
The Roles of Autophagy and the Inflammasome during Environmental Stress-Triggered Skin Inflammation

 ,
,
Abstract
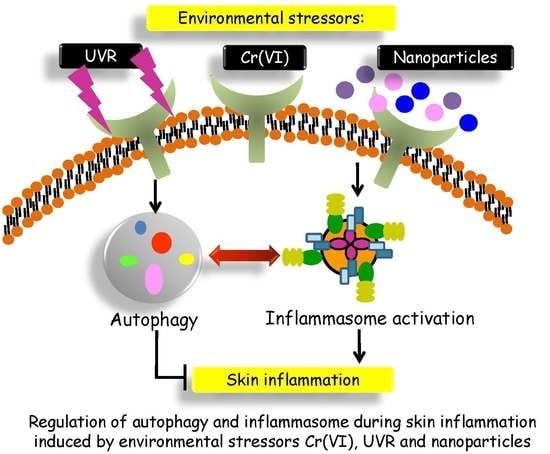
:
{kind=link}
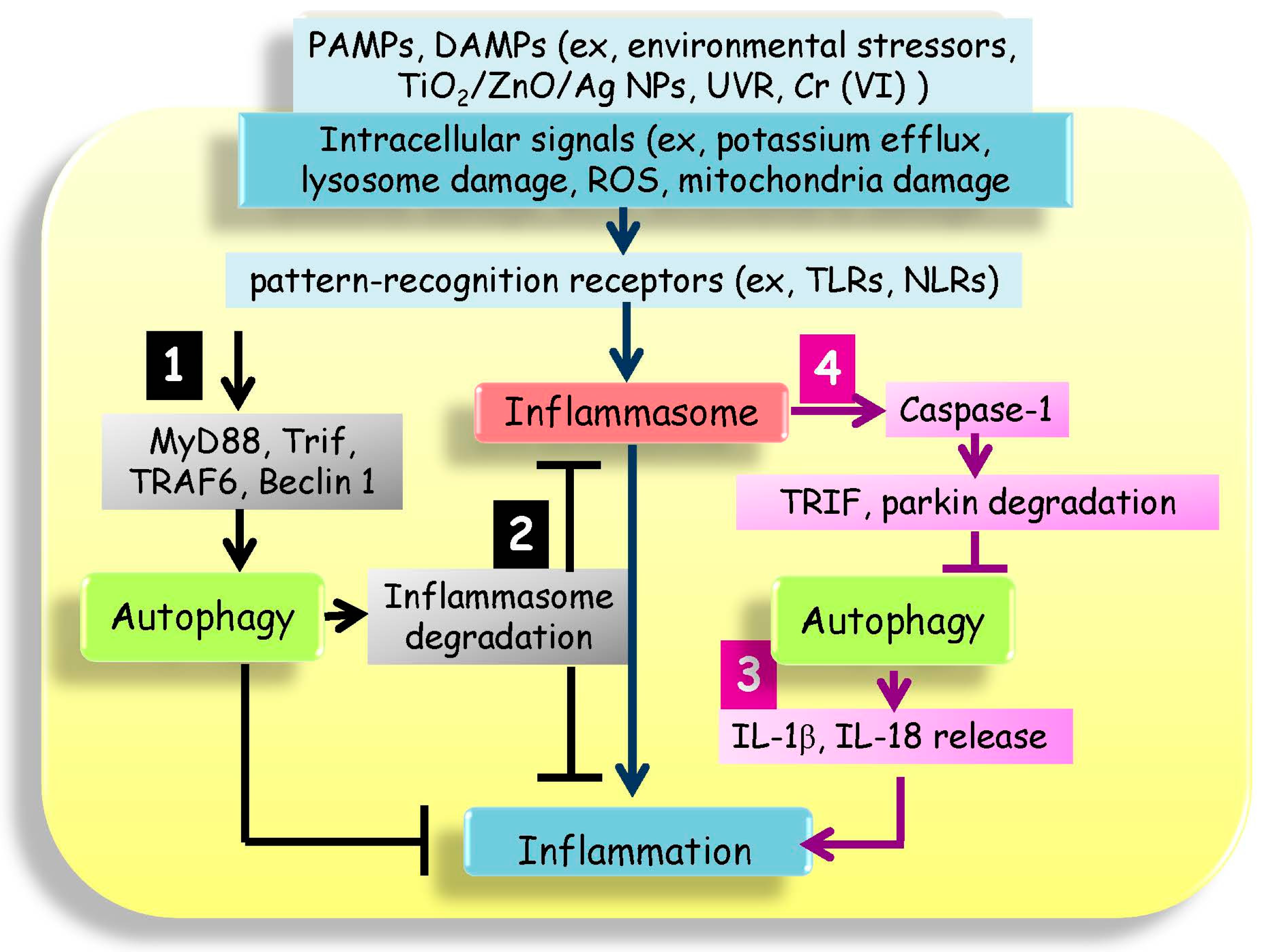{kind=link}
1. Introduction
2. Autophagy Intersects the Inflammasome
2.1. Autophagy Could Be Activated by Specific Inflammasome Sensors
2.2. Autophagy Regulates Inflammasome Inactivation and Degradation
2.3. Autophagy Regulates Biogenesis and Release of Inflammatory Cytokines
2.4. Inflammasome Possesses Inhibitory Effects on Autophagy
3. Cr(VI)-Induced Skin Inflammation
4. The Possible Role of the Inflammasome and Autophagy in UVR-Induced Skin Inflammation
5. Possible Role of Inflammasome and Autophagy in Nanoparticle-Induced Skin Damage
5.1. Titanium Dioxide Nanoparticles (TiO2 NPs)
5.2. Zinc Oxide Nanoparticles (ZnO NPs)
5.3. Silver Nanoparticles (Ag NPs)
6. Conclusions and Perspectives
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Nakatogawa, H.; Suzuki, K.; Kamada, Y.; Ohsumi, Y. Dynamics and diversity in autophagy mechanisms: Lessons from yeast. Nat. Rev. Mol. Cell Biol. 2009, 10, 458–467. [Google Scholar] [CrossRef] [PubMed]
- Yu, T.; Zuber, J.; Li, J. Targeting autophagy in skin diseases. J. Mol. Med. 2015, 93, 31–38. [Google Scholar] [CrossRef] [PubMed]
- Saitoh, T.; Akira, S. Regulation of inflammasomes by autophagy. J. Allergy Clin. Immunol. 2016, 138, 28–36. [Google Scholar] [CrossRef] [PubMed]
- Rathinam, V.A.; Fitzgerald, K.A. Inflammasome complexes: Emerging mechanisms and effector functions. Cell 2016, 165, 792–800. [Google Scholar] [CrossRef] [PubMed]
- Rodgers, M.A.; Bowman, J.W.; Liang, Q.; Jung, J.U. Regulation where autophagy intersects the inflammasome. Antioxid. Redox Signal. 2014, 20, 495–506. [Google Scholar] [CrossRef] [PubMed]
- De Lavera, I.; Pavon, A.D.; Paz, M.V.; Oropesa-Avila, M.; de la Mata, M.; Alcocer-Gomez, E.; Garrido-Maraver, J.; Cotan, D.; Alvarez-Cordoba, M.; Sanchez-Alcazar, J.A. The connections among autophagy, inflammasome and mitochondria. In Current Drug Targets; University of Notre Dame: Notre Dame, IN, USA, 2016. [Google Scholar] [PubMed]
- Martins, J.D.; Liberal, J.; Silva, A.; Ferreira, I.; Neves, B.M.; Cruz, M.T. Autophagy and inflammasome interplay. DNA Cell Biol. 2015, 34, 274–281. [Google Scholar] [CrossRef] [PubMed]
- Larese Filon, F.; Mauro, M.; Adami, G.; Bovenzi, M.; Crosera, M. Nanoparticles skin absorption: New aspects for a safety profile evaluation. Regul. Toxicol. Pharmacol. 2015, 72, 310–322. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Chen, X.; Gu, H. The signaling involving in autophagy machinery in keratinocytes and therapeutic approaches for skin diseases. Oncotarget 2016. [Google Scholar] [CrossRef] [PubMed]
- Kroemer, G.; Marino, G.; Levine, B. Autophagy and the integrated stress response. Mol. Cell 2010, 40, 280–293. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.K.; Shin, J.S.; Nahm, M.H. NOD-like receptors in infection, immunity, and diseases. Yonsei Med. J. 2016, 57, 5–14. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Sun, B. Negative regulation of NLRP3 inflammasome signaling. Protein Cell 2013, 4, 251–258. [Google Scholar] [CrossRef] [PubMed]
- Nasti, T.H.; Timares, L. Inflammasome activation of IL-1 family mediators in response to cutaneous photodamage. Photochem. Photobiol. 2012, 88, 1111–1125. [Google Scholar] [CrossRef] [PubMed]
- Faustin, B.; Reed, J.C. Sunburned skin activates inflammasomes. Trends Cell Biol. 2008, 18, 4–8. [Google Scholar] [CrossRef] [PubMed]
- Jeru, I.; Duquesnoy, P.; Fernandes-Alnemri, T.; Cochet, E.; Yu, J.W.; Lackmy-Port-Lis, M.; Grimprel, E.; Landman-Parker, J.; Hentgen, V.; Marlin, S.; et al. Mutations in NALP12 cause hereditary periodic fever syndromes. Proc. Natl. Acad. Sci. USA 2008, 105, 1614–1619. [Google Scholar] [CrossRef] [PubMed]
- Ebrahimiadib, N.; Samra, K.A.; Domina, A.M.; Stiles, E.R.; Ewer, R.; Bocian, C.P.; Foster, C.S. A novel NOD2-associated mutation and variant blau syndrome: Phenotype and molecular analysis. Ocul. Immunol. Inflamm. 2016, 15, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Macaluso, F.; Nothnagel, M.; Parwez, Q.; Petrasch-Parwez, E.; Bechara, F.G.; Epplen, J.T.; Hoffjan, S. Polymorphisms in NACHT-LRR (NLR) genes in atopic dermatitis. Exp. Dermatol. 2007, 16, 692–698. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Godinez, M.A.; Cruz-Dominguez, M.P.; Jara, L.J.; Dominguez-Lopez, A.; Jarillo-Luna, R.A.; Vera-Lastra, O.; Montes-Cortes, D.H.; Campos-Rodriguez, R.; Lopez-Sanchez, D.M.; Mejia-Barradas, C.M.; et al. Expression of NLRP3 inflammasome, cytokines and vascular mediators in the skin of systemic sclerosis patients. Isr. Med. Assoc. J. 2015, 17, 5–10. [Google Scholar] [PubMed]
- Miller, L.S. Toll-like receptors in skin. Adv. Dermatol. 2008, 24, 71–87. [Google Scholar] [CrossRef] [PubMed]
- Pedraza-Alva, G.; Perez-Martinez, L.; Valdez-Hernandez, L.; Meza-Sosa, K.F.; Ando-Kuri, M. Negative regulation of the inflammasome: Keeping inflammation under control. Immunol. Rev. 2015, 265, 231–257. [Google Scholar] [CrossRef] [PubMed]
- Orlowski, G.M.; Colbert, J.D.; Sharma, S.; Bogyo, M.; Robertson, S.A.; Rock, K.L. Multiple cathepsins promote pro-IL-1β synthesis and NLRP3-mediated IL-1β activation. J. Immunol. 2015, 195, 1685–1697. [Google Scholar] [CrossRef] [PubMed]
- Katsnelson, M.A.; Lozada-Soto, K.M.; Russo, H.M.; Miller, B.A.; Dubyak, G.R. NLRP3 inflammasome signaling is activated by low-level lysosome disruption but inhibited by extensive lysosome disruption: roles for K+ efflux and Ca2+ influx. Am. J. Physiol. Cell Physiol. 2016, 311, C83–C100. [Google Scholar] [CrossRef] [PubMed]
- Douroudis, K.; Kingo, K.; Traks, T.; Reimann, E.; Raud, K.; Ratsep, R.; Mossner, R.; Silm, H.; Vasar, E.; Koks, S. Polymorphisms in the ATG16L1 gene are associated with psoriasis vulgaris. Acta Derm. Venereol. 2012, 92, 85–87. [Google Scholar] [CrossRef] [PubMed]
- Han, J.W.; Zheng, H.F.; Cui, Y.; Sun, L.D.; Ye, D.Q.; Hu, Z.; Xu, J.H.; Cai, Z.M.; Huang, W.; Zhao, G.P.; et al. Genome-wide association study in a Chinese Han population identifies nine new susceptibility loci for systemic lupus erythematosus. Nat. Genet. 2009, 41, 1234–1237. [Google Scholar] [CrossRef] [PubMed]
- English, L.; Chemali, M.; Duron, J.; Rondeau, C.; Laplante, A.; Gingras, D.; Alexander, D.; Leib, D.; Norbury, C.; Lippe, R.; et al. Autophagy enhances the presentation of endogenous viral antigens on MHC class I molecules during HSV-1 infection. Nat. Immunol. 2009, 10, 480–487. [Google Scholar] [CrossRef] [PubMed]
- Shi, C.S.; Kehrl, J.H. MyD88 and Trif target Beclin 1 to trigger autophagy in macrophages. J. Biol. Chem. 2008, 283, 33175–33182. [Google Scholar] [CrossRef] [PubMed]
- Shi, C.S.; Kehrl, J.H. TRAF6 and A20 regulate lysine 63-linked ubiquitination of Beclin-1 to control TLR4-induced autophagy. Sci. Signal. 2010, 3, ra42. [Google Scholar] [CrossRef] [PubMed]
- Byrne, B.G.; Dubuisson, J.F.; Joshi, A.D.; Persson, J.J.; Swanson, M.S. Inflammasome components coordinate autophagy and pyroptosis as macrophage responses to infection. MBio 2013, 4, e00620-12. [Google Scholar] [CrossRef] [PubMed]
- West, A.P.; Brodsky, I.E.; Rahner, C.; Woo, D.K.; Erdjument-Bromage, H.; Tempst, P.; Walsh, M.C.; Choi, Y.; Shadel, G.S.; Ghosh, S. TLR signalling augments macrophage bactericidal activity through mitochondrial ROS. Nature 2011, 472, 476–480. [Google Scholar] [CrossRef] [PubMed]
- Shi, C.S.; Shenderov, K.; Huang, N.N.; Kabat, J.; Abu-Asab, M.; Fitzgerald, K.A.; Sher, A.; Kehrl, J.H. Activation of autophagy by inflammatory signals limits IL-1β production by targeting ubiquitinated inflammasomes for destruction. Nat. Immunol. 2012, 13, 255–263. [Google Scholar] [CrossRef] [PubMed]
- Py, B.F.; Kim, M.S.; Vakifahmetoglu-Norberg, H.; Yuan, J. Deubiquitination of NLRP3 by BRCC3 critically regulates inflammasome activity. Mol. Cell 2013, 49, 331–338. [Google Scholar] [CrossRef] [PubMed]
- Harris, J.; Hartman, M.; Roche, C.; Zeng, S.G.; O′Shea, A.; Sharp, F.A.; Lambe, E.M.; Creagh, E.M.; Golenbock, D.T.; Tschopp, J.; et al. Autophagy controls IL-1β secretion by targeting pro-IL-1β for degradation. J. Biol. Chem. 2011, 286, 9587–9597. [Google Scholar] [CrossRef] [PubMed]
- Dupont, N.; Jiang, S.; Pilli, M.; Ornatowski, W.; Bhattacharya, D.; Deretic, V. Autophagy-based unconventional secretory pathway for extracellular delivery of IL-1β. EMBO J. 2011, 30, 4701–4711. [Google Scholar] [CrossRef] [PubMed]
- Murai, H.; Okazaki, S.; Hayashi, H.; Kawakita, A.; Hosoki, K.; Yasutomi, M.; Sur, S.; Ohshima, Y. Alternaria extract activates autophagy that induces IL-18 release from airway epithelial cells. Biochem. Biophys. Res. Commun. 2015, 464, 969–974. [Google Scholar] [CrossRef] [PubMed]
- Luo, M.X.; Wong, S.H.; Chan, M.T.; Yu, L.; Yu, S.S.; Wu, F.; Xiao, Z.; Wang, X.; Zhang, L.; Cheng, A.S.; et al. Autophagy Mediates HBx-Induced Nuclear Factor-κB Activation and Release of IL-6, IL-8, and CXCL2 in Hepatocytes. J. Cell. Physiol. 2015, 230, 2382–2389. [Google Scholar] [CrossRef] [PubMed]
- Jabir, M.S.; Ritchie, N.D.; Li, D.; Bayes, H.K.; Tourlomousis, P.; Puleston, D.; Lupton, A.; Hopkins, L.; Simon, A.K.; Bryant, C.; et al. Caspase-1 cleavage of the TLR adaptor TRIF inhibits autophagy and β-interferon production during Pseudomonas aeruginosa infection. Cell Host Microbe 2014, 15, 214–227. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Nagasu, H.; Murakami, T.; Hoang, H.; Broderick, L.; Hoffman, H.M.; Horng, T. Inflammasome activation leads to Caspase-1-dependent mitochondrial damage and block of mitophagy. Proc. Natl. Acad. Sci. USA 2014, 111, 15514–15519. [Google Scholar] [CrossRef] [PubMed]
- Jounai, N.; Kobiyama, K.; Shiina, M.; Ogata, K.; Ishii, K.J.; Takeshita, F. NLRP4 negatively regulates autophagic processes through an association with beclin1. J. Immunol. 2011, 186, 1646–1655. [Google Scholar] [CrossRef] [PubMed]
- Caicedo, M.S.; Samelko, L.; McAllister, K.; Jacobs, J.J.; Hallab, N.J. Increasing both CoCrMo-alloy particle size and surface irregularity induces increased macrophage inflammasome activation in vitro potentially through lysosomal destabilization mechanisms. J. Orthop. Res. 2013, 31, 1633–1642. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.J.; Sheu, H.M.; Guo, Y.L.; Lee, Y.H.; Lai, C.S.; Pan, M.H.; Wang, Y.J. Hexavalent chromium induced ROS formation, Akt, NF-κB, and MAPK activation, and TNF-α and IL-1α production in keratinocytes. Toxicol. Lett. 2010, 198, 216–224. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.H.; Su, S.B.; Huang, C.C.; Sheu, H.M.; Tsai, J.C.; Lin, C.H.; Wang, Y.J.; Wang, B.J. N-acetylcysteine attenuates hexavalent chromium-induced hypersensitivity through inhibition of cell death, ROS-related signaling and cytokine expression. PLoS ONE 2014, 9, e108317. [Google Scholar] [CrossRef] [PubMed]
- Caicedo, M.S.; Desai, R.; McAllister, K.; Reddy, A.; Jacobs, J.J.; Hallab, N.J. Soluble and particulate Co-Cr-Mo alloy implant metals activate the inflammasome danger signaling pathway in human macrophages: A novel mechanism for implant debris reactivity. J. Orthop. Res. 2009, 27, 847–854. [Google Scholar] [CrossRef] [PubMed]
- Xie, Y.; Xiao, F.; Luo, L.; Zhong, C. Activation of autophagy protects against ROS-mediated mitochondria-dependent apoptosis in L-02 hepatocytes induced by Cr(VI). Cell. Physiol. Biochem. 2014, 33, 705–716. [Google Scholar] [CrossRef] [PubMed]
- Salzer, S.; Kresse, S.; Hirai, Y.; Koglin, S.; Reinholz, M.; Ruzicka, T.; Schauber, J. Cathelicidin peptide LL-37 increases UVB-triggered inflammasome activation: Possible implications for rosacea. J. Dermatol. Sci. 2014, 76, 173–179. [Google Scholar] [CrossRef] [PubMed]
- Feldmeyer, L.; Keller, M.; Niklaus, G.; Hohl, D.; Werner, S.; Beer, H.D. The inflammasome mediates UVB-induced activation and secretion of interleukin-1β by keratinocytes. Curr. Biol. 2007, 17, 1140–1145. [Google Scholar] [CrossRef] [PubMed]
- Yoon, S.J.; Park, J.Y.; Choi, S.; Lee, J.B.; Jung, H.; Kim, T.D.; Yoon, S.R.; Choi, I.; Shim, S.; Park, Y.J. Ginsenoside Rg3 regulates S-nitrosylation of the NLRP3 inflammasome via suppression of iNOS. Biochem. Biophys. Res. Commun. 2015, 463, 1184–1189. [Google Scholar] [CrossRef] [PubMed]
- Misovic, M.; Milenkovic, D.; Martinovic, T.; Ciric, D.; Bumbasirevic, V.; Kravic-Stevovic, T. Short-term exposure to UV-A, UV-B, and UV-C irradiation induces alteration in cytoskeleton and autophagy in human keratinocytes. Ultrastruct. Pathol. 2013, 37, 241–248. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Quach, C.; Liang, C. Autophagy Modulator Takes A Part in UV Protection. Autophagy 2016, 12, 1677–1678. [Google Scholar] [CrossRef] [PubMed]
- Carroll, B.; Mohd-Naim, N.; Maximiano, F.; Frasa, M.A.; McCormack, J.; Finelli, M.; Thoresen, S.B.; Perdios, L.; Daigaku, R.; Francis, R.E.; et al. The TBC/RabGAP Armus coordinates Rac1 and Rab7 functions during autophagy. Dev. Cell 2013, 25, 15–28. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.A.; Zhou, B.R.; Xu, Y.; Chen, X.; Liu, J.; Gozali, M.; Wu, D.; Yin, Z.Q.; Luo, D. miR-23a-depressed autophagy is a participant in PUVA- and UVB-induced premature senescence. Oncotarget 2016, 7, 37420–37435. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Zhang, C.F.; Rossiter, H.; Eckhart, L.; Konig, U.; Karner, S.; Mildner, M.; Bochkov, V.N.; Tschachler, E.; Gruber, F. Autophagy is induced by UVA and promotes removal of oxidized phospholipids and protein aggregates in epidermal keratinocytes. J. Investig. Dermatol. 2013, 133, 1629–1637. [Google Scholar] [CrossRef] [PubMed]
- Chikh, A.; Sanza, P.; Raimondi, C.; Akinduro, O.; Warnes, G.; Chiorino, G.; Byrne, C.; Harwood, C.A.; Bergamaschi, D. iASPP is a novel autophagy inhibitor in keratinocytes. J. Cell Sci. 2014, 127 Pt 14, 3079–3093. [Google Scholar] [CrossRef] [PubMed]
- Adachi, K.; Yamada, N.; Yoshida, Y.; Yamamoto, O. Subchronic exposure of titanium dioxide nanoparticles to hairless rat skin. Exp. Dermatol. 2013, 22, 278–283. [Google Scholar] [CrossRef] [PubMed]
- Filipe, P.; Silva, J.N.; Silva, R.; Cirne de Castro, J.L.; Marques Gomes, M.; Alves, L.C.; Santus, R.; Pinheiro, T. Stratum corneum is an effective barrier to TiO2 and ZnO nanoparticle percutaneous absorption. Skin Pharmacol. Physiol. 2009, 22, 266–275. [Google Scholar] [CrossRef] [PubMed]
- Peira, E.; Turci, F.; Corazzari, I.; Chirio, D.; Battaglia, L.; Fubini, B.; Gallarate, M. The influence of surface charge and photo-reactivity on skin-permeation enhancer property of nano-TiO2 in ex vivo pig skin model under indoor light. Int. J. Pharm. 2014, 467, 90–99. [Google Scholar] [CrossRef] [PubMed]
- Sadrieh, N.; Wokovich, A.M.; Gopee, N.V.; Zheng, J.; Haines, D.; Parmiter, D.; Siitonen, P.H.; Cozart, C.R.; Patri, A.K.; McNeil, S.E.; et al. Lack of significant dermal penetration of titanium dioxide from sunscreen formulations containing nano- and submicron-size TiO2 particles. Toxicol. Sci. 2010, 115, 156–166. [Google Scholar] [CrossRef] [PubMed]
- Monteiro-Riviere, N.A.; Wiench, K.; Landsiedel, R.; Schulte, S.; Inman, A.O.; Riviere, J.E. Safety evaluation of sunscreen formulations containing titanium dioxide and zinc oxide nanoparticles in UVB sunburned skin: An in vitro and in vivo study. Toxicol. Sci. 2011, 123, 264–280. [Google Scholar] [CrossRef] [PubMed]
- Cross, S.E.; Innes, B.; Roberts, M.S.; Tsuzuki, T.; Robertson, T.A.; McCormick, P. Human skin penetration of sunscreen nanoparticles: In vitro assessment of a novel micronized zinc oxide formulation. Skin Pharmacol. Physiol. 2007, 20, 148–154. [Google Scholar] [CrossRef] [PubMed]
- Gulson, B.; McCall, M.; Korsch, M.; Gomez, L.; Casey, P.; Oytam, Y.; Taylor, A.; McCulloch, M.; Trotter, J.; Kinsley, L.; et al. Small amounts of zinc from zinc oxide particles in sunscreens applied outdoors are absorbed through human skin. Toxicol. Sci. 2010, 118, 140–149. [Google Scholar] [CrossRef] [PubMed]
- Watkinson, A.C.; Bunge, A.L.; Hadgraft, J.; Lane, M.E. Nanoparticles do not penetrate human skin—a theoretical perspective. Pharm. Res. 2013, 30, 1943–1946. [Google Scholar] [CrossRef] [PubMed]
- Wiesenthal, A.; Hunter, L.; Wang, S.; Wickliffe, J.; Wilkerson, M. Nanoparticles: Small and mighty. Int. J. Dermatol. 2011, 50, 247–254. [Google Scholar] [CrossRef] [PubMed]
- Saquib, Q.; Al-Khedhairy, A.A.; Siddiqui, M.A.; Abou-Tarboush, F.M.; Azam, A.; Musarrat, J. Titanium dioxide nanoparticles induced cytotoxicity, oxidative stress and DNA damage in human amnion epithelial (WISH) cells. Toxicol. In Vitro 2012, 26, 351–361. [Google Scholar] [CrossRef] [PubMed]
- Yin, J.J.; Liu, J.; Ehrenshaft, M.; Roberts, J.E.; Fu, P.P.; Mason, R.P.; Zhao, B. Phototoxicity of nano titanium dioxides in HaCaT keratinocytes—Generation of reactive oxygen species and cell damage. Toxicol. Appl. Pharmacol. 2012, 263, 81–88. [Google Scholar] [CrossRef] [PubMed]
- Tyagi, N.; Srivastava, S.K.; Arora, S.; Omar, Y.; Ijaz, Z.M.; Al-Ghadhban, A.; Deshmukh, S.K.; Carter, J.E.; Singh, A.P.; Singh, S. Comparative analysis of the relative potential of silver, Zinc-oxide and titanium-dioxide nanoparticles against UVB-induced DNA damage for the prevention of skin carcinogenesis. Cancer Lett. 2016, 383, 53–61. [Google Scholar] [CrossRef] [PubMed]
- Kang, M.A.; So, E.Y.; Simons, A.L.; Spitz, D.R.; Ouchi, T. DNA damage induces reactive oxygen species generation through the H2AX-Nox1/Rac1 pathway. Cell Death Dis. 2012, 3, e249. [Google Scholar] [CrossRef] [PubMed]
- Peters, K.; Unger, R.E.; Kirkpatrick, C.J.; Gatti, A.M.; Monari, E. Effects of nano-scaled particles on endothelial cell function in vitro: Studies on viability, proliferation and inflammation. J. Mater. Sci. Mater. Med. 2004, 15, 321–325. [Google Scholar] [CrossRef] [PubMed]
- Hiroike, M.; Sakabe, J.; Kobayashi, M.; Shimauchi, T.; Ito, T.; Hirakawa, S.; Inoh, A.; Tokura, Y. Acicular, but not globular, titanium dioxide nanoparticles stimulate keratinocytes to produce pro-inflammatory cytokines. J. Dermatol. 2013, 40, 357–362. [Google Scholar] [CrossRef] [PubMed]
- Gutowska-Owsiak, D.; Ogg, G.S. The epidermis as an adjuvant. J. Investig. Dermatol. 2012, 132 3 Pt 2, 940–948. [Google Scholar] [CrossRef] [PubMed]
- Winter, M.; Beer, H.D.; Hornung, V.; Kramer, U.; Schins, R.P.; Forster, I. Activation of the inflammasome by amorphous silica and TiO2 nanoparticles in murine dendritic cells. Nanotoxicology 2011, 5, 326–340. [Google Scholar] [CrossRef] [PubMed]
- Palomaki, J.; Valimaki, E.; Sund, J.; Vippola, M.; Clausen, P.A.; Jensen, K.A.; Savolainen, K.; Matikainen, S.; Alenius, H. Long, needle-like carbon nanotubes and asbestos activate the NLRP3 inflammasome through a similar mechanism. ACS Nano 2011, 5, 6861–6870. [Google Scholar] [CrossRef] [PubMed]
- Tucci, P.; Porta, G.; Agostini, M.; Dinsdale, D.; Iavicoli, I.; Cain, K.; Finazzi-Agro, A.; Melino, G.; Willis, A. Metabolic effects of TiO2 nanoparticles, a common component of sunscreens and cosmetics, on human keratinocytes. Cell Death Dis. 2013, 4, e549. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, Y.; Howe, J.L.; Yu, Z.; Leong, D.T.; Chu, J.J.; Loo, J.S.; Ng, K.W. Exposure to titanium dioxide nanoparticles induces autophagy in primary human keratinocytes. Small 2013, 9, 387–392. [Google Scholar] [CrossRef] [PubMed]
- Lopes, V.R.; Loitto, V.; Audinot, J.N.; Bayat, N.; Gutleb, A.C.; Cristobal, S. Dose-dependent autophagic effect of titanium dioxide nanoparticles in human HaCaT cells at non-cytotoxic levels. J. Nanobiotechnol. 2016, 14, 22. [Google Scholar] [CrossRef] [PubMed]
- Li, R.; Ji, Z.; Qin, H.; Kang, X.; Sun, B.; Wang, M.; Chang, C.H.; Wang, X.; Zhang, H.; Zou, H.; et al. Interference in autophagosome fusion by rare earth nanoparticles disrupts autophagic flux and regulation of an interleukin-1β producing inflammasome. ACS Nano 2014, 8, 10280–10292. [Google Scholar] [CrossRef] [PubMed]
- Fage, S.W.; Muris, J.; Jakobsen, S.S.; Thyssen, J.P. Titanium: A review on exposure, release, penetration, allergy, epidemiology, and clinical reactivity. Contact Dermat. 2016, 74, 323–345. [Google Scholar] [CrossRef] [PubMed]
- Pinheiro, T.; Pallon, J.; Alves, L.C.; Veríssimo, A.; Filipe, P.; Silva, J.N.; Silva, R. The influence of corneocyte structure on the interpretation of permeation profiles of nanoparticles across skin. Nucl. Instrum. Methods Phys. Res. B 2007, 260, 119–123. [Google Scholar] [CrossRef]
- Jones, N.; Ray, B.; Ranjit, K.T.; Manna, A.C. Antibacterial activity of ZnO nanoparticle suspensions on a broad spectrum of microorganisms. FEMS Microbiol. Lett. 2008, 279, 71–76. [Google Scholar] [CrossRef] [PubMed]
- Scharffetter-Kochanek, K.; Wlaschek, M.; Brenneisen, P.; Schauen, M.; Blaudschun, R.; Wenk, J. UV-induced reactive oxygen species in photocarcinogenesis and photoaging. Biol. Chem. 1997, 378, 1247–1257. [Google Scholar]
- Holmes, A.M.; Song, Z.; Moghimi, H.R.; Roberts, M.S. Relative penetration of zinc oxide and zinc ions into human skin after application of different zinc oxide formulations. ACS Nano 2016, 10, 1810–1819. [Google Scholar] [CrossRef] [PubMed]
- Osmond, M.J.; McCall, M.J. Zinc oxide nanoparticles in modern sunscreens: An analysis of potential exposure and hazard. Nanotoxicology 2010, 4, 15–41. [Google Scholar] [CrossRef] [PubMed]
- Jeong, S.H.; Kim, H.J.; Ryu, H.J.; Ryu, W.I.; Park, Y.H.; Bae, H.C.; Jang, Y.S.; Son, S.W. ZnO nanoparticles induce TNF-α expression via ROS-ERK-Egr-1 pathway in human keratinocytes. J. Dermatol. Sci. 2013, 72, 263–273. [Google Scholar] [CrossRef] [PubMed]
- Heng, B.C.; Zhao, X.; Tan, E.C.; Khamis, N.; Assodani, A.; Xiong, S.; Ruedl, C.; Ng, K.W.; Loo, J.S. Evaluation of the cytotoxic and inflammatory potential of differentially shaped zinc oxide nanoparticles. Arch. Toxicol. 2011, 85, 1517–1528. [Google Scholar] [CrossRef] [PubMed]
- Yazdi, A.S.; Guarda, G.; Riteau, N.; Drexler, S.K.; Tardivel, A.; Couillin, I.; Tschopp, J. Nanoparticles activate the NLR pyrin domain containing 3 (Nlrp3) inflammasome and cause pulmonary inflammation through release of IL-1α and IL-1β. Proc. Natl. Acad. Sci. USA 2010, 107, 19449–19454. [Google Scholar] [CrossRef] [PubMed]
- Johnson, B.M.; Fraietta, J.A.; Gracias, D.T.; Hope, J.L.; Stairiker, C.J.; Patel, P.R.; Mueller, Y.M.; McHugh, M.D.; Jablonowski, L.J.; Wheatley, M.A.; et al. Acute exposure to ZnO nanoparticles induces autophagic immune cell death. Nanotoxicology 2015, 9, 737–748. [Google Scholar] [CrossRef] [PubMed]
- Yu, K.N.; Yoon, T.J.; Minai-Tehrani, A.; Kim, J.E.; Park, S.J.; Jeong, M.S.; Ha, S.W.; Lee, J.K.; Kim, J.S.; Cho, M.H. Zinc oxide nanoparticle induced autophagic cell death and mitochondrial damage via reactive oxygen species generation. Toxicol. In Vitro 2013, 27, 1187–1195. [Google Scholar] [CrossRef] [PubMed]
- Wijnhoven, S.W.P.; Peijnenburg, W.J.G.M.; Herberts, C.A.; Hagens, W.I.; Oomen, A.G.; Heugens, E.H.W.; Roszek, B.; Bisschops, J.; Gosens, I.; van De Meent, D.; et al. Nano-silver—A review of available data and knowledge gaps in human and environmental risk assessment. Nanotoxicology 2009, 3, 109–138. [Google Scholar] [CrossRef]
- Samberg, M.E.; Oldenburg, S.J.; Monteiro-Riviere, N.A. Evaluation of silver nanoparticle toxicity in skin in vivo and keratinocytes in vitro. Environ. Health Perspect. 2010, 118, 407–413. [Google Scholar] [CrossRef] [PubMed]
- Feng, H.; Pyykko, I.; Zou, J. Involvement of ubiquitin-editing protein A20 in modulating inflammation in rat cochlea associated with silver nanoparticle-induced CD68 upregulation and TLR4 activation. Nanoscale Res. Lett. 2016, 11, 240. [Google Scholar] [CrossRef] [PubMed]
- Mishra, A.R.; Zheng, J.; Tang, X.; Goering, P.L. Silver Nanoparticle-induced autophagic-lysosomal disruption and NLRP3-inflammasome activation in HepG2 cells is size-dependent. Toxicol. Sci. 2016, 150, 473–487. [Google Scholar] [CrossRef] [PubMed]
- Murphy, A.; Casey, A.; Byrne, G.; Chambers, G.; Howe, O. Silver nanoparticles induce pro-inflammatory gene expression and inflammasome activation in human monocytes. J. Appl. Toxicol. 2016, 36, 1311–1320. [Google Scholar] [CrossRef] [PubMed]
- Simard, J.C.; Vallieres, F.; de Liz, R.; Lavastre, V.; Girard, D. Silver nanoparticles induce degradation of the endoplasmic reticulum stress sensor activating transcription factor-6 leading to activation of the NLRP-3 inflammasome. J. Biol. Chem. 2015, 290, 5926–5939. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.H.; Cheng, F.Y.; Chiu, H.W.; Tsai, J.C.; Fang, C.Y.; Chen, C.W.; Wang, Y.J. Cytotoxicity, oxidative stress, apoptosis and the autophagic effects of silver nanoparticles in mouse embryonic fibroblasts. Biomaterials 2014, 35, 4706–4715. [Google Scholar] [CrossRef] [PubMed]
- Choi, A.J.; Ryter, S.W. Autophagy in inflammatory diseases. Int. J. Cell Biol. 2011, 2011, 732798. [Google Scholar] [CrossRef] [PubMed]
- Vitale, N.; Kisslinger, A.; Paladino, S.; Procaccini, C.; Matarese, G.; Pierantoni, G.M.; Mancini, F.P.; Tramontano, D. Resveratrol couples apoptosis with autophagy in UVB-irradiated HaCaT cells. PLoS ONE 2013, 8, e80728. [Google Scholar] [CrossRef] [PubMed]
- Bridgeman, B.B.; Wang, P.; Ye, B.; Pelling, J.C.; Volpert, O.V.; Tong, X. Inhibition of mTOR by apigenin in UVB-irradiated keratinocytes: A new implication of skin cancer prevention. Cell Signal. 2016, 28, 460–468. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Li, M.; Li, L.; Xu, S.; Huang, D.; Ju, M.; Huang, J.; Chen, K.; Gu, H. Trehalose, sucrose and raffinose are novel activators of autophagy in human keratinocytes through an mTOR-independent pathway. Sci. Rep. 2016, 6, 28423. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Hu, C.; Wu, X.; Wang, S.; Zhang, A.; Chen, W.; Shen, Y.; Tan, R.; Sun, Y.; Xu, Q. Roseotoxin B Improves allergic contact dermatitis through a unique anti-inflammatory mechanism involving excessive activation of autophagy in activated T lymphocytes. J. Investig. Dermatol. 2016, 136, 1636–1646. [Google Scholar] [CrossRef] [PubMed]
- Zhou, B.R.; Guo, X.F.; Zhang, J.A.; Xu, Y.; Li, W.; Wu, D.; Yin, Z.Q.; Permatasari, F.; Luo, D. Elevated miR-34c-5p mediates dermal fibroblast senescence by ultraviolet irradiation. Int. J. Biol. Sci. 2013, 9, 743–752. [Google Scholar] [CrossRef] [PubMed]

© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, R.-J.; Lee, Y.-H.; Yeh, Y.-L.; Wang, Y.-J.; Wang, B.-J. The Roles of Autophagy and the Inflammasome during Environmental Stress-Triggered Skin Inflammation. Int. J. Mol. Sci. 2016, 17, 2063. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms17122063
Chen R-J, Lee Y-H, Yeh Y-L, Wang Y-J, Wang B-J. The Roles of Autophagy and the Inflammasome during Environmental Stress-Triggered Skin Inflammation. International Journal of Molecular Sciences. 2016; 17(12):2063. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms17122063
Chicago/Turabian StyleChen, Rong-Jane, Yu-Hsuan Lee, Ya-Ling Yeh, Ying-Jan Wang, and Bour-Jr Wang. 2016. "The Roles of Autophagy and the Inflammasome during Environmental Stress-Triggered Skin Inflammation" International Journal of Molecular Sciences 17, no. 12: 2063. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms17122063