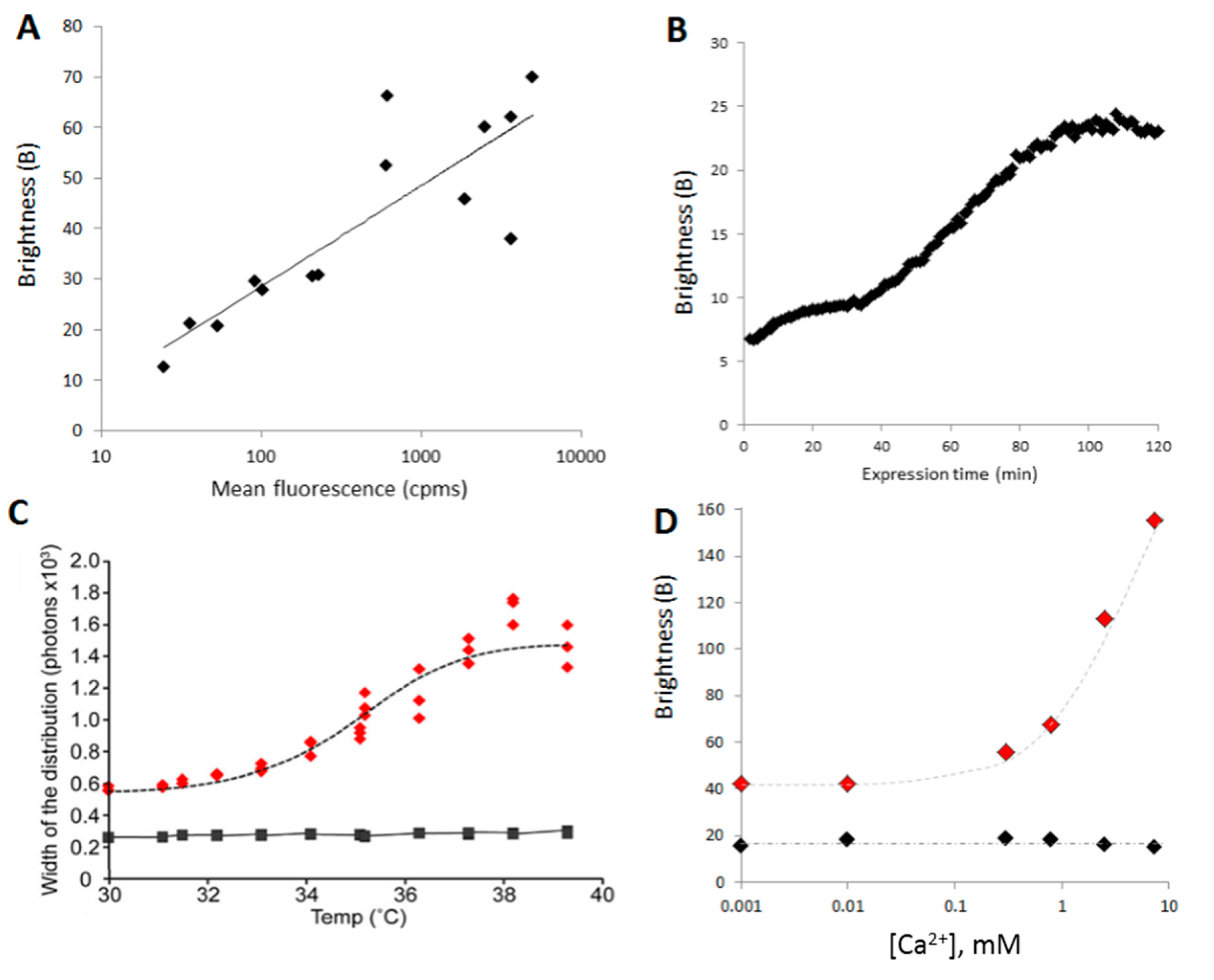
3.1. Aggregation as a Function of Expression
3.2. Time Course of Aggregation
3.3. Determination of Thermal Stability
3.4. Small Molecule Inhibitors of Aggregation
4. Discussions
5. Conclusions
Supplementary Materials
Conflicts of Interest
References
- Ali, M.H.; Imperiali, B. Protein oligomerization: How and why. Bioorg. Med. Chem. 2005, 13, 5013–5020. [Google Scholar] [CrossRef] [PubMed]
- Marianayagam, N.J.; Sunde, M.; Matthews, J.M. The power of two: Protein dimerization in biology. Trends Biochem. Sci. 2004, 29, 618–625. [Google Scholar] [CrossRef] [PubMed]
- Bae, J.H.; Schlessinger, J. Asymmetric tyrosine kinase arrangements in activation or autophosphorylation of receptor tyrosine kinases. Mol. Cells 2010, 29, 443–448. [Google Scholar] [CrossRef] [PubMed]
- Lemmon, M.A.; Schlessinger, J. Cell signaling by receptor tyrosine kinases. Cell 2010, 141, 1117–1134. [Google Scholar] [CrossRef] [PubMed]
- Niemann, H.H. Structural basis of MET receptor dimerization by the bacterial invasion protein InlB and the HGF/SF splice variant NK1. Biochim. Biophys. Acta (BBA)—Prot. Prot. 2013, 1834, 2195–2204. [Google Scholar] [CrossRef] [PubMed]
- Sarabipour, S.; Hristova, K. Mechanism of FGF receptor dimerization and activation. Nat. Commun. 2016, 7. [Google Scholar] [CrossRef] [PubMed]
- Ferguson, K.M. Structure-based view of epidermal growth factor receptor regulation. Annu. Rev. Biophys. 2008, 37, 353–373. [Google Scholar] [CrossRef] [PubMed]
- Endres, N.F.; Engel, K.; Das, R.; Kovacs, E.; Kuriyan, J. Regulation of the catalytic activity of the EGF receptor. Curr. Opin. Struct. Biol. 2011, 21, 777–784. [Google Scholar] [CrossRef] [PubMed]
- Lemmon, M.A.; Schlessinger, J.; Ferguson, K.M. The EGFR family: Not so prototypical receptor tyrosine kinases. Cold Spring Harb. Perspect. Biol. 2014, 6. [Google Scholar] [CrossRef] [PubMed]
- Tynan, C.J.; Schiavo, V.L.; Zanetti-Domingues, L.; Needham, S.R.; Roberts, S.K.; Hirsch, M.; Rolfe, D.J.; Korovesis, D.; Clarke, D.T.; Martin-Fernandez, M.L. A tale of the epidermal growth factor receptor: The quest for structural resolution on cells. Methods 2015, 95, 86–93. [Google Scholar] [CrossRef] [PubMed]
- Zanetti-Domingues, L.C.; Hirsch, M.; Tynan, C.J.; Rolfe, D.J.; Boyadzhiev, T.V.; Scherer, K.M.; Clarke, D.T.; Martin-Fernandez, M.L.; Needham, S.R. Determining the geometry of oligomers of the human epidermal growth factor family on cells with 7 nm resolution. Prog. Biophys. Mol. Biol. 2015, 118, 139–152. [Google Scholar] [CrossRef] [PubMed]
- Tao, Y.; Pinzi, V.; Bourhis, J.; Deutsch, E. Mechanisms of disease: Signaling of the insulin-like growth factor 1 receptor pathway—Therapeutic perspectives in cancer. Nat. Clin. Prac. Oncol. 2007, 4, 591–602. [Google Scholar] [CrossRef] [PubMed]
- Roskoski, R., Jr. ErbB/HER protein-tyrosine kinases: Structures and small molecule inhibitors. Pharmacol. Res. 2014, 87, 42–59. [Google Scholar] [CrossRef] [PubMed]
- Hu, H.; Liu, Y.; Jiang, T. Mutation-introduced dimerization of receptor tyrosine kinases: From protein structure aberrations to carcinogenesis. Tumour Biol. 2015, 36, 1423–1428. [Google Scholar] [CrossRef] [PubMed]
- So, C.W.; Cleary, M.L. Dimerization: A versatile switch for oncogenesis. Blood 2004, 104, 919–922. [Google Scholar] [CrossRef] [PubMed]
- Brooks, A.J.; Dai, W.; O’Mara, M.L.; Abankwa, D.; Chhabra, Y.; Pelekanos, R.A.; Gardon, O.; Tunny, K.A.; Blucher, K.M.; Morton, C.J.; et al. Mechanism of activation of protein kinase JAK2 by the growth hormone receptor. Science 2014, 344. [Google Scholar] [CrossRef] [PubMed]
- Goodsell, D.; Dutta, S.; Zardecki, C.; Voigt, M.; Berman, H.; Burley, S. The RCSB PDB "Molecule of the Month": Inspiring a molecular view of biology. PLoS Biol. 2015, 13, e1002140. [Google Scholar] [CrossRef] [PubMed]
- Schlierf, B.; Ludwig, A.; Klenovsek, K.; Wegner, M. Cooperative binding of Sox10 to DNA: Requirements and consequences. Nucleic Acids Res. 2002, 30, 5509–5516. [Google Scholar] [CrossRef] [PubMed]
- Perumal, K.; Dirr, H.W.; Fanucchi, S. A single amino acid in the hinge loop region of the FOXP forkhead domain is significant for dimerisation. Protein J. 2015, 34, 111–121. [Google Scholar] [CrossRef] [PubMed]
- Hou, C.; Tsodikov, O.V. Structural basis for dimerization and DNA binding of transcription factor FLI1. Biochemistry 2015, 54, 7365–7374. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.H.; Jankowski, A.; Cheah, K.S.; Prabhakar, S.; Jauch, R. SOXE transcription factors form selective dimers on non-compact DNA motifs through multifaceted interactions between dimerization and high-mobility group domains. Sci. Rep. 2015, 5. [Google Scholar] [CrossRef] [PubMed]
- Kamada, R.; Toguchi, Y.; Nomura, T.; Imagawa, T.; Sakaguchi, K. Tetramer formation of tumor suppressor protein p53: Structure, function, and applications. Biopolymers 2015. [Google Scholar] [CrossRef] [PubMed]
- Hass, M.R.; Liow, H.H.; Chen, X.; Sharma, A.; Inoue, Y.U.; Inoue, T.; Reeb, A.; Martens, A.; Fulbright, M.; Raju, S.; et al. SpDamID: Marking DNA bound by protein complexes identifies Notch-dimer responsive enhancers. Mol. Cell 2015, 59, 685–697. [Google Scholar] [CrossRef] [PubMed]
- Bernard, P.; Tang, P.; Liu, S.; Dewing, P.; Harley, V.R.; Vilain, E. Dimerization of SOX9 is required for chondrogenesis, but not for sex determination. Hum. Mol. Genet. 2003, 12, 1755–1765. [Google Scholar] [CrossRef] [PubMed]
- Peirano, R.I.; Wegner, M. The glial transcription factor Sox10 binds to DNA both as monomer and dimer with different functional consequences. Nucleic Acids Res. 2000, 28, 3047–3055. [Google Scholar] [CrossRef] [PubMed]
- Staab, J.; Riebeling, T.; Koch, V.; Herrmann-Lingen, C.; Meyer, T. The two interfaces of the STAT1 N-terminus exhibit opposite functions in IFNγ-regulated gene expression. Mol. Immunol. 2015, 67, 596–606. [Google Scholar] [CrossRef] [PubMed]
- Marston, N.J.; Jenkins, J.R.; Vousden, K.H. Oligomerisation of full length p53 contributes to the interaction with mdm2 but not HPV E6. Oncogene 1995, 10, 1709–1715. [Google Scholar] [PubMed]
- Grosshans, J.; Schnorrer, F.; Nusslein-Volhard, C. Oligomerisation of Tube and Pelle leads to nuclear localisation of dorsal. Mech. Dev. 1999, 81, 127–138. [Google Scholar] [CrossRef]
- Iida, T.; Mutoh, R.; Onai, K.; Morishita, M.; Furukawa, Y.; Namba, K.; Ishiura, M. Importance of the monomer–dimer–tetramer interconversion of the clock protein KaiB in the generation of circadian oscillations in cyanobacteria. Genes Cells 2015, 20, 173–790. [Google Scholar] [CrossRef] [PubMed]
- Bhambhani, C.; Chang, J.L.; Akey, D.L.; Cadigan, K.M. The oligomeric state of CtBP determines its role as a transcriptional co-activator and co-repressor of Wingless targets. EMBO J. 2011, 30, 2031–2043. [Google Scholar] [CrossRef] [PubMed]
- Khan, M.R.; Li, L.; Pérez-Sánchez, C.; Saraf, A.; Florens, L.; Slaughter, B.D.; Unruh, J.R.; Si, K. Amyloidogenic oligomerization transforms Drosophila Orb2 from a translation repressor to an activator. Cell 2015, 163, 1468–1483. [Google Scholar] [CrossRef] [PubMed]
- Parton, R.G.; Simons, K. The multiple faces of caveolae. Nat. Rev. Mol. Cell Biol. 2007, 8, 185–194. [Google Scholar] [CrossRef] [PubMed]
- Pearse, B.M. Clathrin: A unique protein associated with intracellular transfer of membrane by coated vesicles. Proc. Nat. Acad. Sci. USA 1976, 73, 1255–1259. [Google Scholar] [CrossRef] [PubMed]
- Korn, E.D. Actin polymerization and its regulation by proteins from nonmuscle cells. Physiol. Rev. 1982, 62, 672–737. [Google Scholar] [PubMed]
- Cai, X.; Chen, J.; Xu, H.; Liu, S.; Jiang, Q.X.; Halfmann, R.; Chen, Z.J. Prion-like polymerization underlies signal transduction in antiviral immune defense and inflammasome activation. Cell 2014, 156, 1207–1222. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; He, X.; Zheng, H.; Huang, L.J.; Hou, F.; Yu, Z.; de la Cruz, M.J.; Borkowski, B.; Zhang, X.; Chen, Z.J.; et al. Structural basis for the prion-like MAVS filaments in antiviral innate immunity. eLife 2014, 3. [Google Scholar] [CrossRef] [PubMed]
- Lu, A.; Magupalli, V.G.; Ruan, J.; Yin, Q.; Atianand, M.K.; Vos, M.R.; Schröder, G.F.; Fitzgerald, K.A.; Wu, H.; Egelman, E.H. Unified polymerization mechanism for the assembly of ASC-dependent inflammasomes. Cell 2014, 156, 1193–1206. [Google Scholar] [CrossRef] [PubMed]
- Franklin, B.S.; Bossaller, L.; de Nardo, D.; Ratter, J.M.; Stutz, A.; Engels, G.; Brenker, C.; Nordhoff, M.; Mirandola, S.R.; Al-Amoudi, A.; et al. The adaptor ASC has extracellular and ‘prionoid’ activities that propagate inflammation. Nat. Immunol. 2014, 15, 727–737. [Google Scholar] [CrossRef] [PubMed]
- Ruland, J. Inflammasome: Putting the pieces together. Cell 2014, 156, 1127–1129. [Google Scholar] [CrossRef] [PubMed]
- Fioriti, L.; Myers, C.; Huang, Y.Y.; Li, X.; Stephan, J.S.; Trifilieff, P.; Colnaghi, L.; Kosmidis, S.; Drisaldi, B.; Pavlopoulos, E.; et al. The persistence of hippocampal-based memory requires protein synthesis mediated by the prion-like protein CPEB3. Neuron 2015, 86, 1433–1448. [Google Scholar] [CrossRef] [PubMed]
- Cai, X.; Chen, Z.J. Prion-like polymerization as a signaling mechanism. Trends Immunol. 2014, 35, 622–630. [Google Scholar] [CrossRef] [PubMed]
- Daus, M.L. Disease transmission by misfolded prion-protein isoforms, prion-like amyloids, functional amyloids and the Central Dogma. Biology (Basel) 2016, 5. [Google Scholar] [CrossRef] [PubMed]
- Fraser, P.E. Prions and prion-like proteins. J. Biol. Chem. 2014, 289, 19839–19840. [Google Scholar] [CrossRef] [PubMed]
- Murakami, T.; Ishiguro, N.; Higuchi, K. Transmission of systemic AA amyloidosis in animals. Vet. Pathol. 2014, 51, 363–371. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.; Kim, H.J. Prion-like mechanism in amyotrophic lateral sclerosis: Are protein aggregates the key? Exp. Neurobiol. 2015, 24, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Peggion, C.; Sorgato, M.C.; Bertoli, A. Prions and prion-like pathogens in neurodegenerative disorders. Pathogens 2014, 3, 149–163. [Google Scholar] [CrossRef] [PubMed]
- Goedert, M.; Falcon, B.; Clavaguera, F.; Tolnay, M. Prion-like mechanisms in the pathogenesis of tauopathies and synucleinopathies. Curr. Neurol. Neurosci. Rep. 2014, 14. [Google Scholar] [CrossRef] [PubMed]
- Goedert, M. Alzheimer’s and Parkinson’s diseases: The prion concept in relation to assembled Aβ, tau, and α-synuclein. Science 2015, 349. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Chen, W.D.; Wang, Y.D. β-Amyloid: The key peptide in the pathogenesis of Alzheimer’s disease. Front. Pharmacol. 2015, 6. [Google Scholar] [CrossRef] [PubMed]
- Luna, E.; Luk, K.C. Bent out of shape: α-Synuclein misfolding and the convergence of pathogenic pathways in Parkinson’s disease. FEBS Lett. 2015, 589, 3749–3759. [Google Scholar] [CrossRef] [PubMed]
- Dehay, B.; Bourdenx, M.; Gorry, P.; Przedborski, S.; Vila, M.; Hunot, S.; Singleton, A.; Olanow, C.W.; Merchant, K.M.; Bezard, E.; et al. Targeting α-synuclein for treatment of Parkinson’s disease: Mechanistic and therapeutic considerations. Lancet Neurol. 2015, 14, 855–866. [Google Scholar] [CrossRef]
- Simic, G.; Babić Leko, M.; Wray, S.; Harrington, C.; Delalle, I.; Jovanov-Milošević, N.; Bažadona, D.; Buée, L.; de Silva, R.; di Giovanni, G.; et al. Tau protein hyperphosphorylation and aggregation in Alzheimer’s disease and other tauopathies, and possible neuroprotective strategies. Biomolecules 2016, 6. [Google Scholar] [CrossRef] [PubMed]
- Guerrero-Munoz, M.J.; Gerson, J.; Castillo-Carranza, D.L. Tau oligomers: The toxic player at synapses in Alzheimer’s disease. Front. Cell. Neurosci. 2015, 9. [Google Scholar] [CrossRef] [PubMed]
- Hinton, C.; Antony, H.; Hashimi, S.M.; Munn, A.; Wei, M.Q. Significance of prion and prion-like proteins in cancer development, progression and multi-drug resistance. Curr. Cancer Drug. Targets 2013, 13, 895–904. [Google Scholar] [CrossRef] [PubMed]
- De Oliveira, G.A.; Rangel, L.P.; Costa, D.C.; Silva, J.L. Misfolding, aggregation, and disordered segments in c-Abl and p53 in human cancer. Front. Oncol. 2015, 5. [Google Scholar] [CrossRef] [PubMed]
- Silva, J.L.; de Moura Gallo, C.V.; Costa, D.C.; Rangel, L.P. Prion-like aggregation of mutant p53 in cancer. Trends Biochem. Sci. 2014, 39, 260–267. [Google Scholar] [CrossRef] [PubMed]
- Forget, K.J.; Tremblay, G.; Roucou, X. p53 aggregates penetrate cells and induce the co-aggregation of intracellular p53. PLoS ONE 2013, 8, e69242. [Google Scholar] [CrossRef] [PubMed]
- Laborda, F.; Bolea, E.; Cepriá, G.; Gómez, M.T.; Jiménez, M.S.; Pérez-Arantegui, J.; Castillo, J.R. Detection, characterization and quantification of inorganic engineered nanomaterials: A review of techniques and methodological approaches for the analysis of complex samples. Anal. Chim. Acta 2016, 904, 10–32. [Google Scholar] [CrossRef] [PubMed]
- Villar-Pique, A.; Espargaro, A.; Ventura, S.; Sabate, R. Screening for amyloid aggregation: In-silico, in vitro and in vivo detection. Curr. Protein Pept. Sci. 2014, 15, 477–489. [Google Scholar] [CrossRef] [PubMed]
- Kurouski, D.; van Duyne, R.P.; Lednev, I.K. Exploring the structure and formation mechanism of amyloid fibrils by Raman spectroscopy: A review. Analyst 2015, 140, 4967–4980. [Google Scholar] [CrossRef] [PubMed]
- Bronsoms, S.; Trejo, S.A. Applications of mass spectrometry to the study of protein aggregation. Methods Mol. Biol. 2015, 1258, 331–345. [Google Scholar] [PubMed]
- Beeg, M.; Diomede, L.; Stravalaci, M.; Salmona, M.; Gobbi, M. Novel approaches for studying amyloidogenic peptides/proteins. Curr. Opin. Pharmacol. 2013, 13, 797–801. [Google Scholar] [CrossRef] [PubMed]
- Pryor, N.E.; Moss, M.A.; Hestekin, C.N. Unraveling the early events of amyloid-β protein (Aβ) aggregation: Techniques for the determination of Aβ aggregate size. Int. J. Mol. Sci. 2012, 13, 3038–3072. [Google Scholar] [CrossRef] [PubMed]
- Bemporad, F.; Chiti, F. Protein misfolded oligomers: Experimental approaches, mechanism of formation, and structure-toxicity relationships. Chem. Biol. 2012, 19, 315–327. [Google Scholar] [CrossRef] [PubMed]
- Hillger, F.; Nettels, D.; Dorsch, S.; Schuler, B. Detection and analysis of protein aggregation with confocal single molecule fluorescence spectroscopy. J. Fluoresc. 2007, 17, 759–765. [Google Scholar] [CrossRef] [PubMed]
- Zijlstra, N.; Schilderink, N.; Subramaniam, V. Fluorescence methods for unraveling oligomeric amyloid intermediates. Methods Mol. Biol. 2016, 1345, 151–169. [Google Scholar] [PubMed]
- Paredes, J.M.; Casares, S.; Ruedas-Rama, M.J.; Fernandez, E.; Castello, F.; Varela, L.; Orte, A. Early amyloidogenic oligomerization studied through fluorescence lifetime correlation spectroscopy. Int. J. Mol. Sci. 2012, 13, 9400–9418. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, A.; Neupane, K.; Woodside, M.T. Single-molecule assays for investigating protein misfolding and aggregation. Phys. Chem. Chem. Phys. 2013, 15, 7934–7948. [Google Scholar] [CrossRef] [PubMed]
- Van den Wildenberg, S.M.; Prevo, B.; Peterman, E.J. A brief introduction to single-molecule fluorescence methods. Methods Mol. Biol. 2011, 783, 81–99. [Google Scholar] [PubMed]
- Oleg, K.; Grégoire, B. Fluorescence correlation spectroscopy: The technique and its applications. Rep. Prog. Phys. 2002, 65. [Google Scholar] [CrossRef]
- Sengupta, P.; Garai, K.; Balaji, J.; Periasamy, N.; Maiti, S. Measuring size distribution in highly heterogeneous systems with fluorescence correlation spectroscopy. Biophys. J. 2003, 84, 1977–1984. [Google Scholar] [CrossRef]
- Medina, M.Á.; Schwille, P. Fluorescence correlation spectroscopy for the detection and study of single molecules in biology. BioEssays 2002, 24, 758–764. [Google Scholar] [CrossRef] [PubMed]
- Sahoo, B.; Drombosky, K.W.; Wetzel, R. Fluorescence correlation spectroscopy: A tool to study protein oligomerization and aggregation in vitro and in vivo. Methods Mol. Biol. 2016, 1345, 67–87. [Google Scholar] [PubMed]
- Tosatto, L.; Horrocks, M.H.; Dear, A.J.; Knowles, T.P.; Dalla Serra, M.; Cremades, N.; Dobson, C.M.; Klenerman, D. Single-molecule FRET studies on α-synuclein oligomerization of Parkinson’s disease genetically related mutants. Sci. Rep. 2015, 5, 16696. [Google Scholar] [CrossRef] [PubMed]
- Gambin, Y.; Vandelinder, V.; Ferreon, A.C.M.; Lemke, E.A.; Groisman, A.; Deniz, A.A. Visualizing a one-way protein encounter complex by ultrafast single-molecule mixing. Nat. Methods 2011, 8, 239–241. [Google Scholar] [CrossRef] [PubMed]
- Kostka, M.; Högen, T.; Danzer, K.M.; Levin, J.; Habeck, M.; Wirth, A.; Wagner, R.; Glabe, C.G.; Finger, S.; Heinzelmann, U.; et al. Single particle characterization of iron-induced pore-forming α-synuclein oligomers. J. Biol. Chem. 2008, 283, 10992–11003. [Google Scholar] [CrossRef] [PubMed]
- Högen, T.; Levin, J.; Schmidt, F.; Caruana, M.; Vassallo, N.; Kretzschmar, H.; Bötzel, K.; Kamp, F.; Giese, A. Two different binding modes of α-synuclein to lipid vesicles depending on its aggregation state. Biophys. J. 2012, 102, 1646–1655. [Google Scholar] [CrossRef] [PubMed]
- Bieschke, J.; Giese, A.; Schulz-Schaeffer, W.; Zerr, I.; Poser, S.; Eigen, M.; Kretzschmar, H. Ultrasensitive detection of pathological prion protein aggregates by dual-color scanning for intensely fluorescent targets. Proc. Natl. Acad. Sci. USA 2000, 97, 5468–5473. [Google Scholar] [CrossRef] [PubMed]
- Lv, Z.; Krasnoslobodtsev, A.V.; Zhang, Y.; Ysselstein, D.; Rochet, J.C.; Blanchard, S.C.; Lyubchenko, Y.L. Direct detection of α-synuclein dimerization dynamics: Single-molecule fluorescence analysis. Biophys. J. 2015, 108, 2038–2047. [Google Scholar] [CrossRef] [PubMed]
- Kurz, A.; Schmied, J.J.; Grußmayer, K.S.; Holzmeister, P.; Tinnefeld, P.; Herten, D.P. Counting fluorescent dye molecules on DNA origami by means of photon statistics. Small 2013, 9, 4061–4068. [Google Scholar] [CrossRef] [PubMed]
- Zijlstra, N.; Claessens, M.M.; Blum, C.; Subramaniam, V. Elucidating the aggregation number of dopamine-induced α-synuclein oligomeric assemblies. Biophys. J. 2014, 106, 440–446. [Google Scholar] [CrossRef] [PubMed]
- Mogalisetti, P.; Walt, D.R. Stoichiometry of the α-complementation reaction of Escherichia coli β-galactosidase as revealed through single-molecule studies. Biochemistry 2015, 54, 1583–1588. [Google Scholar] [CrossRef] [PubMed]
- Herrick-Davis, K.; Grinde, E.; Lindsley, T.; Cowan, A.; Mazurkiewicz, J.E. Oligomer size of the serotonin 5-hydroxytryptamine 2C (5-HT2C) receptor revealed by fluorescence correlation spectroscopy with photon counting histogram analysis: Evidence for homodimers without monomers or tetramers. J. Biol. Chem. 2012, 287, 23604–23614. [Google Scholar] [CrossRef] [PubMed]
- Gambin, Y.; Ariotti, N.; McMahon, K.A.; Bastiani, M.; Sierecki, E.; Kovtun, O.; Polinkovsky, M.E.; Magenau, A.; Jung, W.; Okano, S.; et al. Single-molecule analysis reveals self assembly and nanoscale segregation of two distinct cavin subcomplexes on caveolae. Elife 2014, 3. [Google Scholar] [CrossRef] [PubMed]
- Qian, H.; Elson, E.L. On the analysis of high order moments of fluorescence fluctuations. Biophys. J. 1990, 57, 375–380. [Google Scholar] [CrossRef]
- Sergeev, M.; Costantino, S.; Wiseman, P.W. Measurement of monomer-oligomer distributions via fluorescence moment image analysis. Biophys. J. 2006, 91, 3884–3896. [Google Scholar] [CrossRef] [PubMed]
- Digman, M.A.; Dalal, R.; Horwitz, A.F.; Gratton, E. Mapping the number of molecules and brightness in the laser scanning microscope. Biophys. J. 2008, 94, 2320–2332. [Google Scholar] [CrossRef] [PubMed]
- Plotegher, N.; Gratton, E.; Bubacco, L. Number and Brightness analysis of α-synuclein oligomerization and the associated mitochondrial morphology alterations in live cells. Biochim. Biophys. Acta 2014, 1840, 2014–2024. [Google Scholar] [CrossRef] [PubMed]
- Adu-Gyamfi, E.; Digman, M.A.; Gratton, E.; Stahelin, R.V. Investigation of Ebola VP40 assembly and oligomerization in live cells using number and brightness analysis. Biophys. J. 2012, 102, 2517–2525. [Google Scholar] [CrossRef] [PubMed]
- James, N.G.; Digman, M.A.; Gratton, E.; Barylko, B.; Ding, X.; Albanesi, J.P.; Goldberg, M.S.; Jameson, D.M. Number and brightness analysis of LRRK2 oligomerization in live cells. Biophys. J. 2012, 102, L41–L43. [Google Scholar] [CrossRef] [PubMed]
- Ross, J.A.; Digman, M.A.; Wang, L.; Gratton, E.; Albanesi, J.P.; Jameson, D.M. Oligomerization state of dynamin 2 in cell membranes using TIRF and number and brightness analysis. Biophys. J. 2011, 100, L15–L17. [Google Scholar] [CrossRef] [PubMed]
- Trullo, A.; Corti, V.; Arza, E.; Caiolfa, V.R.; Zamai, M. Application limits and data correction in number of molecules and brightness analysis. Microsc. Res. Tech. 2013, 76, 1135–1146. [Google Scholar] [CrossRef] [PubMed]
- Bain, L.J.; Engelhardt, M. Introduction to Probability and Mathematical Statistics; Duxbury Press: Boston, MA, USA, 2000. [Google Scholar]
- Mureev, S.; Kovtun, O.; Nguyen, U.T.; Alexandrov, K. Species-independent translational leaders facilitate cell-free expression. Nat. Biotechnol. 2009, 27, 747–752. [Google Scholar] [CrossRef] [PubMed]
- Gambin, Y.; Schug, A.; Lemke, E.A.; Lavinder, J.J.; Ferreon, A.C.M.; Magliery, T.J.; Onuchic, J.N.; Deniz, A.A. Direct single-molecule observation of a protein living in two opposed native structures. Proc. Nat. Acad. Sci. USA 2009, 106, 10153–10158. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meier, S.; Güthe, S.; Kiefhaber, T.; Grzesiek, S. Foldon, The natural trimerization domain of T4 fibritin, dissociates into a monomeric A-state form containing a stable β-hairpin: Atomic details of trimer dissociation and local β-hairpin stability from residual dipolar couplings. J. Mol. Biol. 2004, 344, 1051–1069. [Google Scholar] [CrossRef] [PubMed]
- Han, S.P.; Gambin, Y.; Gomez, G.A.; Verma, S.; Giles, N.; Michael, M.; Wu, S.K.; Guo, Z.; Johnston, W.; Sierecki, E.; et al. Cortactin scaffolds Arp2/3 and WAVE2 at the epithelial zonula adherens. J. Biol. Chem. 2014, 289, 7764–7775. [Google Scholar] [CrossRef] [PubMed]
- Sierecki, E.; Stevers, L.M.; Giles, N.; Polinkovsky, M.E.; Moustaqil, M.; Mureev, S.; Johnston, W.A.; Dahmer-Heath, M.; Skalamera, D.; Gonda, T.J.; et al. Rapid mapping of interactions between human SNX-BAR proteins measured in vitro by AlphaScreen and single-molecule spectroscopy. Mol. Cell. Proteom. 2014, 13, 2233–2245. [Google Scholar] [CrossRef] [PubMed]
- Martin, S.; Papadopulos, A.; Tomatis, V.M.; Sierecki, E.; Malintan, N.T.; Gormal, R.S.; Giles, N.; Johnston, W.A.; Alexandrov, K.; Gambin, Y.; et al. Increased polyubiquitination and proteasomal degradation of a Munc18-1 disease-linked mutant causes temperature-sensitive defect in exocytosis. Cell Rep. 2014, 9, 206–218. [Google Scholar] [CrossRef] [PubMed]
- Appel-Cresswell, S.; Vilarino-Guell, C.; Encarnacion, M.; Sherman, H.; Yu, I.; Shah, B.; Weir, D.; Thompson, C.; Szu-Tu, C.; Trinh, J.; et al. α-synuclein p.H50Q, a novel pathogenic mutation for Parkinson's disease. Mov. Disord. 2013, 28, 811–833. [Google Scholar] [CrossRef] [PubMed]
- Hinde, E.l.; Pandžić, E.; Yang, Z.; Ng, I.H.; Jans, D.A.; Bogoyevitch, M.A.; Gratton, E.; Gaus, K. Quantifying the dynamics of the oligomeric transcription factor STAT3 by pair correlation of molecular brightness. Nat. Commun. 2016, 7. [Google Scholar] [CrossRef] [PubMed]
- Fricke, F.; Beaudouin, J.; Eils, R.; Heilemann, M. One, two or three? Probing the stoichiometry of membrane proteins by single-molecule localization microscopy. Sci. Rep. 2015, 5. [Google Scholar] [CrossRef] [PubMed]










© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gambin, Y.; Polinkovsky, M.; Francois, B.; Giles, N.; Bhumkar, A.; Sierecki, E. Confocal Spectroscopy to Study Dimerization, Oligomerization and Aggregation of Proteins: A Practical Guide. Int. J. Mol. Sci. 2016, 17, 655. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms17050655
Gambin Y, Polinkovsky M, Francois B, Giles N, Bhumkar A, Sierecki E. Confocal Spectroscopy to Study Dimerization, Oligomerization and Aggregation of Proteins: A Practical Guide. International Journal of Molecular Sciences. 2016; 17(5):655. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms17050655
Chicago/Turabian StyleGambin, Yann, Mark Polinkovsky, Bill Francois, Nichole Giles, Akshay Bhumkar, and Emma Sierecki. 2016. "Confocal Spectroscopy to Study Dimerization, Oligomerization and Aggregation of Proteins: A Practical Guide" International Journal of Molecular Sciences 17, no. 5: 655. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms17050655