1. Introduction
Hepatocellular carcinoma (HCC) is the fifth leading cause of tumor related deaths worldwide with increasing incidence in the Western hemisphere [
1]. Main causes are chronic hepatitis B and C virus infection, long-term alcohol abuse, aflatoxins in foodstuff, and rare inherited diseases such as alpha-1 antitrypsin deficiency and hemochromatosis [
2].
Furthermore, hormonal influences like hyperinsulinism in type 2 diabetes mellitus and long-term intake of oral contraceptives are risk factors for hepatocarcinogenesis in the non-cirrhotic liver [
3]. The prognosis and overall survival of HCC patients is poor, with less than 5% five-year survival. Partial resection and liver transplantation remain the only curative options, but most cases of HCC are diagnosed in an advanced stage and are not suitable for surgical therapy. Ablation therapies, including radio-frequency ablation, transarterial chemoembolization, and radioembolization are effective in slowing tumor progression in advanced stages [
4]. HCC is extremely resistant to conventional chemotherapy [
5]. New hopes come from the use of targeted therapies, such as receptor tyrosine kinase inhibitors. Sorafenib (Nexavar
©, Bayer, Karlsruhe, Germany) was the first (and still the only one) of this class of new drugs to be approved by the Food and Drug Administration for the treatment of HCC, as overall survival was improved [
6].
The epidermal growth factor receptor (EGFR) is a transmembraneous protein with intrinsic tyrosine kinase activity and belongs to the family of erbB proteins. Binding of EGF or transforming growth factor α (TGFα) leads to receptor dimerization and autophosphorylation of the intrinsic tyrosine kinase [
7,
8].
Downstream signaling cascades include the rat sarcoma/rat sarcoma-activated factor/mitogen activated protein kinase/extracellular regulated kinase kinase (Ras/Raf/MEK/ERK) and the phosphatidyl-inositide 3 kinase/protein kinase B/mammalian target of rapamycin (PI3K/AKT/mTOR) pathways [
9]. Immunohistochemical analysis often reveals co-localization of EGFR and TGFα in human HCC, supporting the hypothesis of autocrine and paracrine mitogenic action of TGFα to hepatocytes [
10].
Gefitinib (Iressa
®, Astra-Zeneca, Wedel, Germany) is a small molecule designed to block the catalytic domain of the EGFR tyrosine kinase, thus inhibiting downstream signaling [
11]. The results of a contemporary Phase II trial approaching the adjuvant Gefitinib administration after local ablation of HCC are still pending [
12]. Applying these small molecules in models of hepatocarcinogenesis would lead to both a deeper understanding of the molecular pathogenesis of HCC and the evaluation of their efficacy for the treatment of this deadly disease. Continuous oral administration of
N-Nitrosomorpholine (NNM) is a well-established model of chemically-induced hepatocarcinogenesis [
13]. Earliest morphological changes in the liver are pre-neoplastic lesions (foci of altered hepatocytes (FAH)) that are of a clear cell or mixed cell appearance, which progress to hepatocellular adenoma (HCA) and carcinoma (HCC).
FAH can also be induced by hormonal stimuli—after intraportal transplantation of pancreatic islets in diabetic rats (PTx, [
14]), ovarian fragments in ovarectomized rats (OTx, [
15]) and thyroid follicles in thyroidectomized rats (TTx, [
16]). After low-number islet transplantation, insulin related alterations lead to glycogen and fat storing clear cell foci downstream of the islet transplant, which evolve into basophilic foci and progress to HCA and HCC [
14]. The hepatocytes downstream of the transplanted ovarian fragments are characterized by an amphophilic phenotype driven by estrogens [
17], which also evolve to HCA and HCC [
15]. Intraportal transplantation of thyroid follicles and the influence of triiodothyronine (T3) lead to hyperproliferative FAH downstream of the transplants, which are amphophilic-tigroid [
16]. T3 is a direct mitogen for hepatocytes without preceding cell loss [
18]. By contrast to the other models, a progression to HCA or HCC could not be seen in long term experiments after thyroid follicle transplantation.
Overexpression of TGFα is documented in NNM induced hepatocarcinogenesis [
19] and pre-neoplastic lesions of PTx and OTx transplantation models [
14,
15], but not in T3 induced hepatocellular alterations [
16].
Therefore, we investigated the antitumoral effect of Gefitinib in rat models of chemically-induced hepatocarcinogenesis (administration of NNM) and three different intraportal transplantation models of hormonal active tissue (PTx, OTx or TTx), with particular regard to early pre-neoplastic lesions (in the case of NNM, insulin and estrogens, respectively), and hyperproliferative hepatocellular alterations (in the case of triiodothyronine, T3).
3. Discussion
Hepatocarcinogenesis is accompanied by overexpression of multiple growth factors and activation of various signaling pathways, including EGF, Vascular endothelial growth factor (VEGF), Insulin like growth factor (IGF), MAPK, PI3K/AKT/mTOR, and Wnt/β-Catenin [
21,
22,
23,
24,
25,
26]. They might represent valuable targets for systemic molecular-based therapies. EGFR is expressed in most HCC [
10,
27], and EGFR inhibitors such as the monoclonal antibody cetuximab or tyrosine kinase inhibitors such as Gefitinib and Erlotinib are able to suppress HCC growth both in vitro and in vivo [
9,
28,
29].
In this study, we investigated the antiproliferative and antitumoral activity of the EGFR tyrosine kinase inhibitor Gefitinib in various models of hormonally- and chemically-induced hepatocarcinogenesis.
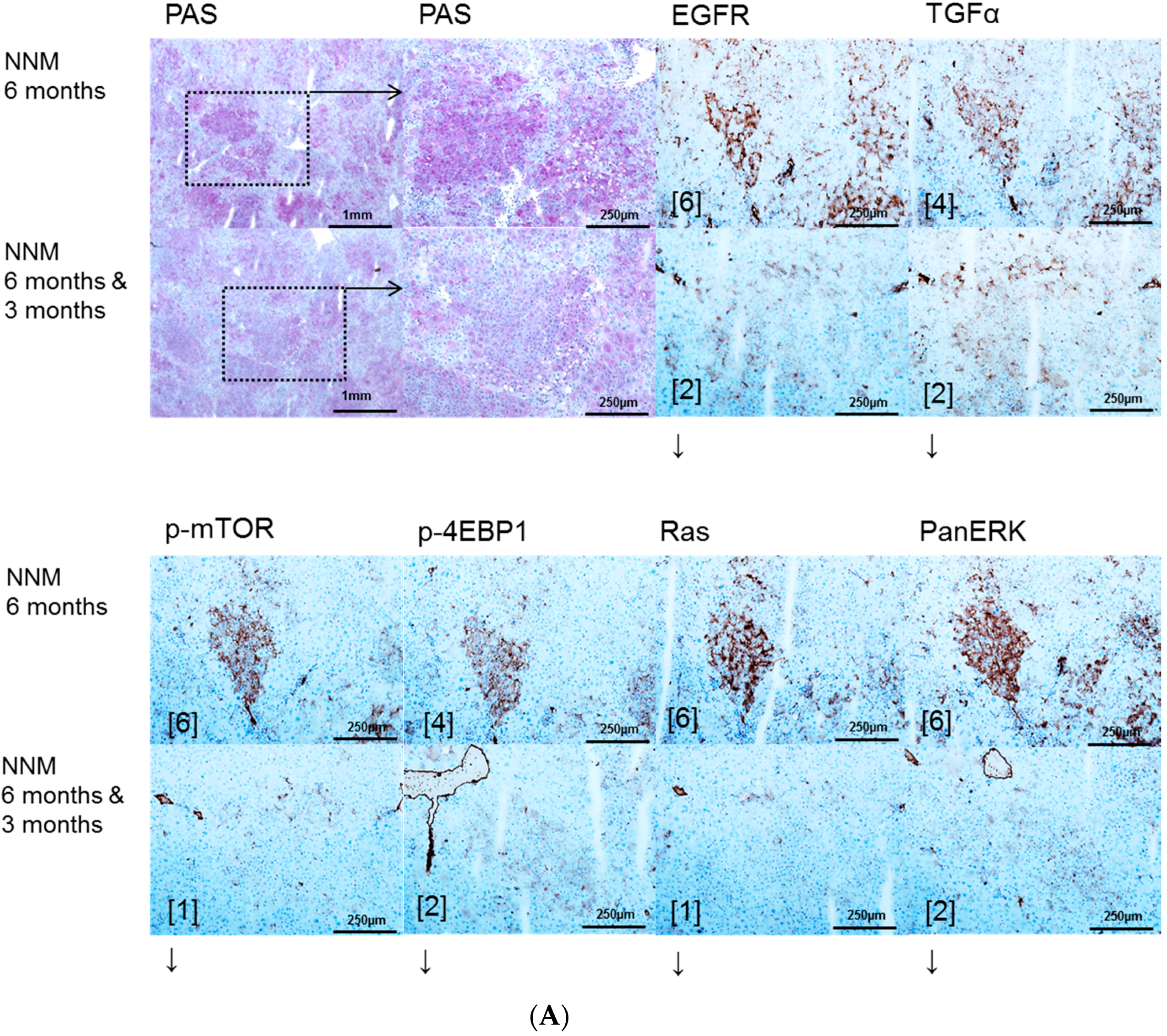
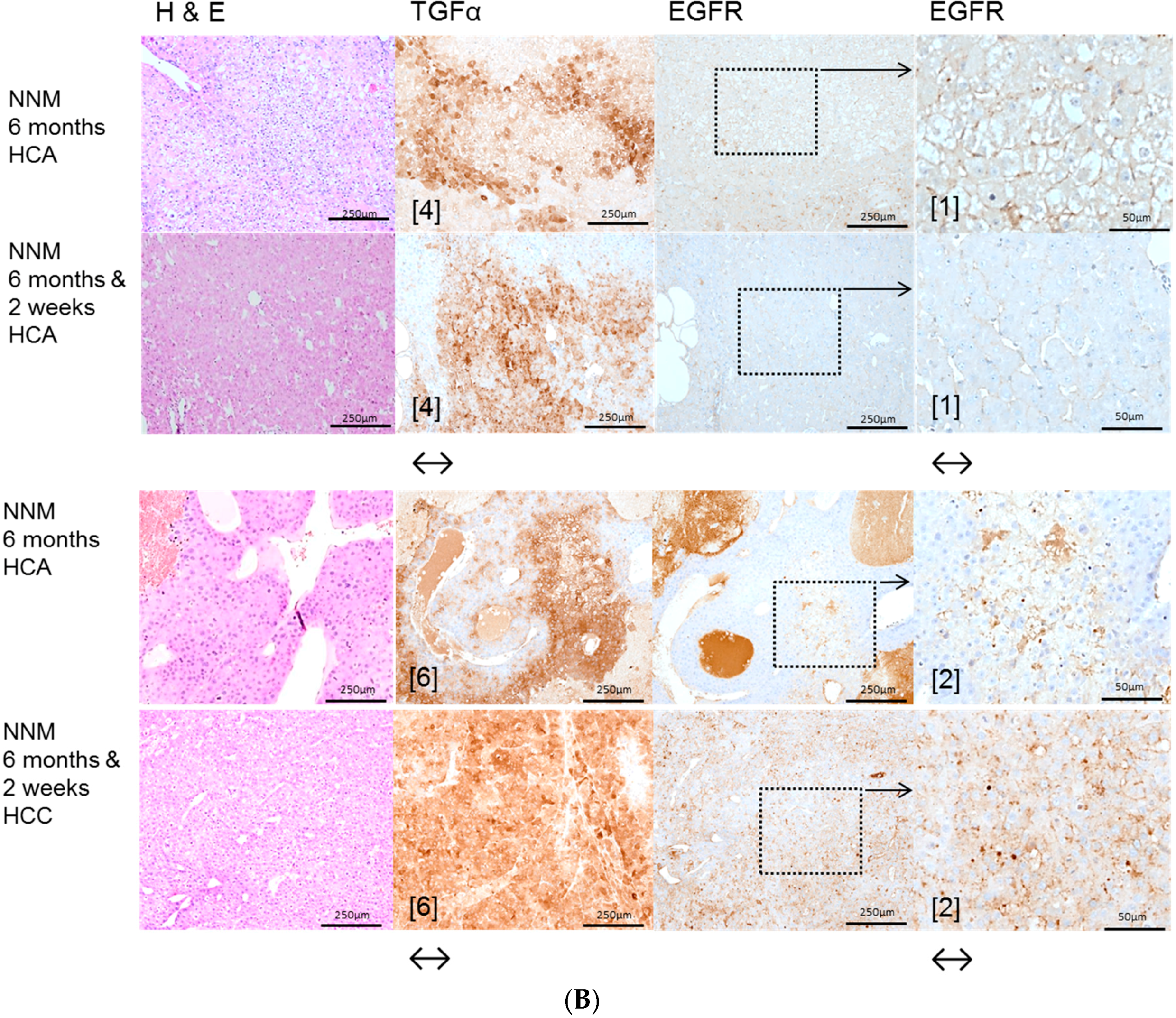
In the NNM model, representing chemical hepatocarcinogenesis without contemporary liver cirrhosis, selective EGFR inhibition by Gefitinib diminished hepatocarcinogenesis at the level of initiation (early time points) and progression (later time points), as the volume fraction of pre-neoplastic liver lesions and HCAs and the number of HCC were reduced. These results are supported by former findings of Gefitinib effects on sequential cirrhosis and HCC development in diethylnitrosamine treated rats [
30], which was interpreted as a chemopreventive option for HCC development. Interestingly, EGFR inhibition by Gefitinib did not influence proliferative activity in NNM induced FAH, but the amount of pre-neoplastic and neoplastic tissue was reduced, accompanied by downregulation of the EGFR and TGFα expression levels and reduction of related protooncogenic signaling. Furthermore, the antitumoral effect of Gefitinib might be directed toward cells other than hepatocytes. In accordance with the latter hypothesis, it has been shown that Gefitinib administration leads to the inhibition of angiogenesis due to apoptosis induction in endothelial cells and decreased production of proangiogenic molecules in pancreatic cancer [
31].
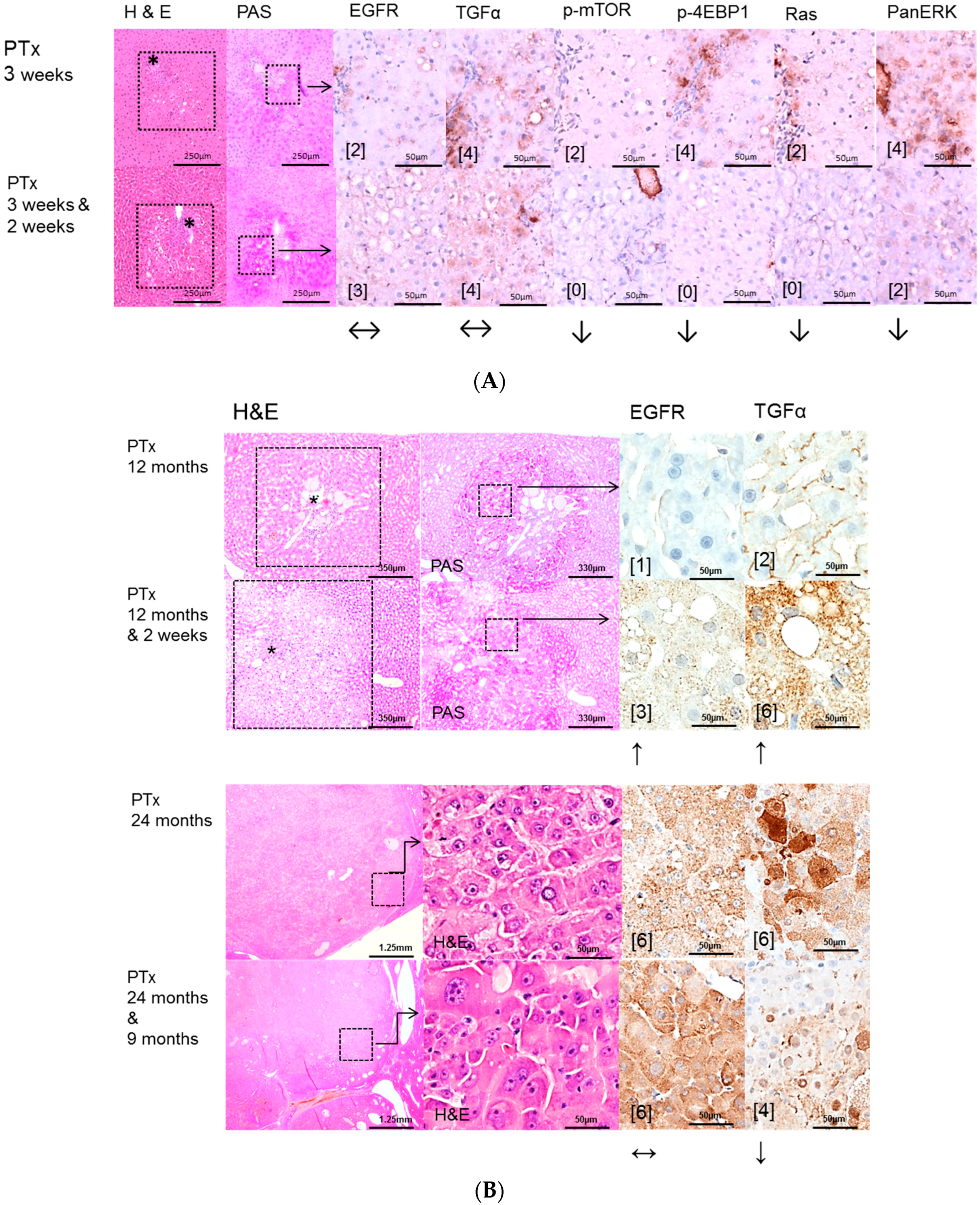
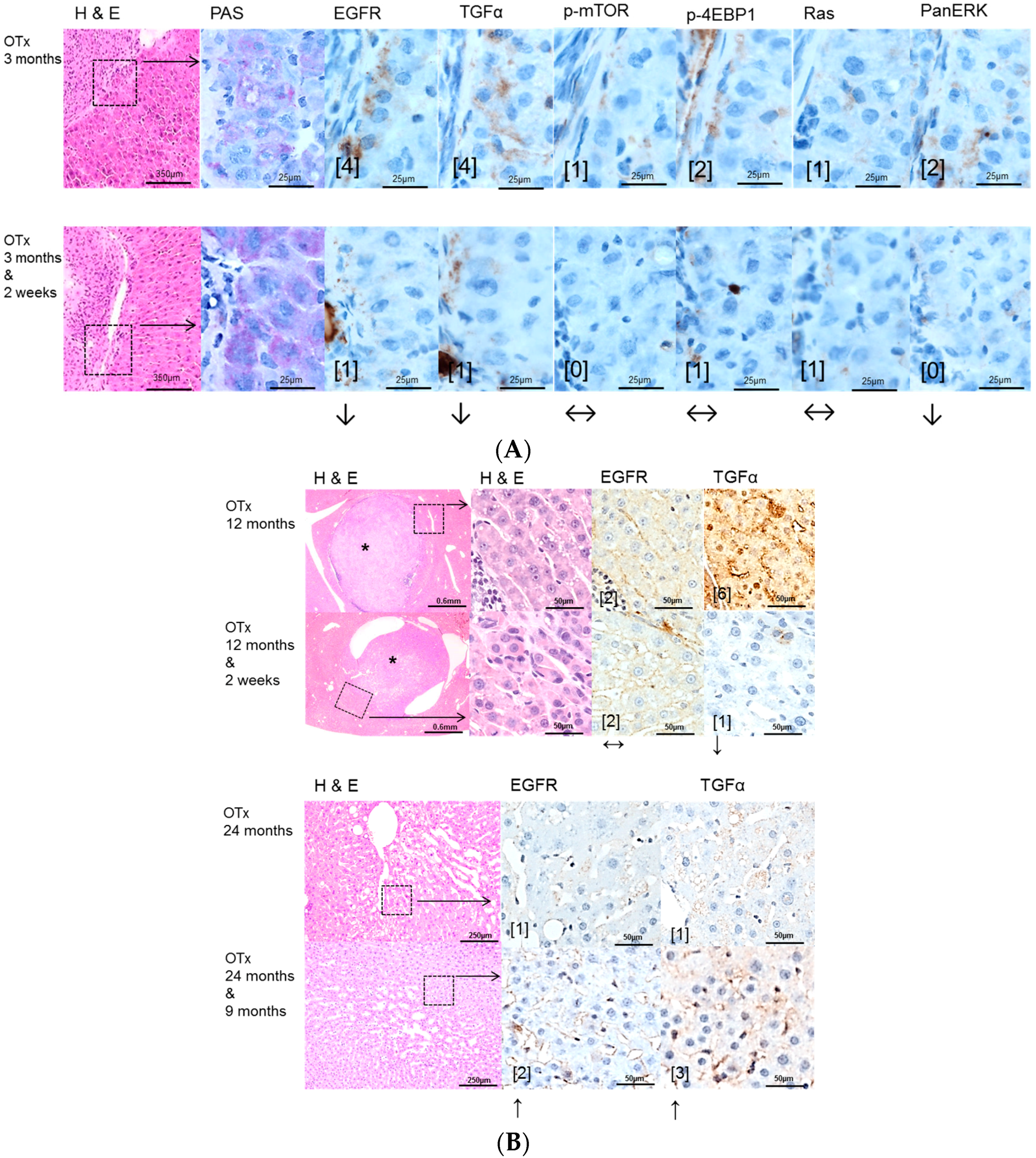
The protooncogenic signaling pathways of AKT/mTOR and Ras/raf-1/MAPK, which are activated in the NNM and the PTx [
32,
33,
34], could also be substantiated to be slightly upregulated after OTx, but not after TTx. At the initiation level of FAH (short time experiments) inhibition of the EGFR by Gefitinib could reduce proliferative activity corresponding to a diminished downstream signaling in FAH after PTx and OTx.
On the contrary, Gefitinib administration did not influence proliferative activity of FAH or the development of hepatocellular neoplasms at the progression level in long-term experiments. Furthermore, downstream signaling pathways were upregulated after Gefitinib administration, suggesting resistance mechanisms in hepatocytes. One has to consider that, due to severe side effects, Gefitinib must be administered at a low dose in long-term experiments, which might partly explain the lack of inhibition of liver tumor development in the various models examined. Nevertheless, resistance to Gefitinib in the late stages of hepatocarcinogenesis is very likely and has been described in the treatment of human non-small-cell lung cancer as well [
35] and in HCCs of diethylnitrosamine treated rats [
30].
Possible resistance of the AKT/mTOR and Ras/raf-1/MAPK oncogenic signaling pathways to EGFR tyrosine kinase inhibitors has been described [
30,
36]. Activation of these pathways by other growth factor receptors like Hepatocyte growth factor/c-Met, IGF-1R and the Insulin receptor could explain the emergence of residual tumors [
37,
38,
39,
40,
41]. Furthermore, other members of the erb-B receptor family, which are also upregulated in human HCC [
22], might also drive hepatocarcinogenesis. The effect of Gefitinib to these receptors remains poorly understood.
Lately, Steinway [
37] found that the EGFR pathway acts as a compensatory survival mechanism upon c-Met inhibition in human c-Met-positive HCC, and combined inhibition of both c-Met and EGFR oncogenic pathways provides superior suppression of HCC tumor growth.
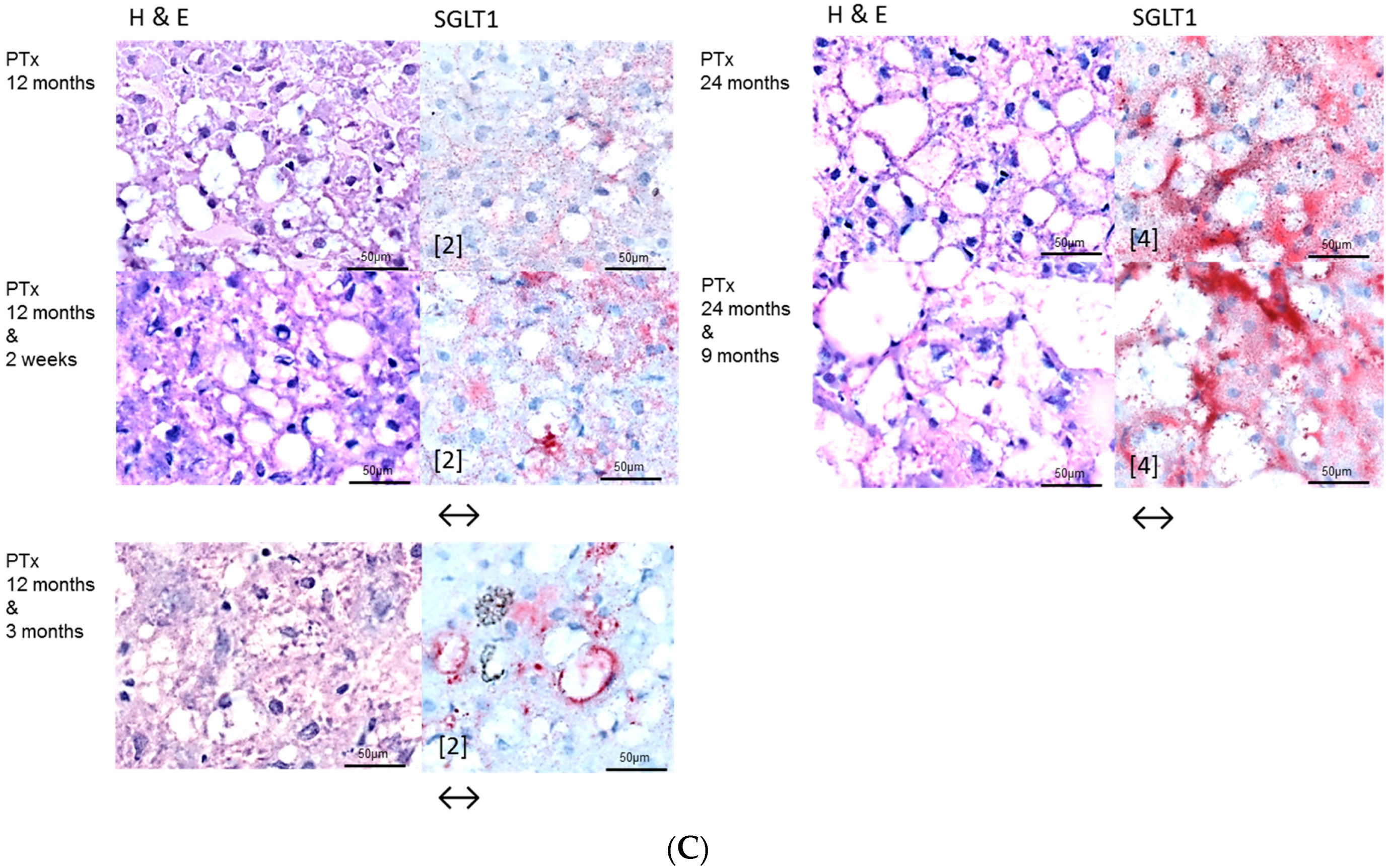
We could substantiate an upregulation of the sodium/glucose cotransporter 1 (SGLT1) in FAH of PTX rats after Gefitinib treatment, another possible explanation of resistance with unrestrained proliferation and cell growth. SGLT1 maintains high intracellular glucose levels leading to cell survival and intracellular homeostasis, is activated by EGFR independent of its kinase activity, and is therefore not influenced by tyrosine kinase inhibitors [
20].
Furthermore, our findings of upregulation of the EGFR expression levels, mainly in liver lesions after PTx, illustrate resistance to Gefitinib after long term application. Nevertheless, this paradoxical conversion of EGFR expression cannot be explained by direct effects of Gefitinib, as its target is only the catalytic domain of the EGFR tyrosine kinase [
11] with inhibition of downstream signaling. Mechanisms of transactivation of the EGFR by other activated receptors in hepatocarcinogenesis, as has been shown for the Insulin-Like Growth Factor Receptor 1 (IGF-1R) in Erlotinib treated HCC cell lines Huh7 [
42], would be a possible explanation.
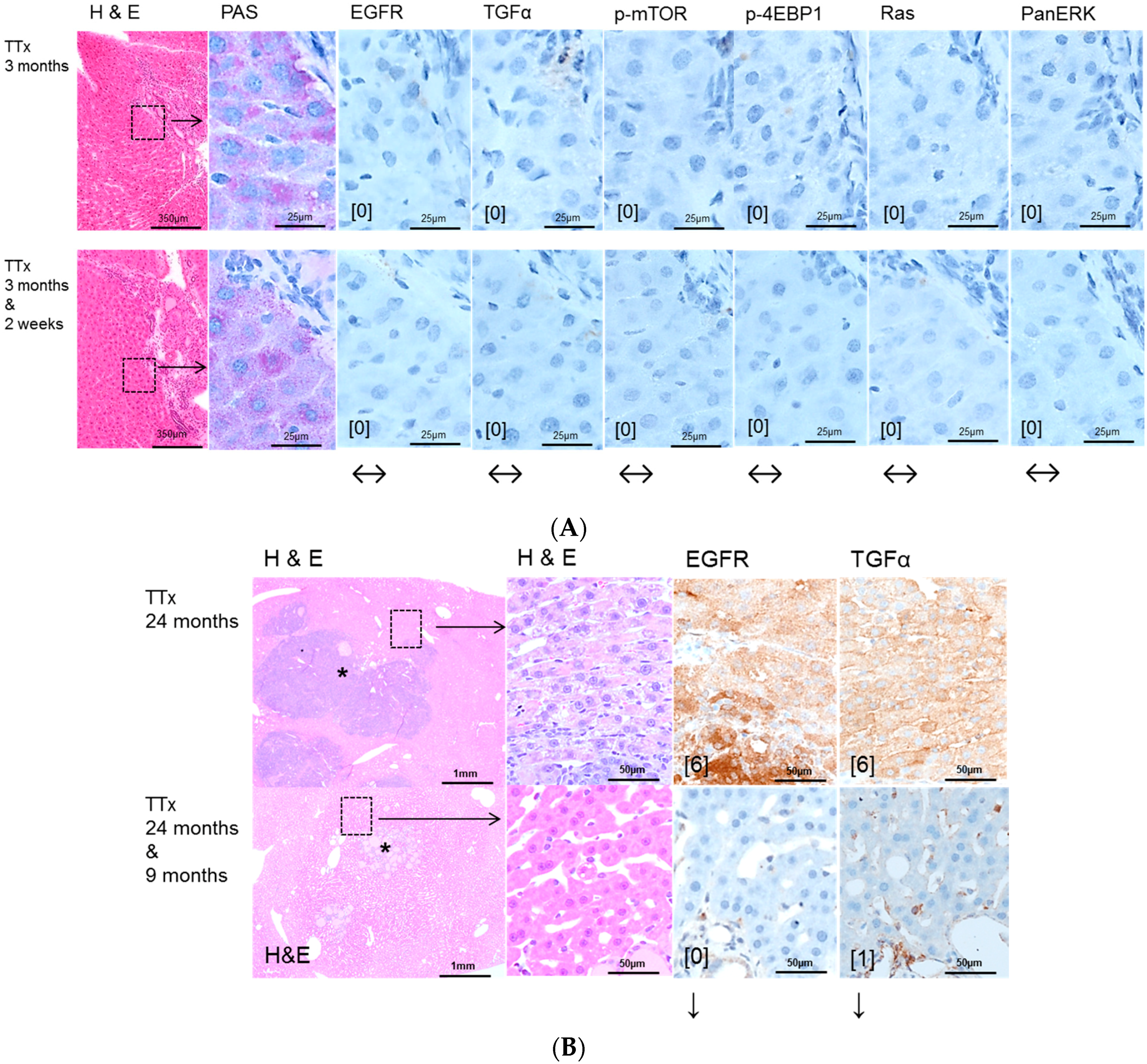
In this study, some HCA could be detected after intraportal transplantation of thyroid follicles (TTx) for the first time in this experimental setting, as manifest hepatocellular neoplasms have not been described by Dombrowski et al. [
16]. Despite one simple HCA near a transplant tumor after TTx, neither the EGFR nor the TGFα were upregulated in hormonal induced alterations of hepatocytes after TTx. Nevertheless, Gefitinib treatment induced a reduction of proliferative activity, suggesting that T3 induced pathways in hepatocytes, like the nuclear thyroid hormone receptor activation or related enzyme activities of i.e., cyclooxygenases, glucose-6-phosphatase or glucose-6-phosphat-dehydrogenase [
16], are influenced by Gefitinib, independently of its EGFR-activity.









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}