
The Role of Deoxycytidine Kinase (dCK) in Radiation-Induced Cell Death


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
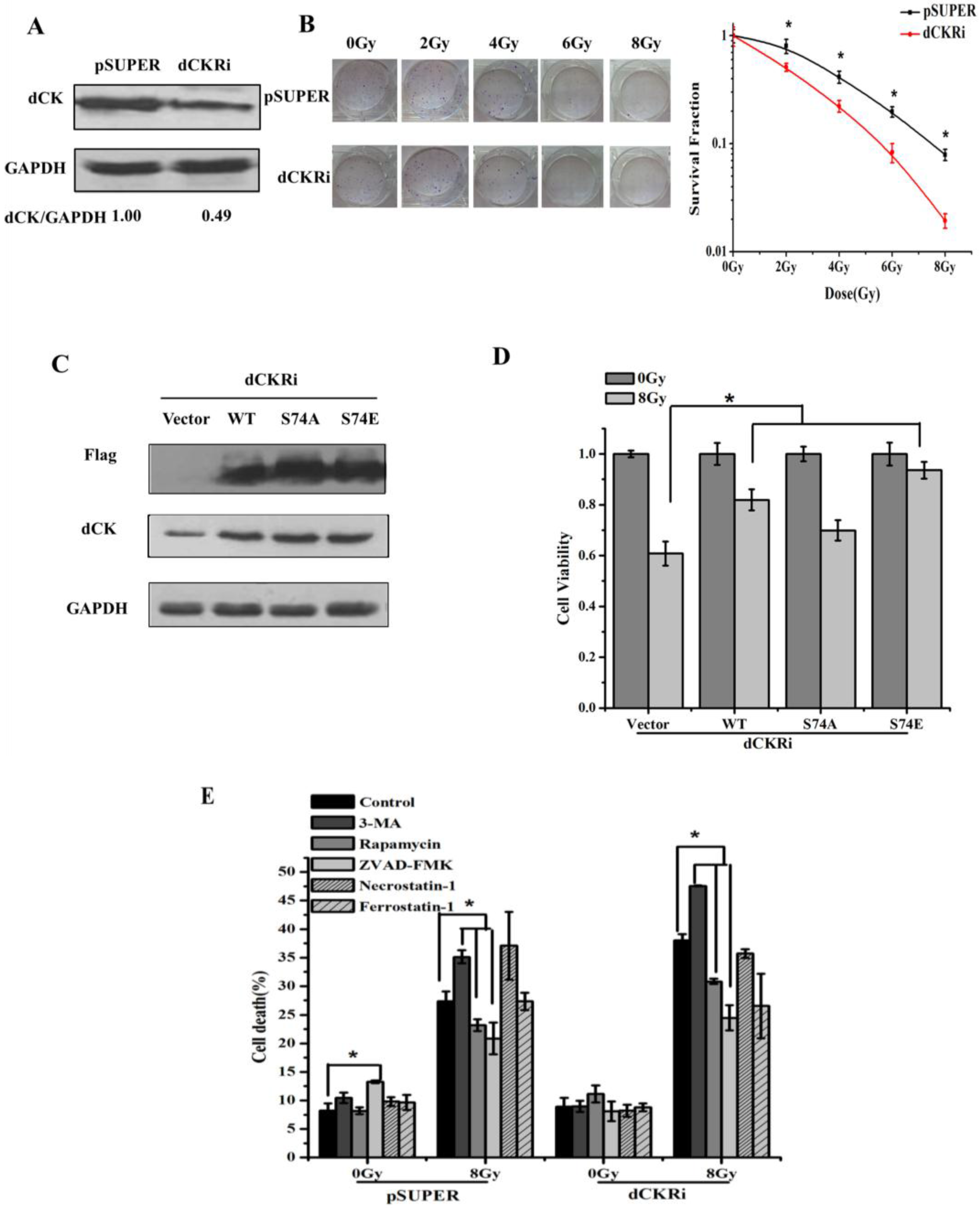
2.1. Deoxycytidine Kinase Decreased Radiation-Induced Cell Death
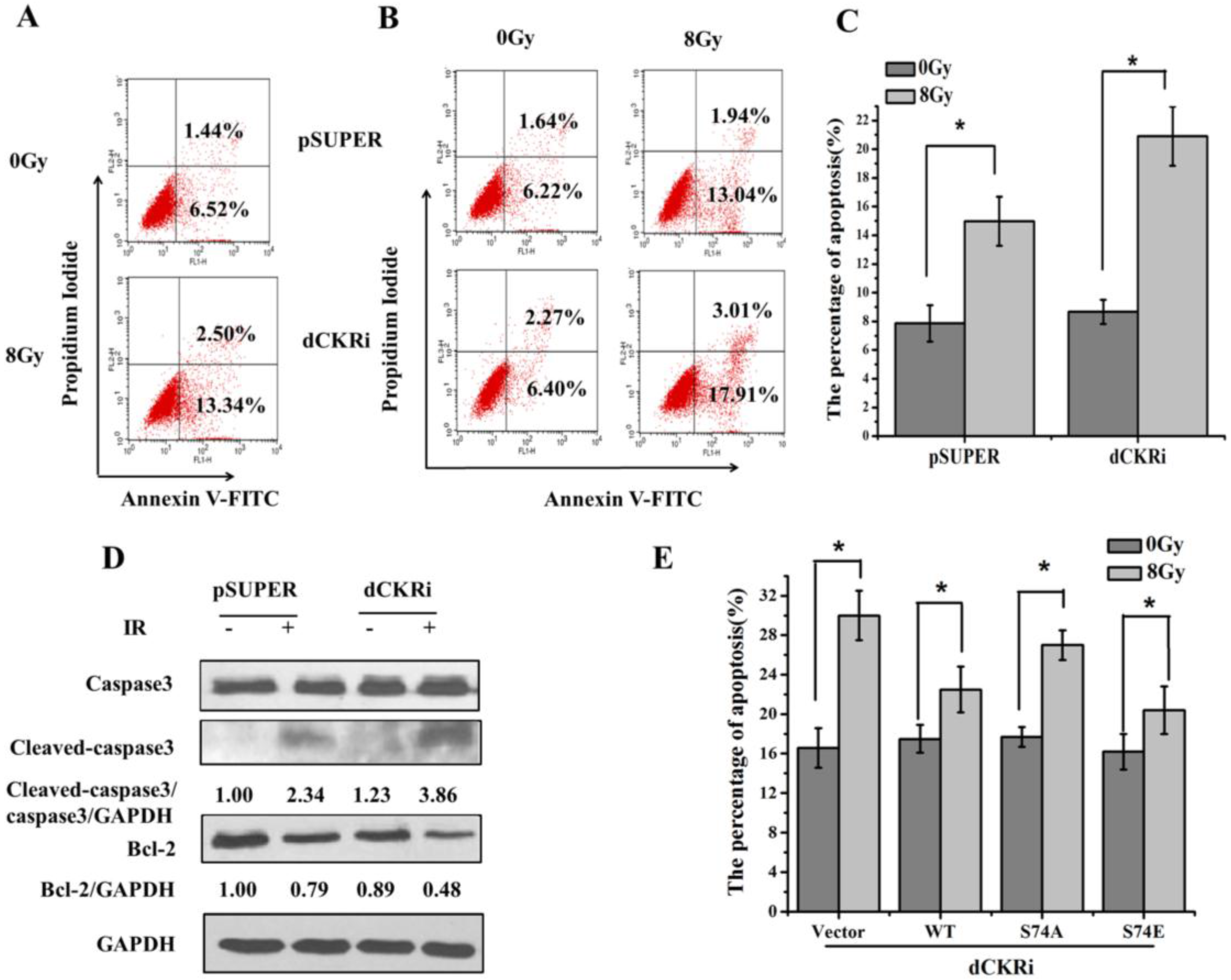
2.2. dCK Suppressed the Ionizing Radiation (IR)-Induced Apoptosis
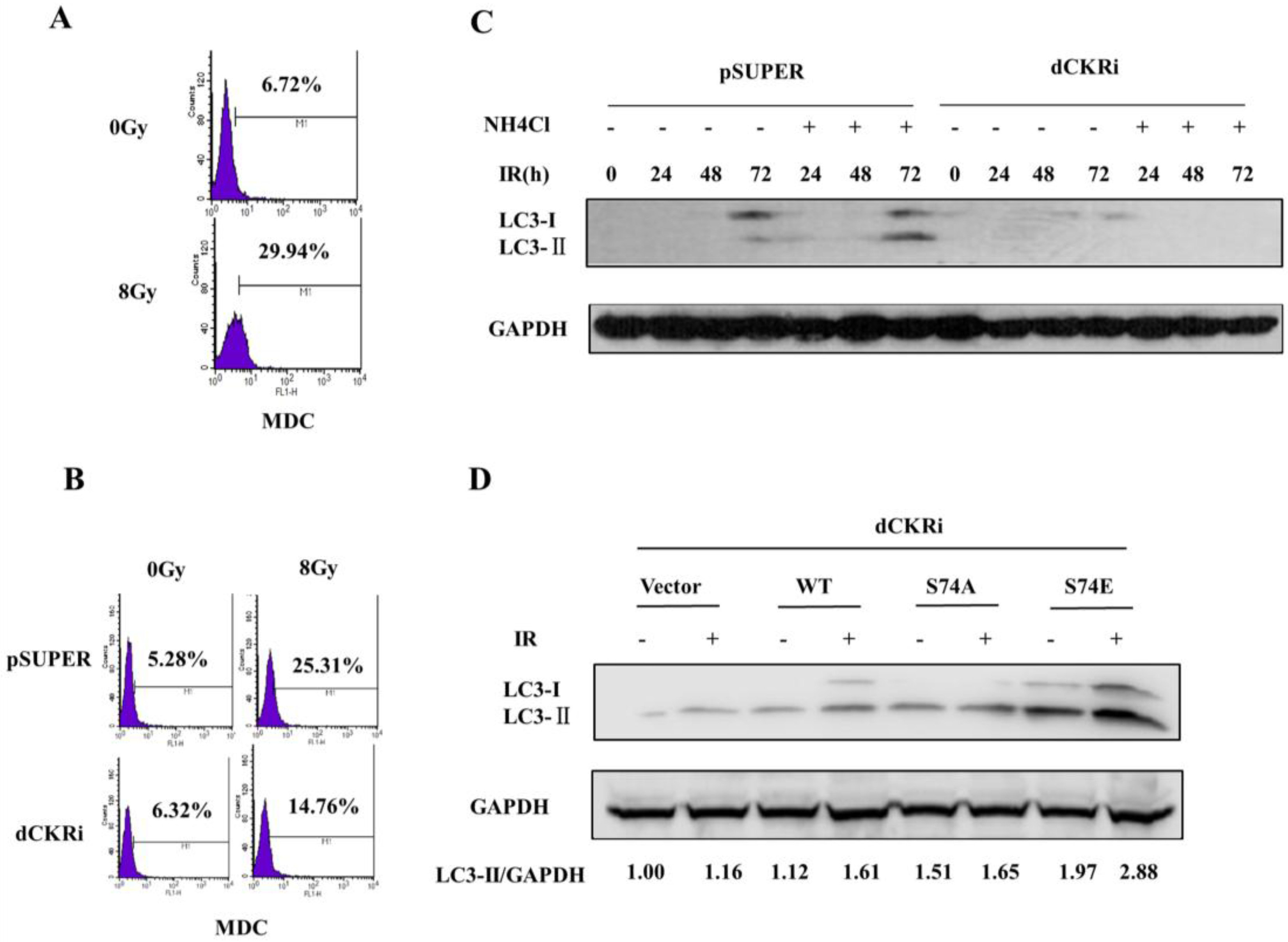
2.3. dCK Promoted the IR-Induced Autophagy
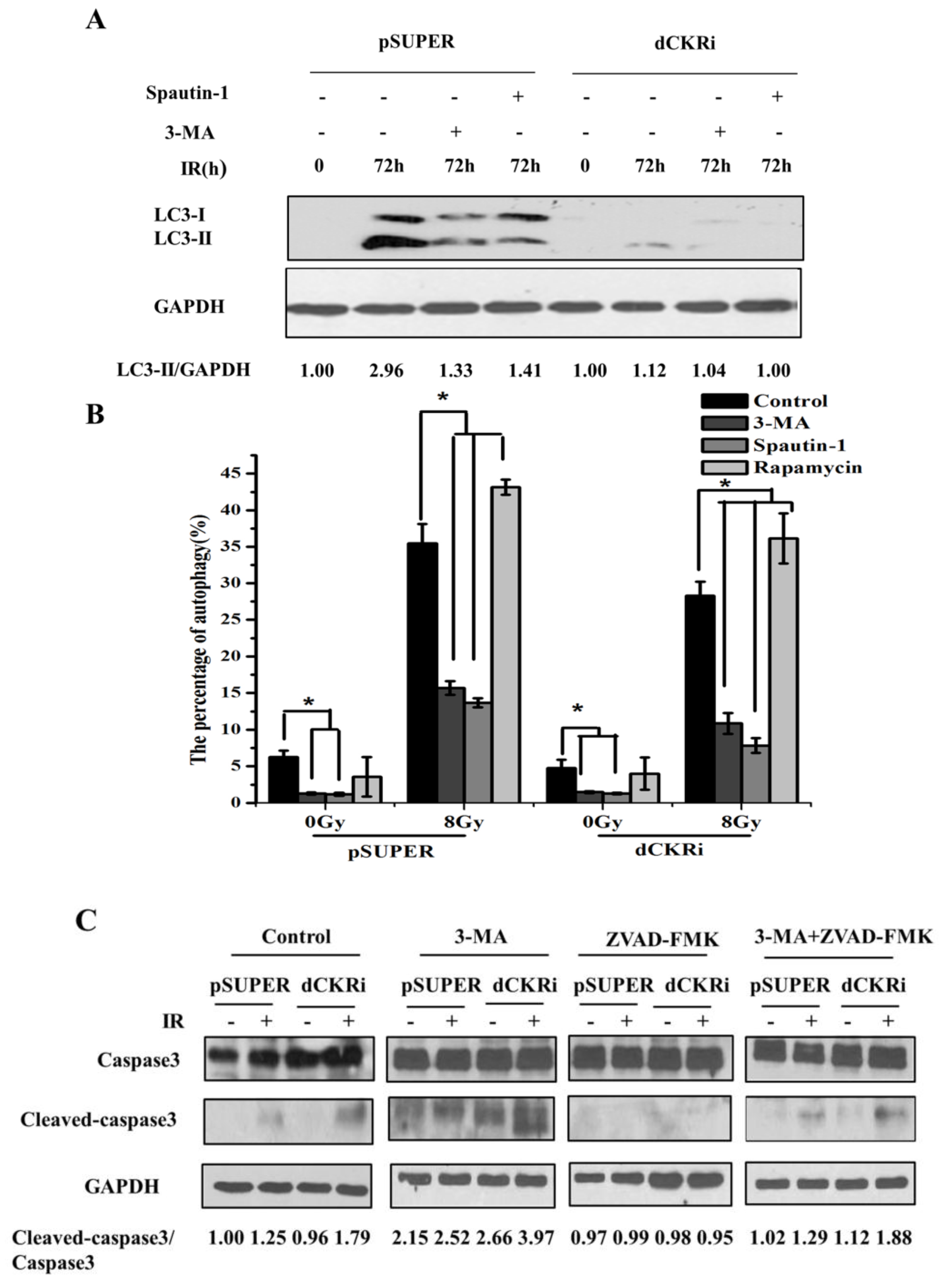
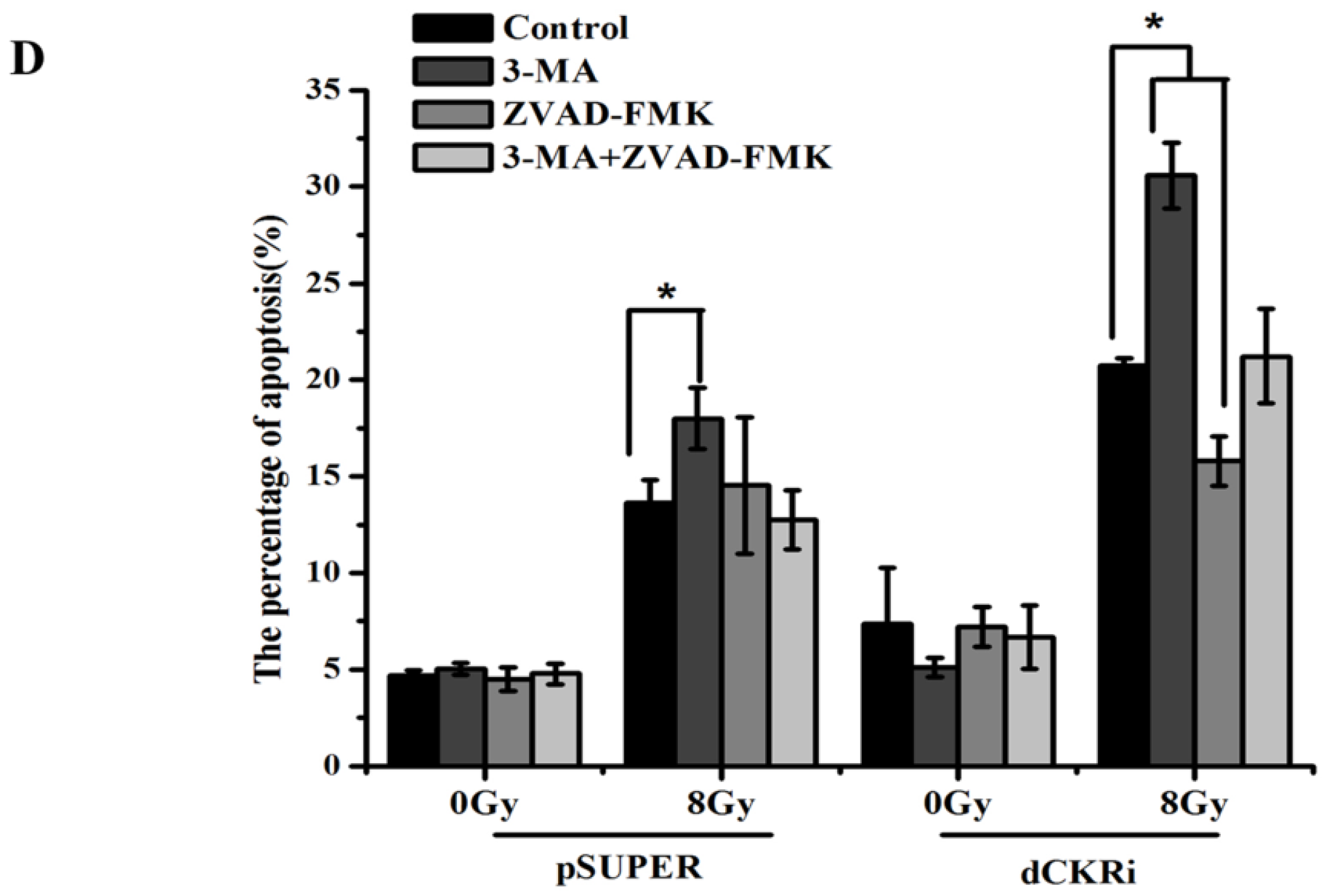
2.4. Suppressing Autophagy Could Increase Apoptosis Induced by ionizing Radiation
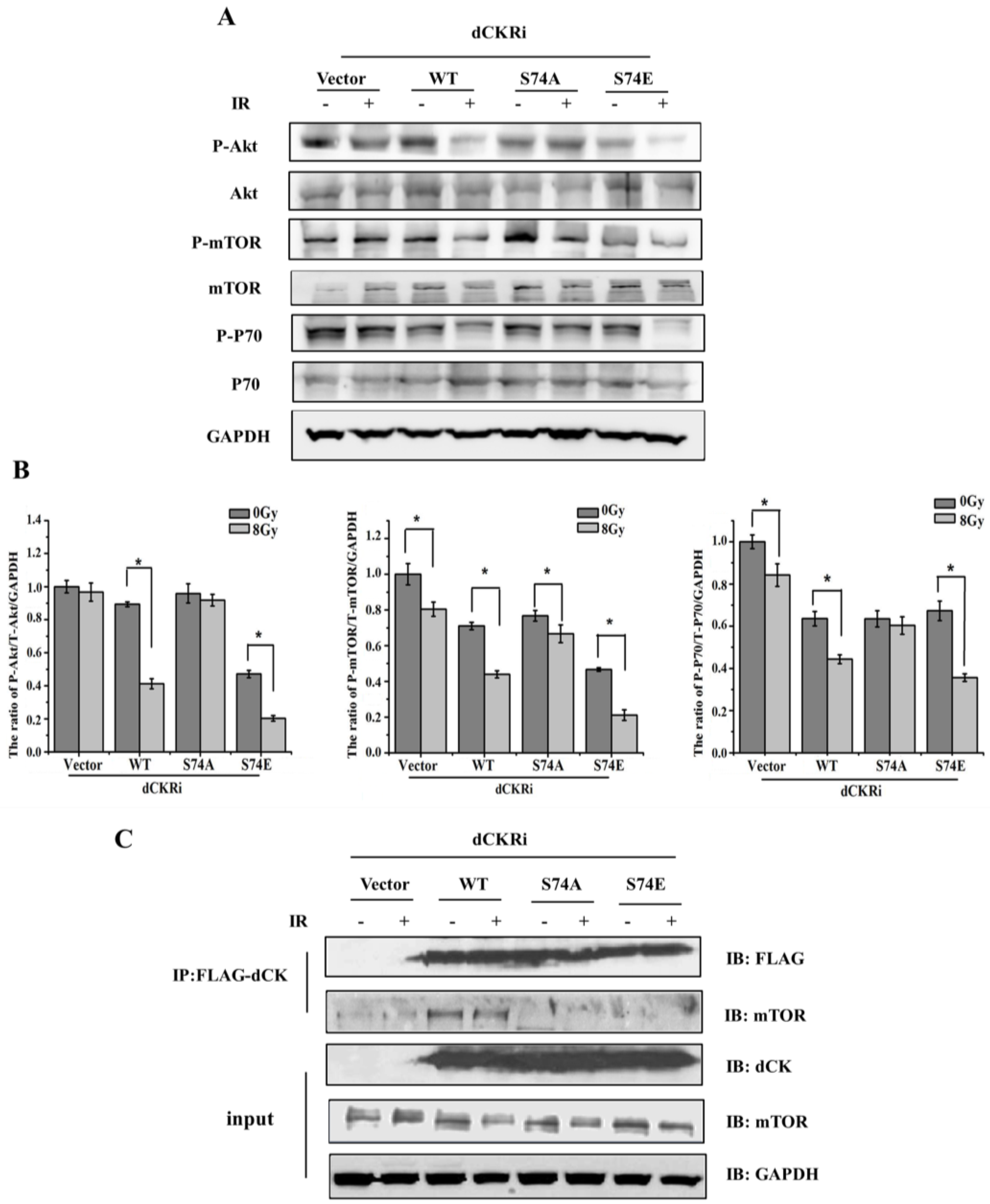
2.5. dCK Regulated IR-Induced Autophagy through mTOR Pathway
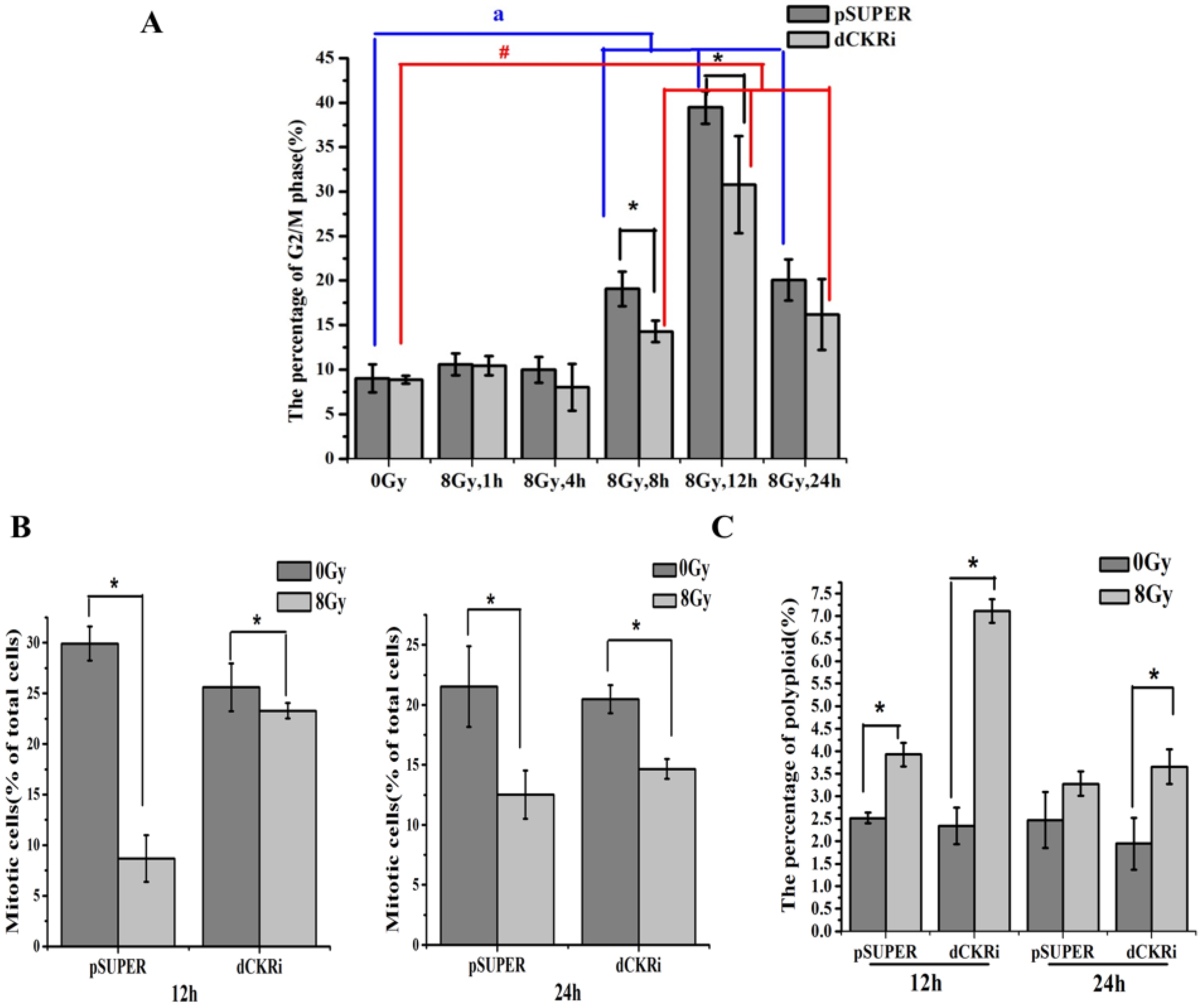
2.6. dCK Is Required for the G2/M Cell Cycle Checkpoint and to Protect Cells against Mitotic Catastrophe
3. Discussion
4. Materials and Methods
4.1. Cell Line, Antibody and Reagents
4.2. Radiation
4.3. Plasmids
4.4. Establishment of Deoxycytidine Kinase (dCK) Silencing Cells
4.5. Western Blot Analysis
4.6. Co-Immunoprecipitation
4.7. Immunofluorescence Microscopy Analysis
4.8. Colony Formation Assay
4.9. Cell Counting Kit-8 (CCK-8) Assay
4.10. Flow Cytometry Analysis
4.11. Statistical Analysis
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Fuchs, Y.; Steller, H. Programmed cell death in animal development and disease. Cell 2011, 147, 742–758. [Google Scholar] [CrossRef] [PubMed]
- Engelberg-Kulka, H.; Amitai, S.; Kolodkin-Gal, I.; Hazan, R. Bacterial programmed cell death and multicellular behavior in bacteria. PLoS Genet. 2006, 2, e135. [Google Scholar] [CrossRef] [PubMed]
- Chaabane, W.; User, S.D.; El-Gazzah, M.; Jaksik, R.; Sajjadi, E.; Rzeszowska-Wolny, J.; Los, M.J. Autophagy, apoptosis, mitoptosis and necrosis: Interdependence between those pathways and effects on cancer. Arch. Immunol. Ther. Exp. 2013, 61, 43–58. [Google Scholar] [CrossRef] [PubMed]
- Fragkos, M.; Beard, P. Mitotic catastrophe occurs in the absence of apoptosis in p53-null cells with a defective G1 checkpoint. PLoS ONE 2011, 6, e22946. [Google Scholar] [CrossRef] [PubMed]
- Kabbage, M.; Williams, B.; Dickman, M.B. Cell death control: The interplay of apoptosis and autophagy in the pathogenicity of sclerotinia sclerotiorum. PLoS Pathog. 2013, 9, e1003287. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liang, B.; Kong, D.; Liu, Y.; Liang, N.; He, M.; Ma, S.; Liu, X. Autophagy inhibition plays the synergetic killing roles with radiation in the multi-drug resistant SKVCR ovarian cancer cells. Radiat. Oncol. 2012, 7, 213–225. [Google Scholar] [CrossRef] [PubMed]
- Czaja, M.J.; Ding, W.X.; Donohue, T.M., Jr.; Friedman, S.L.; Kim, J.S.; Komatsu, M.; Lemasters, J.J.; Lemoine, A.; Lin, J.D.; Ou, J.H.; et al. Functions of autophagy in normal and diseased liver. Autophagy 2013, 9, 1131–1158. [Google Scholar] [CrossRef] [PubMed]
- Yu, L.; Alva, A.; Su, H.; Dutt, P.; Freundt, E.; Welsh, S.; Baehrecke, E.H.; Lenardo, M.J. Regulation of an ATG7-beclin 1 program of autophagic cell death by caspase-8. Science 2004, 304, 1500–1502. [Google Scholar] [CrossRef] [PubMed]
- Kroemer, G.; Galluzzi, L.; Vandenabeele, P.; Abrams, J.; Alnemri, E.S.; Baehrecke, E.H.; Blagosklonny, M.V.; El-Deiry, W.S.; Golstein, P.; Green, D.R.; et al. Classification of cell death: Recommendations of the nomenclature committee on cell death 2009. Cell Death Differ. 2009, 16, 3–11. [Google Scholar] [CrossRef] [PubMed]
- Galluzzi, L.; Vitale, I.; Abrams, J.M.; Alnemri, E.S.; Baehrecke, E.H.; Blagosklonny, M.V.; Dawson, T.M.; Dawson, V.L.; El-Deiry, W.S.; Fulda, S.; et al. Molecular definitions of cell death subroutines: Recommendations of the nomenclature committee on cell death 2012. Cell Death Differ. 2012, 19, 107–120. [Google Scholar] [CrossRef] [PubMed]
- Vitale, I.; Galluzzi, L.; Castedo, M.; Kroemer, G. Mitotic catastrophe: A mechanism for avoiding genomic instability. Nat. Rev. Mol. Cell Biol. 2011, 12, 385–392. [Google Scholar] [CrossRef] [PubMed]
- Castedo, M.; Perfettini, J.L.; Roumier, T.; Andreau, K.; Medema, R.; Kroemer, G. Cell death by mitotic catastrophe: A molecular definition. Oncogene 2004, 23, 2825–2837. [Google Scholar] [CrossRef] [PubMed]
- Ewald, B.; Sampath, D.; Plunkett, W. Nucleoside analogs: Molecular mechanisms signaling cell death. Oncogene 2008, 27, 6522–6537. [Google Scholar] [CrossRef] [PubMed]
- Hazra, S.; Szewczak, A.; Ort, S.; Konrad, M.; Lavie, A. Post-translational phosphorylation of serine 74 of human deoxycytidine kinase favors the enzyme adopting the open conformation making it competent for nucleoside binding and release. Biochemistry 2011, 50, 2870–2880. [Google Scholar] [CrossRef] [PubMed]
- Sigmond, J.; Haveman, J.; Kreder, N.C.; Loves, W.J.; van Bree, C.; Franken, N.A.; Peters, G.J. Enhanced activity of deoxycytidine kinase after pulsed low dose rate and single dose gamma irradiation. Nucleosides Nucleotides Nucleic Acids 2006, 25, 1177–1180. [Google Scholar] [CrossRef] [PubMed]
- Haveman, J.; Sigmond, J.; van Bree, C.; Franken, N.A.; Koedooder, C.; Peters, G.J. Time course of enhanced activity of deoxycytidine kinase and thymidine kinase 1 and 2 in cultured human squamous lung carcinoma cells, SW-1573, induced by gamma-irradiation. Oncol. Rep. 2006, 16, 901–905. [Google Scholar] [PubMed]
- Smal, C.; Vertommen, D.; Bertrand, L.; Rider, M.H.; van den Neste, E.; Bontemps, F. Identification of phosphorylation sites on human deoxycytidine kinase after overexpression in eucaryotic cells. Nucleosides Nucleotides Nucleic Acids 2006, 25, 1141–1146. [Google Scholar] [CrossRef] [PubMed]
- Amsailale, R.; van Den Neste, E.; Arts, A.; Starczewska, E.; Bontemps, F.; Smal, C. Phosphorylation of deoxycytidine kinase on Ser-74: Impact on kinetic properties and nucleoside analog activation in cancer cells. Biochem. Pharmacol. 2012, 84, 43–51. [Google Scholar] [CrossRef] [PubMed]
- Csapo, Z.; Keszler, G.; Safrany, G.; Spasokoukotskaja, T.; Talianidis, I.; Staub, M.; Sasvari-Szekely, M. Activation of deoxycytidine kinase by gamma-irradiation and inactivation by hyperosmotic shock in human lymphocytes. Biochem. Pharmacol. 2003, 65, 2031–2039. [Google Scholar] [CrossRef]
- Van Den Neste, E.; Smal, C.; Cardoen, S.; Delacauw, A.; Frankard, J.; Ferrant, A.; van den Berghe, G.; Bontemps, F. Activation of deoxycytidine kinase by UV-C-irradiation in chronic lymphocytic leukemia B-lymphocytes. Biochem. Pharmacol. 2003, 65, 573–580. [Google Scholar] [CrossRef]
- Lee, M.W.; Parker, W.B.; Xu, B. New insights into the synergism of nucleoside analogs with radiotherapy. Radiat. Oncol. 2013, 8, 223–233. [Google Scholar] [CrossRef] [PubMed]
- Kerr, M.; Scott, H.E.; Groselj, B.; Stratford, M.R.; Karaszi, K.; Sharma, N.L.; Kiltie, A.E. Deoxycytidine kinase expression underpins response to gemcitabine in bladder cancer. Clin. Cancer Res. 2014, 20, 5435–5445. [Google Scholar] [CrossRef] [PubMed]
- Szatmari, T.; Huszty, G.; Desaknai, S.; Spasokoukotskaja, T.; Sasvari-Szekely, M.; Staub, M.; Esik, O.; Safrany, G.; Lumniczky, K. Adenoviral vector transduction of the human deoxycytidine kinase gene enhances the cytotoxic and radiosensitizing effect of gemcitabine on experimental gliomas. Cancer Gene Ther. 2008, 15, 154–164. [Google Scholar] [CrossRef] [PubMed]
- Bunimovich, Y.L.; Nair-Gill, E.; Riedinger, M.; McCracken, M.N.; Cheng, D.; McLaughlin, J.; Radu, C.G.; Witte, O.N. Deoxycytidine kinase augments ATM-mediated DNA repair and contributes to radiation resistance. PLoS ONE 2014, 9, e104125. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.; Lee, M.; Hao, J.; Cui, X.; Guo, X.; Smal, C.; Bontemps, F.; Ma, S.; Liu, X.; Engler, D.; et al. Deoxycytidine kinase regulates the G2/M checkpoint through interaction with cyclin-dependent kinase 1 in response to DNA damage. Nucleic Acids Res. 2012, 40, 9621–9632. [Google Scholar] [CrossRef] [PubMed]
- Liang, N.; Jia, L.L.; Liu, Y.; Liang, B.; Kong, D.J.; Yan, M.M.; Ma, S.M.; Liu, X.D. ATM pathway is essential for ionizing radiation-induced autophagy. Cell Signal. 2013, 25, 2530–2539. [Google Scholar] [CrossRef] [PubMed]
- Martin-Acebes, M.A.; Blazquez, A.B.; de Oya, N.J.; Escribano-Romero, E.; Shi, P.Y.; Saiz, J.C. A single amino acid substitution in the core protein of West Nile virus increases resistance to acidotropic compounds. PLoS ONE 2013, 8, e69479. [Google Scholar] [CrossRef] [PubMed]
- Kinner, A.; Wu, W.; Staudt, C.; Iliakis, G. Gamma-H2AX in recognition and signaling of DNA double-strand breaks in the context of chromatin. Nucleic Acids Res. 2008, 36, 5678–5694. [Google Scholar] [CrossRef] [PubMed]
- Dickey, J.S.; Redon, C.E.; Nakamura, A.J.; Baird, B.J.; Sedelnikova, O.A.; Bonner, W.M. H2AX: Functional roles and potential applications. Chromosoma 2009, 118, 683–692. [Google Scholar] [CrossRef] [PubMed]
- Clingen, P.H.; Wu, J.Y.; Miller, J.; Mistry, N.; Chin, F.; Wynne, P.; Prise, K.M.; Hartley, J.A. Histone H2AX phosphorylation as a molecular pharmacological marker for DNA interstrand crosslink cancer chemotherapy. Biochem. Pharmacol. 2008, 76, 19–27. [Google Scholar] [CrossRef] [PubMed]
- Rosell, R.; Lord, R.V.; Taron, M.; Reguart, N. DNA repair and cisplatin resistance in non-small-cell lung cancer. Lung Cancer 2002, 38, 217–227. [Google Scholar] [CrossRef]
- Park, S.Y.; Hong, Y.C.; Kim, J.H.; Kwak, S.M.; Cho, J.H.; Lee, H.L.; Ryu, J.S. Effect of ERCC1 polymorphisms and the modification by smoking on the survival of non-small cell lung cancer patients. Med. Oncol. 2006, 23, 489–498. [Google Scholar] [CrossRef]
- Rivero, A.; Rapado, I.; Tomas, J.F.; Montalban, C.; de Ona, R.; Paz-Carreira, J.; Canales, M.; Martinez, R.; Sanchez-Godoy, P.; de Sevilla, A.F.; et al. Relationship between deoxycytidine kinase (dCK) genotypic variants and fludarabine toxicity in patients with follicular lymphoma. Leuk. Res. 2011, 35, 431–437. [Google Scholar] [CrossRef] [PubMed]
- Rejiba, S.; Bigand, C.; Parmentier, C.; Hajri, A. Gemcitabine-based chemogene therapy for pancreatic cancer using Ad-dCK::UMK GDEPT and TS/RR siRNA strategies. Neoplasia 2009, 11, 637–650. [Google Scholar] [CrossRef] [PubMed]
- Sigmond, J.; Bergman, A.M.; Leon, L.G.; Loves, W.J.; Hoebe, E.K.; Peters, G.J. Staurosporine increases toxicity of gemcitabine in non-small cell lung cancer cells: Role of protein kinase C, deoxycytidine kinase and ribonucleotide reductase. Anticancer Drugs 2010, 21, 591–599. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Chen, Z.; Zuo, W.; Zhu, X.; Li, Y.; Liu, X.; Wei, X. Omacetaxine mepesuccinate induces apoptosis and cell cycle arrest, promotes cell differentiation, and reduces telomerase activity in diffuse large B-cell lymphoma cells. Mol. Med. Rep. 2016, 13, 3092–3100. [Google Scholar] [CrossRef] [PubMed]
- Weng, T.; Karmouty-Quintana, H.; Garcia-Morales, L.J.; Molina, J.G.; Pedroza, M.; Bunge, R.R.; Bruckner, B.A.; Loebe, M.; Seethamraju, H.; Blackburn, M.R. Hypoxia-induced deoxycytidine kinase expression contributes to apoptosis in chronic lung disease. FASEB J. 2013, 27, 2013–2026. [Google Scholar] [CrossRef] [PubMed]
- Ju, J.; Qi, Z.; Cai, X.; Cao, P.; Huang, Y.; Wang, S.; Liu, N.; Chen, Y. The apoptotic effects of toosendanin are partially mediated by activation of deoxycytidine kinase in HL-60 cells. PLoS ONE 2012, 7, e52536. [Google Scholar] [CrossRef] [PubMed]
- Kirsch, D.G.; Doseff, A.; Chau, B.N.; Lim, D.S.; de Souza-Pinto, N.C.; Hansford, R.; Kastan, M.B.; Lazebnik, Y.A.; Hardwick, J.M. Caspase-3-dependent cleavage of Bcl-2 promotes release of cytochrome c. J. Biol. Chem. 1999, 274, 21155–21161. [Google Scholar] [CrossRef] [PubMed]
- Maiuri, M.C.; Zalckvar, E.; Kimchi, A.; Kroemer, G. Self-eating and self-killing: Crosstalk between autophagy and apoptosis. Nat. Rev. Mol. Cell Biol. 2007, 8, 741–752. [Google Scholar] [CrossRef] [PubMed]
- Kroemer, G.; Marino, G.; Levine, B. Autophagy and the integrated stress response. Mol. Cell 2010, 40, 280–293. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Klionsky, D.J. The regulation of autophagy—Unanswered questions. J. Cell Sci. 2011, 124, 161–170. [Google Scholar] [CrossRef] [PubMed]
- Smal, C.; Vertommen, D.; Amsailale, R.; Arts, A.; Degand, H.; Morsomme, P.; Rider, M.H.; Neste, E.V.; Bontemps, F. Casein kinase 1δ activates human recombinant deoxycytidine kinase by ser-74 phosphorylation, but is not involved in the in vivo regulation of its activity. Arch. Biochem. Biophys. 2010, 502, 44–52. [Google Scholar] [CrossRef] [PubMed]
- Horie, R.; Nakamura, O.; Yamagami, Y.; Mori, M.; Nishimura, H.; Fukuoka, N.; Yamamoto, T. Apoptosis and antitumor effects induced by the combination of an mTOR inhibitor and an autophagy inhibitor in human osteosarcoma MG63 cells. Int. J. Oncol. 2016, 48, 37–44. [Google Scholar] [CrossRef] [PubMed]
- Seglen, P.O.; Gordon, P.B. 3-methyladenine: Specific inhibitor of autophagic/lysosomal protein degradation in isolated rat hepatocytes. Proc. Natl. Acad. Sci. USA 1982, 79, 1889–1892. [Google Scholar] [CrossRef] [PubMed]
- Klionsky, D.J.; Baehrecke, E.H.; Brumell, J.H.; Chu, C.T.; Codogno, P.; Cuervo, A.M.; Debnath, J.; Deretic, V.; Elazar, Z.; Eskelinen, E.L.; et al. A comprehensive glossary of autophagy-related molecules and processes (2nd edition). Autophagy 2011, 7, 1273–1294. [Google Scholar] [CrossRef] [PubMed]
- Fan, J.; Zeng, X.; Li, Y.; Wang, S.; Wang, Z.; Sun, Y.; Gao, H.; Zhang, G.; Feng, M.; Ju, D. Autophagy plays a critical role in ChLym-1-induced cytotoxicity of non-hodgkin’s lymphoma cells. PLoS ONE 2013, 8, e72478. [Google Scholar] [CrossRef] [PubMed]
- Fan, W.; Zhou, Z.Y.; Huang, X.F.; Bao, C.D.; Du, F. Deoxycytidine kinase promotes the migration and invasion of fibroblast-like synoviocytes from rheumatoid arthritis patients. Int. J. Clin. Exp. Pathol. 2013, 6, 2733–2744. [Google Scholar] [PubMed]
- Margottin-Goguet, F.; Hsu, J.Y.; Loktev, A.; Hsieh, H.M.; Reimann, J.D.; Jackson, P.K. Prophase destruction of emi1 by the SCF(βTrCP/Slimb) ubiquitin ligase activates the anaphase promoting complex to allow progression beyond prometaphase. Dev. Cell 2003, 4, 813–826. [Google Scholar] [CrossRef]







© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhong, R.; Xin, R.; Chen, Z.; Liang, N.; Liu, Y.; Ma, S.; Liu, X. The Role of Deoxycytidine Kinase (dCK) in Radiation-Induced Cell Death. Int. J. Mol. Sci. 2016, 17, 1939. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms17111939
Zhong R, Xin R, Chen Z, Liang N, Liu Y, Ma S, Liu X. The Role of Deoxycytidine Kinase (dCK) in Radiation-Induced Cell Death. International Journal of Molecular Sciences. 2016; 17(11):1939. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms17111939
Chicago/Turabian StyleZhong, Rui, Rui Xin, Zongyan Chen, Nan Liang, Yang Liu, Shumei Ma, and Xiaodong Liu. 2016. "The Role of Deoxycytidine Kinase (dCK) in Radiation-Induced Cell Death" International Journal of Molecular Sciences 17, no. 11: 1939. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms17111939