
Omics-Based Strategies in Precision Medicine: Toward a Paradigm Shift in Inborn Errors of Metabolism Investigations



Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
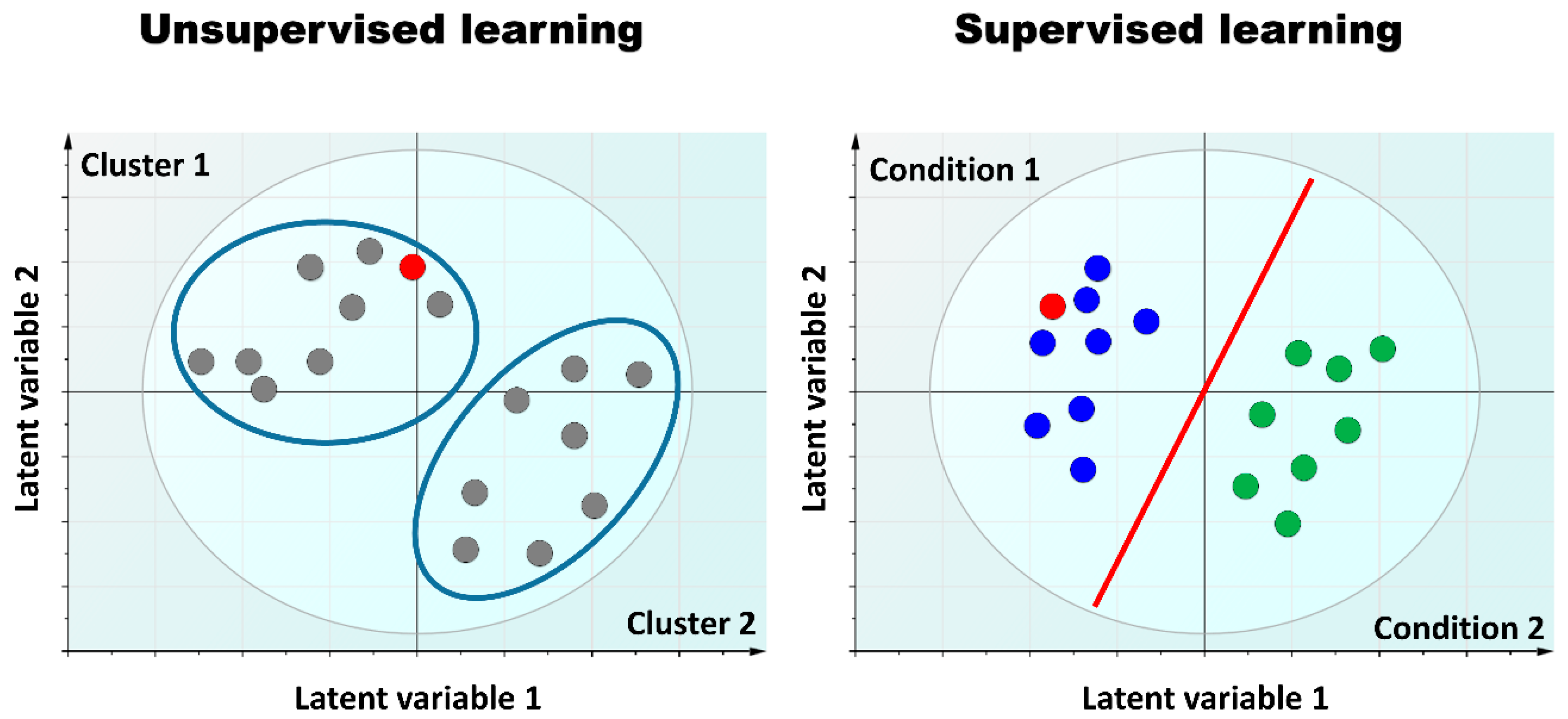{kind=link}
{kind=link}
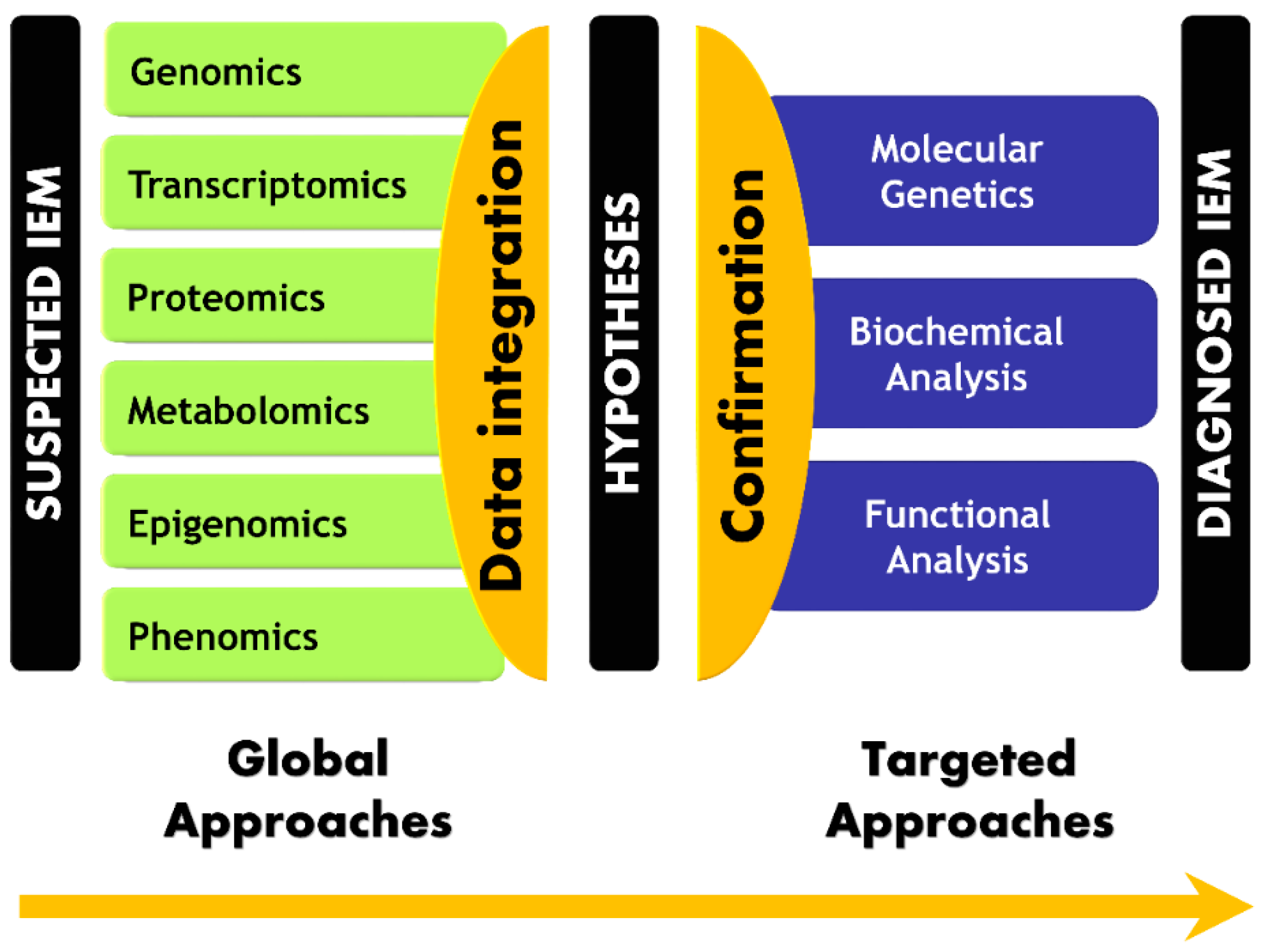{kind=link}
1. Introduction
2. Omics Revolution in Translational and Clinical Contexts
2.1. Omics Technologies
2.1.1. High-Throughput Sequencing (HTS) Technologies
Genomics
Epigenomics
Transcriptomics
2.1.2. Mass Spectrometry-Based Omics
Proteomics
Metabolomics
2.1.3. Phenomics
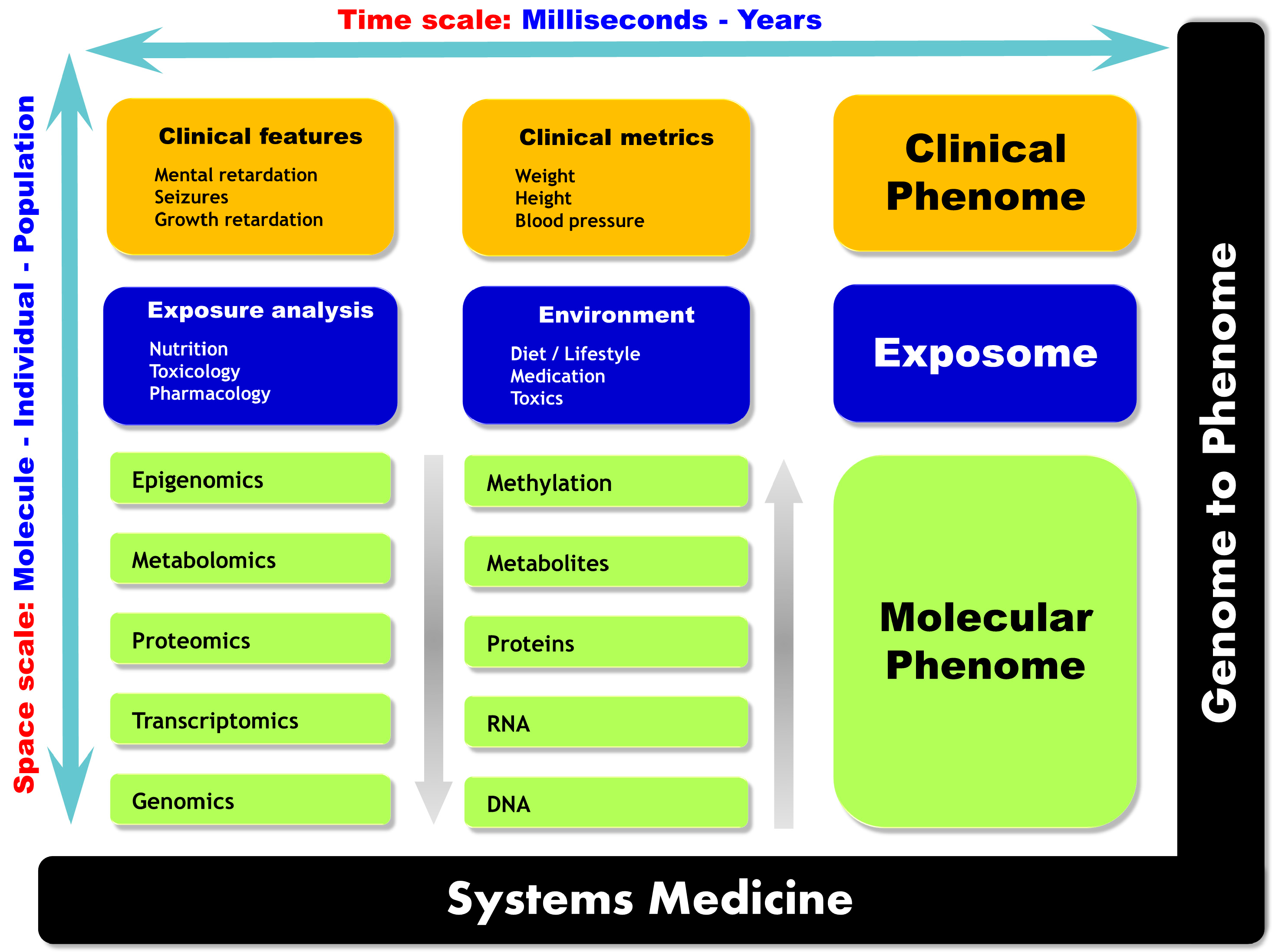
2.2. Multi-Omics Strategies, or When the Whole Is More than the Sum of Its Parts
2.3. Issues and Limitations of Omics Analysis
2.3.1. Technical Limitations
Experimental and Analytical Noise
Analytical Accuracy and Clinical Relevance
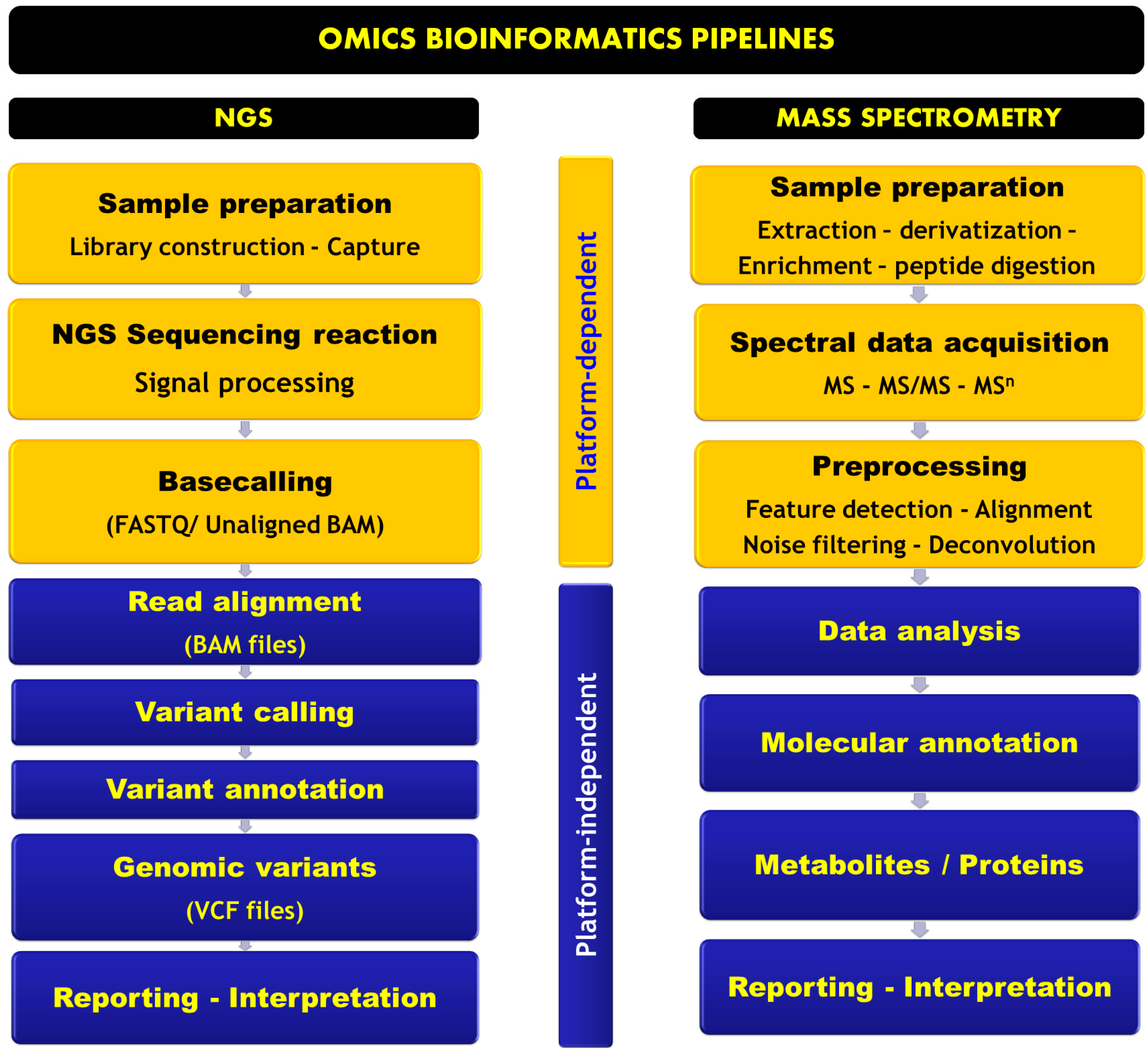
Omics Informatics Pipelines in the Clinical Environment
NGS Informatics Pipeline
Mass Spectrometry-Based Omics Informatics Pipeline
2.3.2. Biological Variation
3. Omics and Biomarkers: From Bench to Bedside
3.1. Definitions
3.2. Biomarker Development
3.3. Criteria for Omics-Based Biomarkers in Clinical Context
3.4. Omics Integration and the Curse of Dimensionality
4. Perspectives and Challenges in Translational and Clinical Contexts
4.1. Data Integrity, Standardization, and Sharing
4.2. Turning Data into Knowledge
4.3. Clinical Research Enterprise and Embracing Multi-Disciplinary Sciences
4.4. Informatics and New Pathways to Clinical Actionability
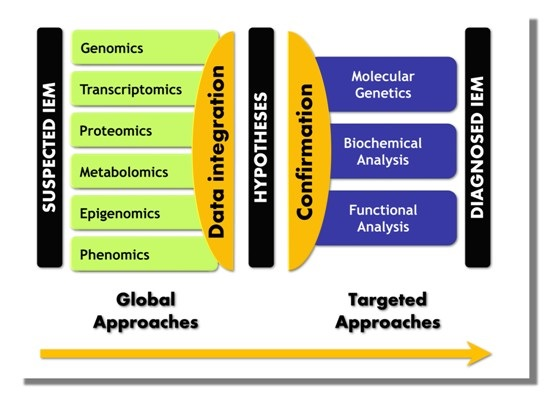
5. Paradigm Shift in IEM Investigations
6. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| ATAC-seq | Assay for transposase-accessible chromatin next-generation sequencing |
| BAM | Binary alignment map |
| ChIP-seq | Chromatin immunoprecipitation next-generation sequencing |
| CT | Computerized tomography |
| DNA | Deoxyribonucleic acid |
| DNase-seq | DNase I digestion of chromatin combined with next-generation sequencing |
| FDA | Food and Drug Administration |
| HTS | High-throughput sequencing |
| ICA | Independent component analysis |
| IEM | Inborn errors of metabolism |
| iPF | Integrative phenotyping framework |
| miRNA | microRNA |
| ML | Machine learning |
| MRI | Magnetic resonance imaging |
| MS | Mass spectrometry |
| MS/MS | Tandem mass spectrometry |
| ncRNA | Non-coding RNA |
| NGS | Next-generation sequencing |
| OPLSDA | Orthogonal partial least squares discriminant analysis |
| PCA | Principal component analysis |
| PLSDA | Partial least squares discriminant analysis |
| PM | Precision medicine |
| QC | Quality control |
| RNA | Ribonucleic acid |
| rRNA | Ribosome RNA |
| SAM | Sequence alignment map |
| SNP | Single-nucleotide polymorphisms |
| SOM | Self-organizing maps |
| SOP | Standard operating procedure |
| SVM | Support vector machines |
| TDA | Topological data analysis |
| tRNA | Transfer RNA |
| VCF | Variant call format |
| WES | Whole-exome sequencing |
| WGS | Whole-genome sequencing |
References
- Collins, F.S.; Varmus, H. A new initiative on precision medicine. N. Engl. J. Med. 2015, 372, 793–795. [Google Scholar] [CrossRef] [PubMed]
- Ahn, A.C.; Tewari, M.; Poon, C.S.; Phillips, R.S. The limits of reductionism in medicine: Could systems biology offer an alternative? PLoS Med. 2006, 3, e208. [Google Scholar] [CrossRef] [PubMed]
- Van Regenmortel, M.H. Reductionism and complexity in molecular biology: Scientists now have the tools to unravel biological and overcome the limitations of reductionism. EMBO Rep. 2004, 5, 1016–1020. [Google Scholar] [CrossRef] [PubMed]
- Aon, M.A. Complex systems biology of networks: The riddle and the challenge. In Systems Biology of Metabolic and Signaling Networks; Springer Berlin Heidelberg: Heidelberg, Germany, 2014; pp. 19–35. [Google Scholar]
- Kitano, H. Systems biology: A brief overview. Science 2002, 295, 1662–1664. [Google Scholar] [CrossRef] [PubMed]
- Lanpher, B.; Brunetti-Pierri, N.; Lee, B. Inborn errors of metabolism: The flux from mendelian to complex diseases. Nat. Rev. Genet. 2006, 7, 449–460. [Google Scholar] [CrossRef] [PubMed]
- Watson, J.D.; Crick, F.H. The structure of DNA. Cold Spring Harb. Symp. Quant. Biol. 1953, 18, 123–131. [Google Scholar] [CrossRef] [PubMed]
- Goodwin, S.; McPherson, J.D.; McCombie, W.R. Coming of age: Ten years of next-generation sequencing technologies. Nat. Rev. Genet. 2016, 17, 333–351. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Muzny, D.M.; Reid, J.G.; Bainbridge, M.N.; Willis, A.; Ward, P.A.; Braxton, A.; Beuten, J.; Xia, F.; Niu, Z.; et al. Clinical whole-exome sequencing for the diagnosis of mendelian disorders. N. Engl. J. Med. 2013, 369, 1502–1511. [Google Scholar] [CrossRef] [PubMed]
- Van Karnebeek, C.D.; Bonafe, L.; Wen, X.Y.; Tarailo-Graovac, M.; Balzano, S.; Royer-Bertrand, B.; Ashikov, A.; Garavelli, L.; Mammi, I.; Turolla, L.; et al. Nans-mediated synthesis of sialic acid is required for brain and skeletal development. Nat. Genet. 2016, 48, 777–784. [Google Scholar] [CrossRef] [PubMed]
- Tarailo-Graovac, M.; Shyr, C.; Ross, C.J.; Horvath, G.A.; Salvarinova, R.; Ye, X.C.; Zhang, L.H.; Bhavsar, A.P.; Lee, J.J.; Drogemoller, B.I.; et al. Exome sequencing and the management of neurometabolic disorders. N. Engl. J. Med. 2016, 374, 2246–2255. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Worthey, E.A.; Mayer, A.N.; Syverson, G.D.; Helbling, D.; Bonacci, B.B.; Decker, B.; Serpe, J.M.; Dasu, T.; Tschannen, M.R.; Veith, R.L.; et al. Making a definitive diagnosis: Successful clinical application of whole exome sequencing in a child with intractable inflammatory bowel disease. Genet. Med. 2011, 13, 255–262. [Google Scholar] [CrossRef] [PubMed]
- Benson, M. Clinical implications of omics and systems medicine: Focus on predictive and individualized treatment. J. Intern. Med. 2016, 279, 229–240. [Google Scholar] [CrossRef] [PubMed]
- Yohe, S.; Hauge, A.; Bunjer, K.; Kemmer, T.; Bower, M.; Schomaker, M.; Onsongo, G.; Wilson, J.; Erdmann, J.; Zhou, Y.; et al. Clinical validation of targeted next-generation sequencing for inherited disorders. Arch. Pathol. Lab. Med. 2015, 139, 204–210. [Google Scholar] [CrossRef] [PubMed]
- Yubero, D.; Brandi, N.; Ormazabal, A.; Garcia-Cazorla, A.; Perez-Duenas, B.; Campistol, J.; Ribes, A.; Palau, F.; Artuch, R.; Armstrong, J.; et al. Targeted next generation sequencing in patients with inborn errors of metabolism. PLoS ONE 2016, 11, e0156359. [Google Scholar] [CrossRef] [PubMed]
- Cirulli, E.T.; Goldstein, D.B. Uncovering the roles of rare variants in common disease through whole-genome sequencing. Nat. Rev. Genet. 2010, 11, 415–425. [Google Scholar] [CrossRef] [PubMed]
- Stranneheim, H.; Wedell, A. Exome and genome sequencing: A revolution for the discovery and diagnosis of monogenic disorders. J. Intern. Med. 2016, 279, 3–15. [Google Scholar] [CrossRef] [PubMed]
- Meienberg, J.; Zerjavic, K.; Keller, I.; Okoniewski, M.; Patrignani, A.; Ludin, K.; Xu, Z.; Steinmann, B.; Carrel, T.; Rothlisberger, B.; et al. New insights into the performance of human whole-exome capture platforms. Nucleic Acids Res. 2015, 43, e76. [Google Scholar] [CrossRef] [PubMed]
- Mortazavi, A.; Williams, B.A.; McCue, K.; Schaeffer, L.; Wold, B. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat. Methods 2008, 5, 621–628. [Google Scholar] [CrossRef] [PubMed]
- Mensaert, K.; Denil, S.; Trooskens, G.; van Criekinge, W.; Thas, O.; de Meyer, T. Next-generation technologies and data analytical approaches for epigenomics. Environ. Mol. Mutagen. 2014, 55, 155–170. [Google Scholar] [CrossRef] [PubMed]
- Sanger, F.; Nicklen, S.; Coulson, A.R. DNA sequencing with chain-terminating inhibitors. Proc. Natl. Acad. Sci. USA 1977, 74, 5463–5467. [Google Scholar] [CrossRef] [PubMed]
- Marsh, M.; Tu, O.; Dolnik, V.; Roach, D.; Solomon, N.; Bechtol, K.; Smietana, P.; Wang, L.; Li, X.; Cartwright, P.; et al. High-throughput DNA sequencing on a capillary array electrophoresis system. J. Capill. Electrophor. 1997, 4, 83–89. [Google Scholar]
- McBride, L.J.; Koepf, S.M.; Gibbs, R.A.; Salser, W.; Mayrand, P.E.; Hunkapiller, M.W.; Kronick, M.N. Automated DNA sequencing methods involving polymerase chain reaction. Clin. Chem. 1989, 35, 2196–2201. [Google Scholar] [PubMed]
- Prober, J.M.; Trainor, G.L.; Dam, R.J.; Hobbs, F.W.; Robertson, C.W.; Zagursky, R.J.; Cocuzza, A.J.; Jensen, M.A.; Baumeister, K. A system for rapid DNA sequencing with fluorescent chain-terminating dideoxynucleotides. Science 1987, 238, 336–341. [Google Scholar] [CrossRef] [PubMed]
- Venter, J.C.; Adams, M.D.; Myers, E.W.; Li, P.W.; Mural, R.J.; Sutton, G.G.; Smith, H.O.; Yandell, M.; Evans, C.A.; Holt, R.A.; et al. The sequence of the human genome. Science 2001, 291, 1304–1351. [Google Scholar] [CrossRef] [PubMed]
- Lander, E.S.; Linton, L.M.; Birren, B.; Nusbaum, C.; Zody, M.C.; Baldwin, J.; Devon, K.; Dewar, K.; Doyle, M.; FitzHugh, W.; et al. Initial sequencing and analysis of the human genome. Nature 2001, 409, 860–921. [Google Scholar] [CrossRef] [PubMed]
- Reuter, J.A.; Spacek, D.V.; Snyder, M.P. High-throughput sequencing technologies. Mol. Cell 2015, 58, 586–597. [Google Scholar] [CrossRef] [PubMed]
- Head, S.R.; Komori, H.K.; LaMere, S.A.; Whisenant, T.; van Nieuwerburgh, F.; Salomon, D.R.; Ordoukhanian, P. Library construction for next-generation sequencing: Overviews and challenges. Biotechniques 2014, 56, 61. [Google Scholar] [CrossRef] [PubMed]
- Mardis, E.R. Next-generation sequencing platforms. Annu. Rev. Anal. Chem. 2013, 6, 287–303. [Google Scholar] [CrossRef] [PubMed]
- Lim, E.C.; Brett, M.; Lai, A.H.; Lee, S.P.; Tan, E.S.; Jamuar, S.S.; Ng, I.S.; Tan, E.C. Next-generation sequencing using a pre-designed gene panel for the molecular diagnosis of congenital disorders in pediatric patients. Hum. Genom. 2015, 9, 33. [Google Scholar] [CrossRef] [PubMed]
- Taylor, R.W.; Pyle, A.; Griffin, H.; Blakely, E.L.; Duff, J.; He, L.; Smertenko, T.; Alston, C.L.; Neeve, V.C.; Best, A.; et al. Use of whole-exome sequencing to determine the genetic basis of multiple mitochondrial respiratory chain complex deficiencies. JAMA 2014, 312, 68–77. [Google Scholar] [CrossRef] [PubMed]
- Howard, H.C.; Knoppers, B.M.; Cornel, M.C.; Wright Clayton, E.; Senecal, K.; Borry, P. Whole-genome sequencing in newborn screening? A statement on the continued importance of targeted approaches in newborn screening programmes. Eur. J. Hum. Genet. 2015, 23, 1593–1600. [Google Scholar] [CrossRef] [PubMed]
- Ashley, E.A. Towards precision medicine. Nat. Rev. Genet. 2016, 17, 507–522. [Google Scholar] [CrossRef] [PubMed]
- Goldfeder, R.L.; Ashley, E.A. A precision metric for clinical genome sequencing. bioRxiv 2016, 051490. [Google Scholar] [CrossRef]
- Bird, A. Perceptions of epigenetics. Nature 2007, 447, 396–398. [Google Scholar] [CrossRef] [PubMed]
- Huang, B.; Jiang, C.; Zhang, R. Epigenetics: The language of the cell? Epigenomics 2014, 6, 73–88. [Google Scholar] [CrossRef] [PubMed]
- Sadakierska-Chudy, A.; Filip, M. A comprehensive view of the epigenetic landscape. Part II: Histone post-translational modification, nucleosome level, and chromatin regulation by ncRNAs. Neurotox. Res. 2015, 27, 172–197. [Google Scholar] [CrossRef] [PubMed]
- Sadakierska-Chudy, A.; Kostrzewa, R.M.; Filip, M. A comprehensive view of the epigenetic landscape part I: DNA methylation, passive and active DNA demethylation pathways and histone variants. Neurotox. Res. 2015, 27, 84–97. [Google Scholar] [CrossRef] [PubMed]
- Kundaje, A.; Meuleman, W.; Ernst, J.; Bilenky, M.; Yen, A.; Heravi-Moussavi, A.; Kheradpour, P.; Zhang, Z.; Wang, J.; Ziller, M.J.; et al. Integrative analysis of 111 reference human epigenomes. Nature 2015, 518, 317–330. [Google Scholar] [CrossRef] [PubMed]
- Barski, A.; Cuddapah, S.; Cui, K.; Roh, T.Y.; Schones, D.E.; Wang, Z.; Wei, G.; Chepelev, I.; Zhao, K. High-resolution profiling of histone methylations in the human genome. Cell 2007, 129, 823–837. [Google Scholar] [CrossRef] [PubMed]
- Yaragatti, M.; Basilico, C.; Dailey, L. Identification of active transcriptional regulatory modules by the functional assay of DNA from nucleosome-free regions. Genome Res. 2008, 18, 930–938. [Google Scholar] [CrossRef] [PubMed]
- Lister, R.; O’Malley, R.C.; Tonti-Filippini, J.; Gregory, B.D.; Berry, C.C.; Millar, A.H.; Ecker, J.R. Highly integrated single-base resolution maps of the epigenome in arabidopsis. Cell 2008, 133, 523–536. [Google Scholar] [CrossRef] [PubMed]
- Buenrostro, J.D.; Wu, B.; Chang, H.Y.; Greenleaf, W.J. Atac-seq: A method for assaying chromatin accessibility genome-wide. Curr. Protoc. Mol. Biol. 2015, 109. [Google Scholar] [CrossRef]
- Meyer, C.A.; Liu, X.S. Identifying and mitigating bias in next-generation sequencing methods for chromatin biology. Nat. Rev. Genet. 2014, 15, 709–721. [Google Scholar] [CrossRef] [PubMed]
- Guay, S.P.; Voisin, G.; Brisson, D.; Munger, J.; Lamarche, B.; Gaudet, D.; Bouchard, L. Epigenome-wide analysis in familial hypercholesterolemia identified new loci associated with high-density lipoprotein cholesterol concentration. Epigenomics 2012, 4, 623–639. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Gerstein, M.; Snyder, M. RNA-seq: A revolutionary tool for transcriptomics. Nat. Rev. Genet. 2009, 10, 57–63. [Google Scholar] [CrossRef] [PubMed]
- Nogales-Gadea, G.; Consuegra-Garcia, I.; Rubio, J.C.; Arenas, J.; Cuadros, M.; Camara, Y.; Torres-Torronteras, J.; Fiuza-Luces, C.; Lucia, A.; Martin, M.A.; et al. A transcriptomic approach to search for novel phenotypic regulators in mcardle disease. PLoS ONE 2012, 7, e31718. [Google Scholar] [CrossRef] [PubMed]
- Mazzoccoli, G.; Tomanin, R.; Mazza, T.; D’Avanzo, F.; Salvalaio, M.; Rigon, L.; Zanetti, A.; Pazienza, V.; Francavilla, M.; Giuliani, F.; et al. Circadian transcriptome analysis in human fibroblasts from hunter syndrome and impact of iduronate-2-sulfatase treatment. BMC Med. Genom. 2013, 6, 37. [Google Scholar] [CrossRef] [PubMed]
- Tringham, M.; Kurko, J.; Tanner, L.; Tuikkala, J.; Nevalainen, O.S.; Niinikoski, H.; Nanto-Salonen, K.; Hietala, M.; Simell, O.; Mykkanen, J. Exploring the transcriptomic variation caused by the finnish founder mutation of lysinuric protein intolerance (LPI). Mol. Genet. Metab. 2012, 105, 408–415. [Google Scholar] [CrossRef] [PubMed]
- Dauphinot, L.; Mockel, L.; Cahu, J.; Jinnah, H.A.; Ledroit, M.; Potier, M.C.; Ceballos-Picot, I. Transcriptomic approach to Lesch–Nyhan disease. Nucleosides Nucleotides Nucleic Acids 2014, 33, 208–217. [Google Scholar] [CrossRef] [PubMed]
- Cluzeau, C.V.; Watkins-Chow, D.E.; Fu, R.; Borate, B.; Yanjanin, N.; Dail, M.K.; Davidson, C.D.; Walkley, S.U.; Ory, D.S.; Wassif, C.A.; et al. Microarray expression analysis and identification of serum biomarkers for niemann-pick disease, type c1. Hum. Mol. Genet. 2012, 21, 3632–3646. [Google Scholar] [CrossRef] [PubMed]
- Cajka, T.; Fiehn, O. Toward merging untargeted and targeted methods in mass spectrometry-based metabolomics and lipidomics. Anal. Chem. 2015, 88, 524–545. [Google Scholar] [CrossRef] [PubMed]
- Scherl, A. Clinical protein mass spectrometry. Methods 2015, 81, 3–14. [Google Scholar] [CrossRef] [PubMed]
- Kusebauch, U.; Campbell, D.S.; Deutsch, E.W.; Chu, C.S.; Spicer, D.A.; Brusniak, M.-Y.; Slagel, J.; Sun, Z.; Stevens, J.; Grimes, B.; et al. Human srmatlas: A resource of targeted assays to quantify the complete human proteome. Cell 2016, 166, 766–778. [Google Scholar] [CrossRef] [PubMed]
- May, J.C.; McLean, J.A. Advanced multidimensional separations in mass spectrometry: Navigating the big data deluge. Annu. Rev. Anal. Chem. 2016, 9, 387–409. [Google Scholar] [CrossRef] [PubMed]
- Tebani, A.; Schmitz-Afonso, I.; Rutledge, D.N.; Gonzalez, B.J.; Bekri, S.; Afonso, C. Optimization of a liquid chromatography ion mobility-mass spectrometry method for untargeted metabolomics using experimental design and multivariate data analysis. Anal. Chim. Acta 2016, 913, 55–62. [Google Scholar] [CrossRef] [PubMed]
- James, P. Protein identification in the post-genome era: The rapid rise of proteomics. Quart. Rev. Biophys. 1997, 30, 279–331. [Google Scholar] [CrossRef]
- Khoury, G.A.; Baliban, R.C.; Floudas, C.A. Proteome-wide post-translational modification statistics: Frequency analysis and curation of the swiss-prot database. Sci. Rep. 2011, 1, 90. [Google Scholar] [CrossRef] [PubMed]
- Betzen, C.; Alhamdani, M.S.S.; Lueong, S.; Schröder, C.; Stang, A.; Hoheisel, J.D. Clinical proteomics: Promises, challenges and limitations of affinity arrays. Proteom. Clin. Appl. 2015, 9, 342–347. [Google Scholar] [CrossRef] [PubMed]
- Sabbagh, B.; Mindt, S.; Neumaier, M.; Findeisen, P. Clinical applications of ms-based protein quantification. Proteom. Clin. Appl. 2016, 10, 323–345. [Google Scholar] [CrossRef] [PubMed]
- Lassman, M.E.; McAvoy, T.; Chappell, D.L.; Lee, A.Y.; Zhao, X.X.; Laterza, O.F. The clinical utility of mass spectrometry based protein assays. Clin. Chim. Acta 2016, 459, 155–161. [Google Scholar] [CrossRef] [PubMed]
- Kovacevic, L.; Lu, H.; Goldfarb, D.S.; Lakshmanan, Y.; Caruso, J.A. Urine proteomic analysis in cystinuric children with renal stones. J. Pediatr. Urol. 2015, 11, 217.e1–217.e6. [Google Scholar] [CrossRef] [PubMed]
- Heywood, W.E.; Camuzeaux, S.; Doykov, I.; Patel, N.; Preece, R.L.; Footitt, E.; Cleary, M.; Clayton, P.; Grunewald, S.; Abulhoul, L.; et al. Proteomic discovery and development of a multiplexed targeted mrm-lc-ms/ms assay for urine biomarkers of extracellular matrix disruption in mucopolysaccharidoses I, II, and VI. Anal. Chem. 2015, 87, 12238–12244. [Google Scholar] [CrossRef] [PubMed]
- Williams, E.G.; Wu, Y.; Jha, P.; Dubuis, S.; Blattmann, P.; Argmann, C.A.; Houten, S.M.; Amariuta, T.; Wolski, W.; Zamboni, N.; et al. Systems proteomics of liver mitochondria function. Science 2016, 352, aad0189. [Google Scholar] [CrossRef] [PubMed]
- Martens, L. Bringing proteomics into the clinic: The need for the field to finally take itself seriously. Proteom. Clin. Appl. 2013, 7, 388–391. [Google Scholar] [CrossRef] [PubMed]
- Holmes, E.; Wilson, I.D.; Nicholson, J.K. Metabolic phenotyping in health and disease. Cell 2008, 134, 714–717. [Google Scholar] [CrossRef] [PubMed]
- Oliver, S.G.; Winson, M.K.; Kell, D.B.; Baganz, F. Systematic functional analysis of the yeast genome. Trends Biotechnol. 1998, 16, 373–378. [Google Scholar] [CrossRef]
- Nicholson, J.K.; Lindon, J.C.; Holmes, E. “Metabonomics”: Understanding the metabolic responses of living systems to pathophysiological stimuli via multivariate statistical analysis of biological NMR spectroscopic data. Xenobiotica 1999, 29, 1181–1189. [Google Scholar] [CrossRef] [PubMed]
- Nicholson, J.K.; Holmes, E.; Kinross, J.M.; Darzi, A.W.; Takats, Z.; Lindon, J.C. Metabolic phenotyping in clinical and surgical environments. Nature 2012, 491, 384–392. [Google Scholar] [CrossRef] [PubMed]
- Suhre, K.; Raffler, J.; Kastenmüller, G. Biochemical insights from population studies with genetics and metabolomics. Arch. Biochem. Biophys. 2016, 589, 168–176. [Google Scholar] [CrossRef] [PubMed]
- Alonso, A.; Marsal, S.; Julia, A. Analytical methods in untargeted metabolomics: State of the art in 2015. Front. Bioeng. Biotechnol. 2015, 3, 23. [Google Scholar] [CrossRef] [PubMed]
- Therrell, B.L.; Padilla, C.D.; Loeber, J.G.; Kneisser, I.; Saadallah, A.; Borrajo, G.J.; Adams, J. Current status of newborn screening worldwide: 2015. Semin. Perinatol. 2015, 39, 171–187. [Google Scholar] [CrossRef] [PubMed]
- Denes, J.; Szabo, E.; Robinette, S.L.; Szatmari, I.; Szonyi, L.; Kreuder, J.G.; Rauterberg, E.W.; Takats, Z. Metabonomics of newborn screening dried blood spot samples: A novel approach in the screening and diagnostics of inborn errors of metabolism. Anal. Chem. 2012, 84, 10113–10120. [Google Scholar] [CrossRef] [PubMed]
- Aygen, S.; Durr, U.; Hegele, P.; Kunig, J.; Spraul, M.; Schafer, H.; Krings, D.; Cannet, C.; Fang, F.; Schutz, B.; et al. NMR-based screening for inborn errors of metabolism: Initial results from a study on turkish neonates. JIMD Rep. 2014, 16, 101–111. [Google Scholar] [PubMed]
- Miller, M.; Kennedy, A.; Eckhart, A.; Burrage, L.; Wulff, J.; Miller, L.D.; Milburn, M.; Ryals, J.; Beaudet, A.; Sun, Q.; et al. Untargeted metabolomic analysis for the clinical screening of inborn errors of metabolism. J. Inherit. Metab. Dis. 2015, 38, 1029–1039. [Google Scholar] [CrossRef] [PubMed]
- Tebani, A.; Abily-Donval, L.; Afonso, C.; Marret, S.; Bekri, S. Clinical metabolomics: The new metabolic window for inborn errors of metabolism investigations in the post-genomic era. Int. J. Mol. Sci. 2016, 17, 1167. [Google Scholar] [CrossRef] [PubMed]
- Houle, D.; Govindaraju, D.R.; Omholt, S. Phenomics: The next challenge. Nat. Rev. Genet. 2010, 11, 855–866. [Google Scholar] [CrossRef] [PubMed]
- Plomin, R.; Haworth, C.M.; Davis, O.S. Common disorders are quantitative traits. Nat. Rev. Genet. 2009, 10, 872–878. [Google Scholar] [CrossRef] [PubMed]
- Bush, W.S.; Oetjens, M.T.; Crawford, D.C. Unravelling the human genome-phenome relationship using phenome-wide association studies. Nat. Rev. Genet. 2016, 17, 129–145. [Google Scholar] [CrossRef] [PubMed]
- Bilder, R.M.; Sabb, F.W.; Cannon, T.D.; London, E.D.; Jentsch, J.D.; Parker, D.S.; Poldrack, R.A.; Evans, C.; Freimer, N.B. Phenomics: The systematic study of phenotypes on a genome-wide scale. Neuroscience 2009, 164, 30–42. [Google Scholar] [CrossRef] [PubMed]
- Freimer, N.; Sabatti, C. The human phenome project. Nat. Genet. 2003, 34, 15–21. [Google Scholar] [CrossRef] [PubMed]
- Gerlai, R. Phenomics: Fiction or the future? Trends Neurosci. 2002, 25, 506–509. [Google Scholar] [CrossRef]
- Oetting, W.S.; Robinson, P.N.; Greenblatt, M.S.; Cotton, R.G.; Beck, T.; Carey, J.C.; Doelken, S.C.; Girdea, M.; Groza, T.; Hamilton, C.M.; et al. Getting ready for the human phenome project: The 2012 forum of the human variome project. Hum. Mutat. 2013, 34, 661–666. [Google Scholar] [CrossRef] [PubMed]
- Groza, T.; Kohler, S.; Moldenhauer, D.; Vasilevsky, N.; Baynam, G.; Zemojtel, T.; Schriml, L.M.; Kibbe, W.A.; Schofield, P.N.; Beck, T.; et al. The human phenotype ontology: Semantic unification of common and rare disease. Am. J. Hum. Genet. 2015, 97, 111–124. [Google Scholar] [CrossRef] [PubMed]
- Ritchie, M.D.; Holzinger, E.R.; Li, R.; Pendergrass, S.A.; Kim, D. Methods of integrating data to uncover genotype-phenotype interactions. Nat. Rev. Genet. 2015, 16, 85–97. [Google Scholar] [CrossRef] [PubMed]
- Tracy, R.P. “Deep phenotyping”: Characterizing populations in the era of genomics and systems biology. Curr. Opin. Lipidol. 2008, 19, 151–157. [Google Scholar] [CrossRef] [PubMed]
- Shameer, K.; Badgeley, M.A.; Miotto, R.; Glicksberg, B.S.; Morgan, J.W.; Dudley, J.T. Translational bioinformatics in the era of real-time biomedical, health care and wellness data streams. Brief. Bioinform. 2016. [Google Scholar] [CrossRef] [PubMed]
- Kochinke, K.; Zweier, C.; Nijhof, B.; Fenckova, M.; Cizek, P.; Honti, F.; Keerthikumar, S.; Oortveld Merel, A.W.; Kleefstra, T.; Kramer, J.M.; et al. Systematic phenomics analysis deconvolutes genes mutated in intellectual disability into biologically coherent modules. Am. J. Hum. Genet. 2016, 98, 149–164. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Herazo-Maya, J.D.; Kang, D.D.; Juan-Guardela, B.M.; Tedrow, J.; Martinez, F.J.; Sciurba, F.C.; Tseng, G.C.; Kaminski, N. Integrative phenotyping framework (iPF): Integrative clustering of multiple omics data identifies novel lung disease subphenotypes. BMC Genom. 2015, 16, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Mungall, C.J.; Washington, N.L.; Nguyen-Xuan, J.; Condit, C.; Smedley, D.; Kohler, S.; Groza, T.; Shefchek, K.; Hochheiser, H.; Robinson, P.N.; et al. Use of model organism and disease databases to support matchmaking for human disease gene discovery. Hum. Mutat. 2015, 36, 979–984. [Google Scholar] [CrossRef] [PubMed]
- Argmann, C.A.; Houten, S.M.; Zhu, J.; Schadt, E.E. A next generation multiscale view of inborn errors of metabolism. Cell Metab. 2016, 23, 13–26. [Google Scholar] [CrossRef] [PubMed]
- Gligorijevic, V.; Przulj, N. Methods for biological data integration: Perspectives and challenges. J. R. Soc. Interface 2015, 12, 112. [Google Scholar] [CrossRef] [PubMed]
- Wanichthanarak, K.; Fahrmann, J.F.; Grapov, D. Genomic, proteomic, and metabolomic data integration strategies. Biomark. Insights 2015, 10, 1–6. [Google Scholar] [PubMed]
- Wahl, S.; Vogt, S.; Stuckler, F.; Krumsiek, J.; Bartel, J.; Kacprowski, T.; Schramm, K.; Carstensen, M.; Rathmann, W.; Roden, M.; et al. Multi-omic signature of body weight change: Results from a population-based cohort study. BMC Med. 2015, 13, 48. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Bai, X.; Liu, Y.; Wang, W.; Han, J.; Wang, Q.; Xu, Y.; Zhang, C.; Zhang, S.; Li, X.; et al. Topologically inferring pathway activity toward precise cancer classification via integrating genomic and metabolomic data: Prostate cancer as a case. Sci. Rep. 2015, 5, 13192. [Google Scholar] [CrossRef] [PubMed]
- Chen, R.; Mias, G.I.; Li-Pook-Than, J.; Jiang, L.; Lam, H.Y.K.; Chen, R.; Miriami, E.; Karczewski, K.J.; Hariharan, M.; Dewey, F.E.; et al. Personal omics profiling reveals dynamic molecular and medical phenotypes. Cell 2012, 148, 1293–1307. [Google Scholar] [CrossRef] [PubMed]
- Bartel, J.; Krumsiek, J.; Schramm, K.; Adamski, J.; Gieger, C.; Herder, C.; Carstensen, M.; Peters, A.; Rathmann, W.; Roden, M.; et al. The human blood metabolome-transcriptome interface. PLoS Genet. 2015, 11, e1005274. [Google Scholar] [CrossRef] [PubMed]
- Shin, S.Y.; Fauman, E.B.; Petersen, A.K.; Krumsiek, J.; Santos, R.; Huang, J.; Arnold, M.; Erte, I.; Forgetta, V.; Yang, T.P.; et al. An atlas of genetic influences on human blood metabolites. Nat. Genet. 2014, 46, 543–550. [Google Scholar] [CrossRef] [PubMed]
- Petersen, A.K.; Zeilinger, S.; Kastenmuller, G.; Romisch-Margl, W.; Brugger, M.; Peters, A.; Meisinger, C.; Strauch, K.; Hengstenberg, C.; Pagel, P.; et al. Epigenetics meets metabolomics: An epigenome-wide association study with blood serum metabolic traits. Hum. Mol. Genet. 2014, 23, 534–545. [Google Scholar] [CrossRef] [PubMed]
- Ioannidis, J.P.; Khoury, M.J. Improving validation practices in “omics” research. Science 2011, 334, 1230–1232. [Google Scholar] [CrossRef] [PubMed]
- Kolker, E.; Ozdemir, V.; Martens, L.; Hancock, W.; Anderson, G.; Anderson, N.; Aynacioglu, S.; Baranova, A.; Campagna, S.R.; Chen, R.; et al. Toward more transparent and reproducible omics studies through a common metadata checklist and data publications. Omics 2014, 18, 10–14. [Google Scholar] [CrossRef] [PubMed]
- Tenenbaum, J.D.; Sansone, S.A.; Haendel, M. A sea of standards for omics data: Sink or swim? JAMIA 2014, 21, 200–203. [Google Scholar] [CrossRef] [PubMed]
- Chitayat, S.; Rudan, J.F. Chapter 10—Phenome centers and global harmonization. In Metabolic Phenotyping in Personalized and Public Healthcare; Academic Press: Boston, MA, USA, 2016; pp. 291–315. [Google Scholar]
- Rocca-Serra, P.; Salek, R.M.; Arita, M.; Correa, E.; Dayalan, S.; Gonzalez-Beltran, A.; Ebbels, T.; Goodacre, R.; Hastings, J.; Haug, K.; et al. Data standards can boost metabolomics research, and if there is a will, there is a way. Metabolomics 2016, 12, 14. [Google Scholar] [CrossRef] [PubMed]
- Dunn, W.B.; Wilson, I.D.; Nicholls, A.W.; Broadhurst, D. The importance of experimental design and QC samples in large-scale and MS-driven untargeted metabolomic studies of humans. Bioanalysis 2012, 4, 2249–2264. [Google Scholar] [CrossRef] [PubMed]
- Walzer, M.; Pernas, L.E.; Nasso, S.; Bittremieux, W.; Nahnsen, S.; Kelchtermans, P.; Pichler, P.; van den Toorn, H.W.; Staes, A.; Vandenbussche, J.; et al. Qcml: An exchange format for quality control metrics from mass spectrometry experiments. Mol. Cell. Proteom. 2014, 13, 1905–1913. [Google Scholar] [CrossRef] [PubMed]
- Issaq, H.J.; Waybright, T.J.; Veenstra, T.D. Cancer biomarker discovery: Opportunities and pitfalls in analytical methods. Electrophoresis 2011, 32, 967–975. [Google Scholar] [CrossRef] [PubMed]
- Jonsson, P.; Wuolikainen, A.; Thysell, E.; Chorell, E.; Stattin, P.; Wikstrom, P.; Antti, H. Constrained randomization and multivariate effect projections improve information extraction and biomarker pattern discovery in metabolomics studies involving dependent samples. Metabolomics 2015, 11, 1667–1678. [Google Scholar] [CrossRef] [PubMed]
- Scherer, A. Batch Effects and Noise in Microarray Experiments: Sources and Solutions; John Wiley & Sons: Chichester, UK, 2009; Volume 868. [Google Scholar]
- Vivian, J.; Rao, A.; Nothaft, F.A.; Ketchum, C.; Armstrong, J.; Novak, A.; Pfeil, J.; Narkizian, J.; Deran, A.D.; Musselman-Brown, A.; et al. Rapid and efficient analysis of 20,000 RNA-Seq samples with toil. bioRxiv 2016. [Google Scholar] [CrossRef]
- Church, D.M.; Schneider, V.A.; Steinberg, K.M.; Schatz, M.C.; Quinlan, A.R.; Chin, C.-S.; Kitts, P.A.; Aken, B.; Marth, G.T.; Hoffman, M.M.; et al. Extending reference assembly models. Genome Biol. 2015, 16, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Goldfeder, R.L.; Priest, J.R.; Zook, J.M.; Grove, M.E.; Waggott, D.; Wheeler, M.T.; Salit, M.; Ashley, E.A. Medical implications of technical accuracy in genome sequencing. Genome Med. 2016, 8, 24. [Google Scholar] [CrossRef] [PubMed]
- Tewhey, R.; Bansal, V.; Torkamani, A.; Topol, E.J.; Schork, N.J. The importance of phase information for human genomics. Nat. Rev. Genet. 2011, 12, 215–223. [Google Scholar] [CrossRef] [PubMed]
- Zheng, G.X.; Lau, B.T.; Schnall-Levin, M.; Jarosz, M.; Bell, J.M.; Hindson, C.M.; Kyriazopoulou-Panagiotopoulou, S.; Masquelier, D.A.; Merrill, L.; Terry, J.M.; et al. Haplotyping germline and cancer genomes with high-throughput linked-read sequencing. Nat. Biotechnol. 2016, 34, 303–311. [Google Scholar] [CrossRef] [PubMed]
- Chaisson, M.J.; Huddleston, J.; Dennis, M.Y.; Sudmant, P.H.; Malig, M.; Hormozdiari, F.; Antonacci, F.; Surti, U.; Sandstrom, R.; Boitano, M. Resolving the complexity of the human genome using single-molecule sequencing. Nature 2015, 517, 608–611. [Google Scholar] [CrossRef] [PubMed]
- Foquet, M.; Samiee, K.T.; Kong, X.; Chauduri, B.P.; Lundquist, P.M.; Turner, S.W.; Freudenthal, J.; Roitman, D.B. Improved fabrication of zero-mode waveguides for single-molecule detection. J. Appl. Phys. 2008, 103, 034301. [Google Scholar] [CrossRef]
- Clarke, J.; Wu, H.-C.; Jayasinghe, L.; Patel, A.; Reid, S.; Bayley, H. Continuous base identification for single-molecule nanopore DNA sequencing. Nat. Nanotechnol. 2009, 4, 265–270. [Google Scholar] [CrossRef] [PubMed]
- Vinaixa, M.; Schymanski, E.L.; Neumann, S.; Navarro, M.; Salek, R.M.; Yanes, O. Mass spectral databases for LC/MS- and GC/MS-based metabolomics: State of the field and future prospects. TrAC Trends Anal. Chem. 2016, 78, 23–35. [Google Scholar] [CrossRef]
- Wu, L.; Candille, S.I.; Choi, Y.; Xie, D.; Jiang, L.; Li-Pook-Than, J.; Tang, H.; Snyder, M. Variation and genetic control of protein abundance in humans. Nature 2013, 499, 79–82. [Google Scholar] [CrossRef] [PubMed]
- Vogel, C.; Marcotte, E.M. Insights into the regulation of protein abundance from proteomic and transcriptomic analyses. Nat. Rev. Genet. 2012, 13, 227–232. [Google Scholar] [CrossRef] [PubMed]
- Bittremieux, W.; Valkenborg, D.; Martens, L.; Laukens, K. Computational quality control tools for mass spectrometry proteomics. Proteomics 2016. [Google Scholar] [CrossRef] [PubMed]
- Deutsch, E.W.; Overall, C.M.; van Eyk, J.E.; Baker, M.S.; Paik, Y.-K.; Weintraub, S.T.; Lane, L.; Martens, L.; Vandenbrouck, Y.; Kusebauch, U.; et al. Human proteome project mass spectrometry data interpretation guidelines 2.1. J. Proteome Res. 2016. [Google Scholar] [CrossRef] [PubMed]
- Whiteaker, J.R.; Zhao, L.; Anderson, L.; Paulovich, A.G. An automated and multiplexed method for high throughput peptide immunoaffinity enrichment and multiple reaction monitoring mass spectrometry-based quantification of protein biomarkers. Mol. Cell. Proteom. 2010, 9, 184–196. [Google Scholar] [CrossRef] [PubMed]
- Fehniger, T.E.; Boja, E.S.; Rodriguez, H.; Baker, M.S.; Marko-Varga, G. Four areas of engagement requiring strengthening in modern proteomics today. J. Proteome Res. 2014, 13, 5310–5318. [Google Scholar] [CrossRef] [PubMed]
- Shang, J.; Zhu, F.; Vongsangnak, W.; Tang, Y.; Zhang, W.; Shen, B. Evaluation and comparison of multiple aligners for next-generation sequencing data analysis. BioMed Res. Int. 2014, 2014, 309650. [Google Scholar] [CrossRef] [PubMed]
- Pabinger, S.; Dander, A.; Fischer, M.; Snajder, R.; Sperk, M.; Efremova, M.; Krabichler, B.; Speicher, M.R.; Zschocke, J.; Trajanoski, Z. A survey of tools for variant analysis of next-generation genome sequencing data. Brief. Bioinform. 2014, 15, 256–278. [Google Scholar] [CrossRef] [PubMed]
- Evani, U.S.; Challis, D.; Yu, J.; Jackson, A.R.; Paithankar, S.; Bainbridge, M.N.; Jakkamsetti, A.; Pham, P.; Coarfa, C.; Milosavljevic, A. Atlas2 cloud: A framework for personal genome analysis in the cloud. BMC Genom. 2012, 13, S19. [Google Scholar] [CrossRef] [PubMed]
- Cibulskis, K.; Lawrence, M.S.; Carter, S.L.; Sivachenko, A.; Jaffe, D.; Sougnez, C.; Gabriel, S.; Meyerson, M.; Lander, E.S.; Getz, G. Sensitive detection of somatic point mutations in impure and heterogeneous cancer samples. Nat. Biotechnol. 2013, 31, 213–219. [Google Scholar] [CrossRef] [PubMed]
- Koboldt, D.C.; Zhang, Q.; Larson, D.E.; Shen, D.; McLellan, M.D.; Lin, L.; Miller, C.A.; Mardis, E.R.; Ding, L.; Wilson, R.K. Varscan 2: Somatic mutation and copy number alteration discovery in cancer by exome sequencing. Genome Res. 2012, 22, 568–576. [Google Scholar] [CrossRef] [PubMed]
- McKenna, A.; Hanna, M.; Banks, E.; Sivachenko, A.; Cibulskis, K.; Kernytsky, A.; Garimella, K.; Altshuler, D.; Gabriel, S.; Daly, M. The genome analysis toolkit: A mapreduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010, 20, 1297–1303. [Google Scholar] [CrossRef] [PubMed]
- Spencer, D.H.; Tyagi, M.; Vallania, F.; Bredemeyer, A.J.; Pfeifer, J.D.; Mitra, R.D.; Duncavage, E.J. Performance of common analysis methods for detecting low-frequency single nucleotide variants in targeted next-generation sequence data. J. Mol. Diagn. 2014, 16, 75–88. [Google Scholar] [CrossRef] [PubMed]
- Manrai, A.K.; Funke, B.H.; Rehm, H.L.; Olesen, M.S.; Maron, B.A.; Szolovits, P.; Margulies, D.M.; Loscalzo, J.; Kohane, I.S. Genetic misdiagnoses and the potential for health disparities. N. Engl. J. Med. 2016, 375, 655–665. [Google Scholar] [CrossRef] [PubMed]
- Gullapalli, R.R.; Desai, K.V.; Santana-Santos, L.; Kant, J.A.; Becich, M.J. Next generation sequencing in clinical medicine: Challenges and lessons for pathology and biomedical informatics. J. Pathol. Inform. 2012, 3, 40. [Google Scholar] [PubMed]
- Deutsch, E.W. File formats commonly used in mass spectrometry proteomics. Mol. Cell. Proteom. 2012, 11, 1612–1621. [Google Scholar] [CrossRef] [PubMed]
- Misra, B.B.; van der Hooft, J.J. Updates in metabolomics tools and resources: 2014–2015. Electrophoresis 2016, 37, 86–110. [Google Scholar] [CrossRef] [PubMed]
- Annesley, T.; Diamandis, E.; Bachmann, L.; Hanash, S.; Hart, B.; Javahery, R.; Singh, R.; Smith, R. A spectrum of views on clinical mass spectrometry. Clin. Chem. 2016, 62, 30–36. [Google Scholar] [CrossRef] [PubMed]
- Lathrop, J.T.; Jeffery, D.A.; Shea, Y.R.; Scholl, P.F.; Chan, M.M. US food and drug administration perspectives on clinical mass spectrometry. Clin. Chem. 2016, 62, 41–47. [Google Scholar] [CrossRef] [PubMed]
- Levin, N.; Salek, R.M.; Steinbeck, C. Chapter 11—From databases to big data. In Metabolic Phenotyping in Personalized and Public Healthcare; Academic Press: Boston, MA, USA, 2016; pp. 317–331. [Google Scholar]
- GTEx Consortium. The genotype-tissue expression (GTEX) pilot analysis: Multitissue gene regulation in humans. Science 2015, 348, 648–660. [Google Scholar]
- Torell, F.; Bennett, K.; Cereghini, S.; Rannar, S.; Lundstedt-Enkel, K.; Moritz, T.; Haumaitre, C.; Trygg, J.; Lundstedt, T. Multi-organ contribution to the metabolic plasma profile using hierarchical modelling. PLoS ONE 2015, 10, e0129260. [Google Scholar] [CrossRef] [PubMed]
- Do, K.T.; Kastenmüller, G.; Mook-Kanamori, D.O.; Yousri, N.A.; Theis, F.J.; Suhre, K.; Krumsiek, J. Network-based approach for analyzing intra- and interfluid metabolite associations in human blood, urine, and saliva. J. Proteome Res. 2015, 14, 1183–1194. [Google Scholar] [CrossRef] [PubMed]
- McGregor, K.; Bernatsky, S.; Colmegna, I.; Hudson, M.; Pastinen, T.; Labbe, A.; Greenwood, C.M.T. An evaluation of methods correcting for cell-type heterogeneity in DNA methylation studies. Genome Biol. 2016, 17, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Buettner, F.; Natarajan, K.N.; Casale, F.P.; Proserpio, V.; Scialdone, A.; Theis, F.J.; Teichmann, S.A.; Marioni, J.C.; Stegle, O. Computational analysis of cell-to-cell heterogeneity in single-cell RNA-Sequencing data reveals hidden subpopulations of cells. Nat. Biotechnol. 2015, 33, 155–160. [Google Scholar] [CrossRef] [PubMed]
- Houseman, E.A.; Molitor, J.; Marsit, C.J. Reference-free cell mixture adjustments in analysis of DNA methylation data. Bioinformatics 2014, 30, 1431–1439. [Google Scholar] [CrossRef] [PubMed]
- Bock, C.; Farlik, M.; Sheffield, N.C. Multi-omics of single cells: Strategies and applications. Trends Biotechnol. 2016, 34, 605–608. [Google Scholar] [CrossRef] [PubMed]
- Biomarkers Definitions Working Group. Biomarkers and surrogate endpoints: Preferred definitions and conceptual framework. Clin. Pharmacol. Ther. 2001, 69, 89–95. [Google Scholar]
- Halim, A.-B. Biomarkers in Drug Development: A Useful Tool but Discrepant Results May Have a Major Impact; INTECH Open Access Publisher: Rijeka, Croatia, 2011. [Google Scholar]
- Micheel, C.M.; Nass, S.J.; Omenn, G.S. Evolution of Translational Omics: Lessons Learned and the Path Forward; National Academies Press: Washington, DC, USA, 2012. [Google Scholar]
- Feuerstein, G.; Dormer, C.; Ruffolo, R.; Stiles, G.; Walsh, F.; Rutkowski, J. Translational medicine perspectives of biomarkers in drug discovery and development. Part I. Target selection and validation-biomarkers take center stage. Int. Drug Discov. 2007, 2, 36–43. [Google Scholar]
- Brünner, N. What is the difference between “predictive and prognostic biomarkers”? Can you give some examples. Connection 2009, 13, 18. [Google Scholar]
- Frank, R.; Hargreaves, R. Clinical biomarkers in drug discovery and development. Nat. Rev. Drug Discov. 2003, 2, 566–580. [Google Scholar] [CrossRef] [PubMed]
- Horvath, A.R.; Lord, S.J.; StJohn, A.; Sandberg, S.; Cobbaert, C.M.; Lorenz, S.; Monaghan, P.J.; Verhagen-Kamerbeek, W.D.J.; Ebert, C.; Bossuyt, P.M.M. From biomarkers to medical tests: The changing landscape of test evaluation. Clin. Chim. Acta 2014, 427, 49–57. [Google Scholar] [CrossRef] [PubMed]
- Hotelling, H. Analysis of a complex of statistical variables into principal components. J. Educ. Psychol. 1933, 24, 417. [Google Scholar] [CrossRef]
- Rutledge, D.N.; Bouveresse, D.J.-R. Independent components analysis with the jade algorithm. TrAC Trends Anal. Chem. 2013, 50, 22–32. [Google Scholar] [CrossRef]
- Hartigan, J.A.; Wong, M.A. Algorithm as 136: A k-means clustering algorithm. J. R. Stat. Soc. Ser. C 1979, 28, 100–108. [Google Scholar] [CrossRef]
- Johnson, S.C. Hierarchical clustering schemes. Psychometrika 1967, 32, 241–254. [Google Scholar] [CrossRef] [PubMed]
- Kohonen, T. The self-organizing map. Proc. IEEE 1990, 78, 1464–1480. [Google Scholar] [CrossRef]
- Wold, S.; Sjöström, M.; Eriksson, L. Pls-regression: A basic tool of chemometrics. Chemom. Intell. Lab. Syst. 2001, 58, 109–130. [Google Scholar] [CrossRef]
- Trygg, J.; Wold, S. Orthogonal projections to latent structures (O-PLS). J. Chemom. 2002, 16, 119–128. [Google Scholar] [CrossRef]
- Cortes, C.; Vapnik, V. Support-vector networks. Mach. Learn. 1995, 20, 273–297. [Google Scholar] [CrossRef]
- McShane, L.M.; Cavenagh, M.M.; Lively, T.G.; Eberhard, D.A.; Bigbee, W.L.; Williams, P.M.; Mesirov, J.P.; Polley, M.-Y.C.; Kim, K.Y.; Tricoli, J.V.; et al. Criteria for the use of omics-based predictors in clinical trials. Nature 2013, 502, 317–320. [Google Scholar] [CrossRef] [PubMed]
- Satagopam, V.; Gu, W.; Eifes, S.; Gawron, P.; Ostaszewski, M.; Gebel, S.; Barbosa-Silva, A.; Balling, R.; Schneider, R. Integration and visualization of translational medicine data for better understanding of human diseases. Big Data 2016, 4, 97–108. [Google Scholar] [CrossRef] [PubMed]
- Offroy, M.; Duponchel, L. Topological data analysis: A promising big data exploration tool in biology, analytical chemistry and physical chemistry. Anal. Chim. Acta 2016, 910, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Lum, P.; Singh, G.; Lehman, A.; Ishkanov, T.; Vejdemo-Johansson, M.; Alagappan, M.; Carlsson, J.; Carlsson, G. Extracting insights from the shape of complex data using topology. Sci. Rep. 2013, 3, 1236. [Google Scholar] [CrossRef] [PubMed]
- Carlsson, G. Topology and data. Bull. Am. Math. Soc. 2009, 46, 255–308. [Google Scholar] [CrossRef]
- Nielson, J.L.; Paquette, J.; Liu, A.W.; Guandique, C.F.; Tovar, C.A.; Inoue, T.; Irvine, K.-A.; Gensel, J.C.; Kloke, J.; Petrossian, T.C.; et al. Topological data analysis for discovery in preclinical spinal cord injury and traumatic brain injury. Nat. Commun. 2015, 6, 8581. [Google Scholar] [CrossRef] [PubMed]
- Salazar, J.; Amri, H.; Noursi, D.; Abu-Asab, M. Computational tools for parsimony phylogenetic analysis of omics data. Omics J. Integr. Biol. 2015, 19, 471–477. [Google Scholar] [CrossRef] [PubMed]
- Altman, R.B.; Khuri, N.; Salit, M.; Giacomini, K.M. Unmet needs: Research helps regulators do their jobs. Sci. Transl. Med. 2015, 7, 315ps22. [Google Scholar] [CrossRef] [PubMed]
- Zerhouni, E.; Hamburg, M. The need for global regulatory harmonization: A public health imperative. Sci. Transl. Med. 2016, 8, 338ed6. [Google Scholar] [CrossRef] [PubMed]
- Jiang, X.; Zhao, Y.; Wang, X.; Malin, B.; Wang, S.; Ohno-Machado, L.; Tang, H. A community assessment of privacy preserving techniques for human genomes. BMC Med. Inform. Decis. Mak. 2014, 14, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Shoenbill, K.; Fost, N.; Tachinardi, U.; Mendonca, E.A. Genetic data and electronic health records: A discussion of ethical, logistical and technological considerations. J. Am. Med. Inform. Assoc. 2014, 21, 171–180. [Google Scholar] [CrossRef] [PubMed]
- Poste, G. Bring on the biomarkers. Nature 2011, 469, 156–157. [Google Scholar] [CrossRef] [PubMed]
- Gligorijevic, V.; Malod-Dognin, N.; Przulj, N. Integrative methods for analyzing big data in precision medicine. Proteomics 2016, 16, 741–758. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Cheng, W.Y.; Glicksberg, B.S.; Gottesman, O.; Tamler, R.; Chen, R.; Bottinger, E.P.; Dudley, J.T. Identification of type 2 diabetes subgroups through topological analysis of patient similarity. Sci. Transl. Med. 2015, 7, 311ra174. [Google Scholar] [CrossRef] [PubMed]
- Asai, Y.; Abe, T.; Li, L.; Oka, H.; Nomura, T.; Kitano, H. Databases for multilevel biophysiology research available at physiome.jp. Front. Physiol. 2015, 6, 251. [Google Scholar] [CrossRef] [PubMed]
- Garny, A.; Cooper, J.; Hunter, P.J. Toward a VPH/physiome toolkit. Wiley Interdiscip. Rev. Syst. Biol. Med. 2010, 2, 134–147. [Google Scholar] [CrossRef] [PubMed]
- Clancy, C.E.; An, G.; Cannon, W.R.; Liu, Y.; May, E.E.; Ortoleva, P.; Popel, A.S.; Sluka, J.P.; Su, J.; Vicini, P.; et al. Multiscale modeling in the clinic: Drug design and development. Ann. Biomed. Eng. 2016, 44, 2591–2610. [Google Scholar] [CrossRef] [PubMed]
- Henricks, W.H.; Karcher, D.S.; Harrison, J.H.; Sinard, J.H.; Riben, M.W.; Boyer, P.J.; Plath, S.; Thompson, A.; Pantanowitz, L. Pathology informatics essentials for residents: A flexible informatics curriculum linked to accreditation council for graduate medical education milestones. J. Pathol. Inform. 2016, 7, 27. [Google Scholar] [PubMed]
- Louis, D.N.; Feldman, M.; Carter, A.B.; Dighe, A.S.; Pfeifer, J.D.; Bry, L.; Almeida, J.S.; Saltz, J.; Braun, J.; Tomaszewski, J.E. Computational pathology: A path ahead. Arch. Pathol. Lab. Med. 2015, 140, 41–50. [Google Scholar] [CrossRef] [PubMed]
- Louis, D.N.; Gerber, G.K.; Baron, J.M.; Bry, L.; Dighe, A.S.; Getz, G.; Higgins, J.M.; Kuo, F.C.; Lane, W.J.; Michaelson, J.S. Computational pathology: An emerging definition. Arch. Pathol. Lab. Med. 2014, 138, 1133–1138. [Google Scholar] [CrossRef] [PubMed]
- Sirintrapun, S.J.; Zehir, A.; Syed, A.; Gao, J.; Schultz, N.; Cheng, D.T. Translational bioinformatics and clinical research (biomedical) informatics. Clin. Lab. Med. 2016, 36, 153–181. [Google Scholar] [CrossRef] [PubMed]
- Miotto, R.; Li, L.; Kidd, B.A.; Dudley, J.T. Deep patient: An unsupervised representation to predict the future of patients from the electronic health records. Sci. Rep. 2016, 6, 26094. [Google Scholar] [CrossRef] [PubMed]
- Soualmia, L.F.; Lecroq, T. Bioinformatics methods and tools to advance clinical care. Findings from the yearbook 2015 section on bioinformatics and translational informatics. Yearb. Med. Inform. 2015, 10, 170–173. [Google Scholar] [CrossRef] [PubMed]
- Tenenbaum, J.D.; Avillach, P.; Benham-Hutchins, M.; Breitenstein, M.K.; Crowgey, E.L.; Hoffman, M.A.; Jiang, X.; Madhavan, S.; Mattison, J.E.; Nagarajan, R.; et al. An informatics research agenda to support precision medicine: Seven key areas. JAMIA 2016, 23, 791–795. [Google Scholar] [CrossRef] [PubMed]
- Altman, R.B.; Prabhu, S.; Sidow, A.; Zook, J.M.; Goldfeder, R.; Litwack, D.; Ashley, E.; Asimenos, G.; Bustamante, C.D.; Donigan, K.; et al. A research roadmap for next-generation sequencing informatics. Sci. Transl. Med. 2016, 8, 335ps310. [Google Scholar] [CrossRef] [PubMed]
- Sahoo, S.; Franzson, L.; Jonsson, J.J.; Thiele, I. A compendium of inborn errors of metabolism mapped onto the human metabolic network. Mol. BioSyst. 2012, 8, 2545–2558. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cho, D.-Y.; Kim, Y.-A.; Przytycka, T.M. Chapter 5: Network biology approach to complex diseases. PLoS Comput. Biol. 2012, 8, e1002820. [Google Scholar] [CrossRef] [PubMed]
- Hood, L.; Balling, R.; Auffray, C. Revolutionizing medicine in the 21st century through systems approaches. Biotechnol. J. 2012, 7, 992–1001. [Google Scholar] [CrossRef] [PubMed]






© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tebani, A.; Afonso, C.; Marret, S.; Bekri, S. Omics-Based Strategies in Precision Medicine: Toward a Paradigm Shift in Inborn Errors of Metabolism Investigations. Int. J. Mol. Sci. 2016, 17, 1555. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms17091555
Tebani A, Afonso C, Marret S, Bekri S. Omics-Based Strategies in Precision Medicine: Toward a Paradigm Shift in Inborn Errors of Metabolism Investigations. International Journal of Molecular Sciences. 2016; 17(9):1555. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms17091555
Chicago/Turabian StyleTebani, Abdellah, Carlos Afonso, Stéphane Marret, and Soumeya Bekri. 2016. "Omics-Based Strategies in Precision Medicine: Toward a Paradigm Shift in Inborn Errors of Metabolism Investigations" International Journal of Molecular Sciences 17, no. 9: 1555. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms17091555