
Recent Methods for Purification and Structure Determination of Oligonucleotides


Abstract
:1. Introduction
2. Separation and Purification of Oligonucleotides
2.1. Purification of Oligonucleotides by PAGE
2.2. Purification of Oligonucleotides by Chromatographic Approaches
2.2.1. RP-HPLC
2.2.2. Ion-Exchange Liquid Chromatography
2.2.3. Ion-Paired Reversed-Phase HPLC (IP-RP-HPLC)
3. Methods for Studying Nucleic Acid Structure
3.1. Structural Determination by X-ray Crystallography Methods
3.2. Structural Determination by NMR
3.2.1. Overview
3.2.2. Chemical Shift Distribution of Oligonucleotide Protons
3.2.3. NMR Experiment Considerations
Sample Preparation
Gradient Suppression Technique
NMR Experiments
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| PAGE | polyacrylamide gel electrophoresis |
| NMR | nuclear magnetic resonance |
| RP-HPLC | reverse-phase HPLC |
| UPLC | ultra-performance liquid chromatography |
| IP-RP-HPLC | Ion-Paired Reversed-Phase HPLC |
| TEAA | Triethylammonium acetate |
| TBAB | tetrabutylammonium bromide |
| TBAA | tributylammonium acetate |
| TEAB | triethylammonium acetate |
| HAA | hexylammonium acetate |
| HFIP | hexafluoroisopropanol |
| DMBAA | dimethylbutylammonium acetate |
| AEC-HPLC | anion-exchange liquid chromatography |
| IEC-HPLC | ion exclusion chromatography |
| SeNA | selenium-derivatized nucleic acids |
| CARC | chaperone assisted RNA crystallography |
| NOE | Overhauser effect |
| TROSY | transverse relaxation-optimized spectroscopy |
| FID | free induction decay |
| COSY | Correlation Spectroscopy |
| TOCSY | Total Correlation Spectroscopy |
| HSQC | Heteronuclear Single Quantum Coherence |
| HETCOR | Heteronuclear Correlation Spectroscopy |
| SAM | Se atom-specific mutagenesis |
| CPMG | Carr-Purcell-Meiboom-Gill |
| AEC-HPLC | anion-exchange liquid chromatography |
| IEC-HPLC | ion exclusion chromatography |
| SAD | Single-wavelength anomalous diffraction |
References
- Sun, H.; Zhu, X.; Lu, P.Y.; Zu, L.Y. Oligonucleotide aptamers: New tools for targeted cancer therapy. Mol. Ther. Nucleic Acids 2014, 3, 182. [Google Scholar] [CrossRef] [PubMed]
- Zhao, N.; You, J.; Zeng, Z.; Zu, L.Y. An ultra pH-sensitive and aptamer-equipped nanoscale drug—Delivery system for selective killing of tumor cells. Small 2013, 9, 3477–3484. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.W.; Kim, H.J.; Heo, K. Therapeutic aptamers: Developmental potential as anticancer drugs. BMB Rep. 2015, 48, 234–237. [Google Scholar] [CrossRef] [PubMed]
- Zhu, G.; Ye, M.; Donovan, M.J. Nucleic acid aptamers: An emerging frontier in cancer therapy. Chem. Commun. (Camb.) 2012, 48, 10472–10480. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Gomollon, S.; Nicolas, F.E. Purification of DNA oligos by denaturing polyacrylamide gel electrophoresis (PAGE). Methods Enzymol. 2013, 529, 65–83. [Google Scholar]
- Cramer, H.; Finn, K.J.; Herzberg, E. Purity analysis and impurities determination by reversed-phase high-performance liquid chromatography. In Handbook of Analysis of Oligonucleotides and Related Products; CRC Press: Boca Raton, FL, USA, 2011; pp. 1–46. [Google Scholar]
- Petrov, A.; Tsa, A.; Puglisi, J.D. Analysis of RNA by analytical polyacrylamide gel electrophoresis. Methods Enzymol. 2013, 530, 301–313. [Google Scholar]
- Petrov, A.; Wu, T.; Puglisi, E.V. RNA purification by preparative polyacrylamide gel electrophoresis. Methods Enzymol. 2013, 530, 315–330. [Google Scholar]
- Holmes, D.L.; Stellwagen, N.C. Estimation of polyacrylamide gel pore size from Ferguson plots of linear DNA fragments. II. Comparison of gels with different crosslinker concentrations, added agarose and added linear polyacrylamide. Electrophoresis 1991, 12, 612–619. [Google Scholar] [CrossRef] [PubMed]
- Ali, W.H.; Pichon, V. Characterization of oligosorbents and application to the purification of ochratoxin A from wheat extracts. Anal. Bioanal. Chem. 2014, 406, 1233–1240. [Google Scholar] [PubMed]
- Abeydeera, N.D.; Egli, M.; Cox, N.; Yang, Y.B. Evoking picomolar binding in RNA by a single phosphorodithioate linkage. Nucleic Acids Res. 2016, 44, 8052–8064. [Google Scholar] [CrossRef] [PubMed]
- Gilar, M. Analysis and purification of synthetic oligonucleotides by reversed-phase high-performance liquid chromatography with photodiode array and mass spectrometry detection. Anal. Biochem. 2001, 298, 196–206. [Google Scholar] [CrossRef] [PubMed]
- Zlobina, M. Efficient large-scale preparation and purification of short single-stranded RNA oligonucleotides. Biotechniques 2016, 60, 75–83. [Google Scholar] [CrossRef] [PubMed]
- Gilar, M.; Bouvier, E.S.P. Purification of crude DNA oligonucleotides by solid-phase extraction and reversed-phase high-performance liquid chromatography. J. Chromatogr. A 2000, 890, 167–177. [Google Scholar] [CrossRef]
- Gilar, M.; Fountain, K.J.; Budman, Y. Ion-pair reversed-phase high-performance liquid chromatography analysis of oligonucleotides. Retention prediction. J. Chromatogr. A 2002, 958, 167–182. [Google Scholar] [CrossRef]
- Westhof, E. Perspectives and pitfalls in nucleic acids crystallography. In Nucleic Acid Crystallography: Methods and Protocols; Ennifar, E., Ed.; Humana Press: New York, NY, USA, 2016; pp. 3–8. [Google Scholar]
- Arnott, S. Polynucleotide secondary structures: An historical perspective. In Oxford Handbook of Nucleic Acid Structure; Neidle, S., Ed.; Oxford University Press: New York, NY, USA, 1999; pp. 1–38. [Google Scholar]
- Neidle, S. DNA structure as observed in fibers and crystals. In Principles of Nucleic Acid Structure, 2nd ed.; Academic Press: London, UK, 2008; pp. 38–74. [Google Scholar]
- Messerschmidt, A. X-ray Crystallography of Biomacromolecules: A Practical Guide; Wiley-VCH: Weinheim, Germany, 2007; pp. 45–79. [Google Scholar]
- Blackburn, G.M. Nucleic Acids in Chemistry and Biology; Royal Society of Chemistry: Cambridge, UK, 2006; pp. 1–10. [Google Scholar]
- Holmes, D.L.; Stellwagen, N.C. Estimation of polyacrylamide gel pore size from Ferguson plots of normal and anomalously migrating DNA fragments I. Gels containing 3% N,N′-methylenebisacrylamide. Electrophoresis 1991, 12, 253–263. [Google Scholar] [CrossRef] [PubMed]
- Greenough, L. Adapting capillary gel electrophoresis as a sensitive, high-throughput method to accelerate characterization of nucleic acid metabolic enzymes. Nucleic Acids Res. 2016, 44, e15. [Google Scholar] [CrossRef] [PubMed]
- Stellwagen, N.C. Electrophoresis of DNA in agarose gels, polyacrylamide gels and in free solution. Electrophoresis 2009, 30, 188–195. [Google Scholar] [CrossRef] [PubMed]
- Greene, J.M.; Struhl, K. S1 analysis of messenger RNA using single-stranded DNA probes. Curr. Protoc. Mol. Biol. 2001, 4–6. [Google Scholar]
- Gilman, M. Ribonuclease protection assay. Curr. Protoc. Mol. Biol. 1993, 4–7. [Google Scholar]
- Andrus, A.; Bloch, W. HPLC of oligonucleotides and polynucleotides. HPLC Macromol. 1998, 872, 141. [Google Scholar]
- Gilar, M.; Neue, U.D. Peak capacity in gradient reversed-phase liquid chromatography of biopolymers. Theoretical and practical implications for the separation of oligonucleotides. J. Chromatogr. A 2007, 1169, 139–150. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.Y.; Huang, Z.; Jaremko, W. High-performance liquid chromatography purification of chemically modified RNA aptamers. Anal. Biochem. 2014, 449, 106–108. [Google Scholar] [CrossRef] [PubMed]
- Biba, M.; Mao, B.; Welch, C.J. Liquid chromatography methods for the separation of short RNA oligonucleotides. LCGC N. Am. 2014, 32, 42–50. [Google Scholar]
- Fountain, K.J.; Gilar, M.; Budman, Y. Purification of dye-labeled oligonucleotides by ion-pair reversed-phase high-performance liquid chromatography. J. Chromatogr. B 2003, 783, 61–72. [Google Scholar] [CrossRef]
- Dickman, M.J. Ion pair reverse-phase chromatography: A versatile platform for the analysis of RNA. Chromatogr. Today 2011, 4, 22. [Google Scholar]
- Hoffman, N.E.; Liao, J.C. Reversed phase high performance liquid chromatographic separations of nucleotides in the presence of solvophobic ions. Anal. Chem. 1977, 49, 2231–2234. [Google Scholar] [CrossRef] [PubMed]
- Andrus, A.; Kuimelis, R.G. Analysis and purification of synthetic nucleic acids using HPLC. Curr. Protoc. Nucleic Acid Chem. 2015, 61, 10–15. [Google Scholar]
- Bidlingmeyer, B.A.; Deming, S.N.; Price, W.P. Retention mechamism for reversed-phase ion-pair liquid chromatography. J. Chromatogr. A 1979, 186, 419–434. [Google Scholar] [CrossRef]
- Melander, W.R.; Horváth, C. Mechanistic study on ion-pair reversed-phase chromatography. J. Chromatogr. A 1980, 201, 211–224. [Google Scholar] [CrossRef]
- Cecchi, T. Ion pairing chromatography. Crit. Rev. Anal. Chem. 2008, 38, 161–213. [Google Scholar] [CrossRef]
- Agrawal, S.; Tang, J.Y.; Brown, D.M. Analytical study of phosphorothioate analogues of oligodeoxynucleotides using high-performance liquid chromatography. J. Chromatogr. A 1990, 509, 396–399. [Google Scholar] [CrossRef]
- Haupt, W.; Pingoud, A. Comparison of several high-performance liquid chromatography techniques for the separation of oligodeoxynucleotides according to their chain lengths. J. Chromatogr. A 1983, 260, 419–427. [Google Scholar] [CrossRef]
- McKeown, A.P.; Shaw, P.N.; Barrett, D.A. Retention behaviour of an homologous series of oligodeoxythymidilic acids using reversed-phase ion-pair chromatography. Chromatographia 2002, 55, 271–277. [Google Scholar] [CrossRef]
- Ikuta, S.; Chattopadhyaya, R.; Dickerson, R.E. Reverse-phase polystyrene column for purification and analysis of DNA oligomers. Anal. Chem. 1984, 56, 2253–2256. [Google Scholar] [CrossRef] [PubMed]
- McCarthy, S.M.; Warren, W.J.; Dubey, A.; Gilar, M. Ion-pairing systems for reversed-phase chromatography separation of oligonucleotides. In Presented at TIDES Conference, Las Vegas, NV, USA, 2008.
- Huang, Z.; Jayaseelan, S.; Hebert, J. Single-nucleotide resolution of RNAs up to 59 nucleotides by high-performance liquid chromatography. Anal. Biochem. 2013, 435, 35–43. [Google Scholar] [CrossRef] [PubMed]
- McCarthy, S.M.; Gilar, M.; Gebler, J. Reversed-phase ion-pair liquid chromatography analysis and purification of small interfering RNA. Anal. Biochem. 2009, 390, 181–188. [Google Scholar] [CrossRef] [PubMed]
- Arrhenius, G. X-ray diffraction procedures for polycrystalline and amorphous materials. J. Chem. Educ. 1955, 32, 228. [Google Scholar] [CrossRef]
- Dauter, Z.; Adamiak, D.A. Anomalous signal of phosphorus used for phasing DNA oligomer: Importance of data redundancy. Acta Crystallogr. D Biol. Crystallogr. 2001, 57, 990–995. [Google Scholar] [CrossRef] [PubMed]
- Abdur, R.; Gerlits, O.O.; Gan, J. Novel complex MAD phasing and RNase H structural insights using selenium oligonucleotides. Acta Crystallogr. D Biol. Crystallogr. 2014, 70, 354–361. [Google Scholar] [CrossRef] [PubMed]
- Buschmann, D.; Haberberger, A.; Kirchner, B. Toward reliable biomarker signatures in the age of liquid biopsies—How to standardize the small RNA-Seq workflow. Nucleic Acids Res. 2016, 44, 5995–6018. [Google Scholar] [CrossRef] [PubMed]
- Mandal, P.K.; Chandrasekaran, A.R.; Madhanagopal, B.R. Ring crystals of oligonucleotides: Growth stages and X-ray diffraction studies. J. Cryst. Growth 2012, 354, 20–26. [Google Scholar] [CrossRef]
- Narayanan, B.C.; Westbrook, J.; Ghosh, S. The nucleic acid database: New features and capabilities. Nucleic Acids Res. 2014, 42, D114–D122. [Google Scholar] [CrossRef] [PubMed]
- Daubner, G.M.; Cléry, A.; Allain, F.H.T. RRM-RNA recognition: NMR or crystallography and new findings. Curr. Opin. Struct. Biol. 2013, 23, 100–108. [Google Scholar] [CrossRef] [PubMed]
- Lipfert, J. Understanding nucleic acid-ion interactions. Annu. Rev. Biochem. 2014, 83, 813–841. [Google Scholar] [CrossRef] [PubMed]
- Carrasco, N. Synthesis of selenium-derivatized nucleosides and oligonucleotides for X-ray crystallography. Nucleos Nucleot Nucl. 2001, 20, 1723–1734. [Google Scholar] [CrossRef] [PubMed]
- Du, Q.; Carrasco, N.; Teplova, M. Internal derivatization of oligonucleotides with selenium for X-ray crystallography using MAD. J. Am. Chem. Soc. 2002, 124, 24–25. [Google Scholar] [CrossRef] [PubMed]
- Mandal, P.K.; Collie, G.W.; Kauffmann, B. Racemic DNA crystallography. Angew. Chem. 2014, 126, 14652–14655. [Google Scholar] [CrossRef]
- Vasilyev, N.; Serganov, A. Preparation of short 5′-triphosphorylated oligoribonucleotides for crystallographic and biochemical studies. Nucleic Acid Crystallogr. Methods Protoc. 2016, 11–20. [Google Scholar]
- Zhang, W.; Szostak, J.W.; Huang, Z. Nucleic acid crystallization and X-ray crystallography facilitated by single selenium atom. Front. Chem. Sci. Eng. 2016, 10, 196–202. [Google Scholar] [CrossRef]
- Peselis, A.; Gao, A.; Serganov, A. Preparation and crystallization of riboswitches. Nucleic Acid Crystallogr. Methods Protoc. 2015, 21–36. [Google Scholar]
- Dégut, C. In vitro/in vivo production of tRNA for X-ray studies. Methods Mol. Biol. 2015, 1320, 37–57. [Google Scholar]
- Ferré-D’Amaré, A.R. Use of the U1A Protein to facilitate crystallization and structure determination of large RNAs. Methods Mol. Biol. 2016, 1320, 67–76. [Google Scholar] [PubMed]
- Sherman, E.; Archer, J.; Ye, J.D. Fab chaperone—Assisted RNA crystallography (Fab CARC). Methods Mol. Biol. 2016, 1320, 77–109. [Google Scholar] [PubMed]
- Egli, M.; Pallan, P.S. Generating crystallographic models of DNA dodecamers from structures of RNase H:DNA complexes. Nucleic Acid Crystallogr. Methods Protoc. 2016, 1320, 111–126. [Google Scholar]
- Diederichs, K. Crystallographic data and model quality. Nucleic Acid Crystallogr. Methods Protoc. 2016, 1320, 147–173. [Google Scholar]
- Finke, A.D.; Panepucci, E.; Vonrhein, C. Advanced crystallographic data collection protocols for experimental phasing. Nucleic Acid Crystallogr. Methods Protoc. 2016, 1320, 175–191. [Google Scholar]
- Zubieta, C.; Nanao, M.H. Practical radiation damage-induced phasing. Nucleic Acid Crystallogr. Methods Protoc. 2016, 1320, 205–218. [Google Scholar]
- Batey, R.T.; Kieft, J.S. Soaking hexammine cations into RNA crystals to obtain derivatives for phasing diffraction data. Nucleic Acid Crystallogr. Methods Protoc. 2016, 1320, 219–232. [Google Scholar]
- Hare, D.R.; Wemmer, D.E.; Chou, S.H. Assignment of the non-exchangeable proton resonances of d(CGCGAATTCGCG) using two-dimensional nuclear magnetic resonance methods. J. Mol. Biol. 1983, 171, 319–336. [Google Scholar] [CrossRef]
- Wuthrich, K. NMR of Proteins and Nucleic Acids; Wiley: Hoboken, NJ, USA, 1986; pp. 20–25. [Google Scholar]
- Dingley, A.J.; Grzesiek, S. Direct observation of hydrogen bonds in nucleic acid base pairs byInternucleotide 2 J NN couplings. J. Am. Chem. Soc. 1998, 120, 8293–8297. [Google Scholar] [CrossRef]
- Pervushin, K.; Ono, A.; Fernández, C. NMR scalar couplings across Watson–Crick base pair hydrogen bonds in DNA observed by transverse relaxation-optimized spectroscopy. Proc. Natl. Acad. Sci. USA 1998, 95, 14147–14151. [Google Scholar] [CrossRef] [PubMed]
- Liu, A.; Majumdar, A.; Hu, W. NMR detection of NHOC hydrogen bonds in 13C,15N-labeled nucleic acids. J. Am. Chem. Soc. 2000, 122, 3206–3210. [Google Scholar] [CrossRef]
- Zwahlen, C.; Legault, P.; Vincent, S.J.F. Methods for measurement of intermolecular NOEs by multinuclear NMR spectroscopy: Application to a bacteriophage λ N-Peptide/boxB RNA complex. J. Am. Chem. Soc. 1997, 119, 6711–6721. [Google Scholar] [CrossRef]
- Lam, S.L.; Ip, L.N. Low temperature solution structures and base pair stacking of double helical d(CGTACG) 2. J. Biomol. Struct. Dyn. 2002, 19, 907–917. [Google Scholar] [CrossRef] [PubMed]
- Parella, T.; Espinosa, J.F. Long-range proton-carbon coupling constants: NMR methods and applications. Prog. Nucl. Magn. Reson. Spectrosc. 2013, 73, 17–55. [Google Scholar] [CrossRef] [PubMed]
- Anderson, K.M.; Nguyen, D.; Esadze, A. A chemical approach for site-specific identification of NMR signals from protein side-chain NH3+ groups forming intermolecular ion pairs in protein–nucleic acid complexes. J. Biomol. NMR 2015, 62, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Wolter, A.C.; Duchardt-Ferner, E.; Nasiri, A.H. NMR resonance assignments for the class II GTP binding RNA aptamer in complex with GTP. Biomol. NMR Assign. 2016, 10, 101–105. [Google Scholar] [CrossRef] [PubMed]
- DeRider, M.L.; Brooks, D.; Burt, G. Structural Determination by NMR. Handbook of Analysis of Oligonucleotides and Related Products; CRC Press: New York, NY, USA, 2011; pp. 361–385. [Google Scholar]
- Souard, F.; Perrier, S.; Noël, V. Optimization of experimental parameters to explore small-ligand/aptamer interactions through use of 1H NMR spectroscopy and molecular modeling. Chemistry 2015, 21, 15740–15748. [Google Scholar] [CrossRef] [PubMed]
- Latham, M.P.; Brown, D.J.; McCallum, S.A. NMR methods for studying the structure and dynamics of RNA. Chembiochem 2005, 6, 1492–1505. [Google Scholar] [CrossRef] [PubMed]
- Zheng, G.; Price, W.S. Solvent signal suppression in NMR. Prog. Nucl. Magn. Reson. Spectrosc. 2010, 56, 267–288. [Google Scholar] [CrossRef] [PubMed]
- Markley, J.L.; Bax, A.; Arata, Y. Recommendations for the presentation of NMR structures of proteins and nucleic acids. J. Mol. Biol. 1998, 280, 933–952. [Google Scholar] [CrossRef] [PubMed]
- Thibaudeau, C.; Chattopadhyaya, J. The discovery of intramolecular stereoelectronic forces that drive the sugar conformation in nucleosides and nucleotides. Nucleos Nucleot Nucl. 2006, 16, 523–529. [Google Scholar] [CrossRef]
- Gossert, A.; Jahnke, W. NMR in drug discovery: A practical guide to identification and validation of ligands interacting with biological macromolecules. Prog. Nucl. Magn. Reson. Spectrosc. 2016, 97, 82–125. [Google Scholar] [CrossRef] [PubMed]
- Mukhopadhyay, R. Liquid NMR probes: Oh so many choices. Anal. Chem. 2007, 79, 7959–7963. [Google Scholar] [CrossRef] [PubMed]
- Imeddourene, A.B.; Xu, X.; Zargarian, L. The intrinsic mechanics of B-DNA in solution characterized by NMR. Nucleic Acids Res. 2016, 44, 3432–3447. [Google Scholar] [CrossRef] [PubMed]
- Feigon, J.; Skelenář, V.; Wang, E. 1H NMR spectroscopy of DNA. Methods Enzymol. 1992, 211, 235–253. [Google Scholar]
- Scott, L.G.; Hennig, M. F-site-specific-labeled nucleotides for nucleic acid structural analysis by NMR. Methods Enzymol. 2016, 566, 59–87. [Google Scholar]
- Graber, D.; Moroder, H.; Micura, R. 19F NMR spectroscopy for the analysis of RNA secondary structure populations. J. Am. Chem. Soc. 2008, 130, 17230–17231. [Google Scholar] [CrossRef] [PubMed]
- Shajani, Z.; Varani, G. NMR studies of dynamics in RNA and DNA by 13C relaxation. Biopolymers 2007, 86, 348–359. [Google Scholar] [CrossRef] [PubMed]
- Schmitz, U.; Ulyanov, N.B.; Kumar, A. Molecular dynamics with weighted time-averaged restraints for a DNA octamer, dynamic interpretation of nuclear magnetic resonance data. J. Mol. Biol. 1993, 234, 373–389. [Google Scholar] [CrossRef] [PubMed]
- Borden, K.L.B.; Jenkins, T.C.; Skelly, J.V. Conformational properties of the G.G mismatch in d(CGC-GAATTGGCG) 2 determined by NMR. Biochemistry 1992, 31, 5411–5422. [Google Scholar] [CrossRef] [PubMed]
- Ulyanov, N.B.; Bauer, W.R.; James, T.L. High resolution NMR structure of an AT-rich DNA sequence. J. Biomol. NMR 2002, 22, 265–280. [Google Scholar] [CrossRef] [PubMed]
- Lipari, G.; Szabo, A. Model-free approach to the interpretation of nuclear magnetic resonance relaxation in macro-molecules. 1. Theory and range of validity. J. Am. Chem. Soc. 1982, 104, 4546–4559. [Google Scholar] [CrossRef]
- Kojima, C.; Ono, A.; Kainosho, M. DNA duplex dynamics: NMR relaxation studies of a decamer with uniformly 13C labeled purine nucleotides. J. Magn. Reson. 1998, 135, 310–333. [Google Scholar] [CrossRef] [PubMed]
- Borgias, B.A.; James, T.L. Procedure for matrix analysis of relaxation for discerning geometry of an aqueous structure. J. Magn. Reson. 1990, 87, 475–487. [Google Scholar] [CrossRef]
- Wang, E.; Malek, S.; Feigon, J. Structure of a G.T.A triplet in an intramolecular DNA triplex. Biochemistry 1992, 31, 4838–4846. [Google Scholar] [CrossRef] [PubMed]
- Bax, A.; Kontaxis, G.; Tjandra, N. Dipolar couplings in macromolecular structure determination. Methods Enzymol. 2001, 339, 127–174. [Google Scholar]
- Spielmann, H.P. Dynamics in psoralen-damaged DNA by 1H detected natural abundance 13C NMR spectroscopy. Biochemistry 1998, 37, 5426–5438. [Google Scholar] [CrossRef] [PubMed]
- Eimer, W.; Williamson, J.R.; Boxer, S.G. Characterization of the overall and internal dynamics of short oligonu-cleotides by depolarized dynamic light-scattering and NMR relaxation measurements. Biochemistry 1990, 29, 799–811. [Google Scholar] [CrossRef] [PubMed]
- Breeze, A.L. Isotope-filtered NMR methods for the study of biomolecular structure and interactions. Prog. Nucl. Magn. Reson. Spectrosc. 2000, 36, 323–372. [Google Scholar] [CrossRef]
- Wang, Y.S.; Liu, D.; Wyss, D.F. Competition STD NMR for the detection of high-affinity ligands and NMR-based screening. Magn. Reson. Chem. 2004, 42, 485–489. [Google Scholar] [CrossRef] [PubMed]
- Schmitz, U.; James, T.L. How to generate accurate solution structures of double-helical nucleic acid fragments using nuclear magnetic resonance and restrained molecular dynamics. Methods Enzymol. 1995, 261, 3–44. [Google Scholar]
- Briinger, A.T.; Adams, P.D.; Clore, G.M. Crystallography & NMR system: A new software suite for macromolecular structure determination. Acta Crystallogr. D Biol. Crystallogr. 1998, 54, 905–921. [Google Scholar]
- Hirao, I.; Kawai, G.; Yoshizawa, S. Most compact hairpin-turn structure exerted by a short DNA fragment, d(GCGAAGC) in solution: An extraordinarilystable structure resistant to nucleases and heat. Nucleic Acids Res. 1994, 22, 576–582. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
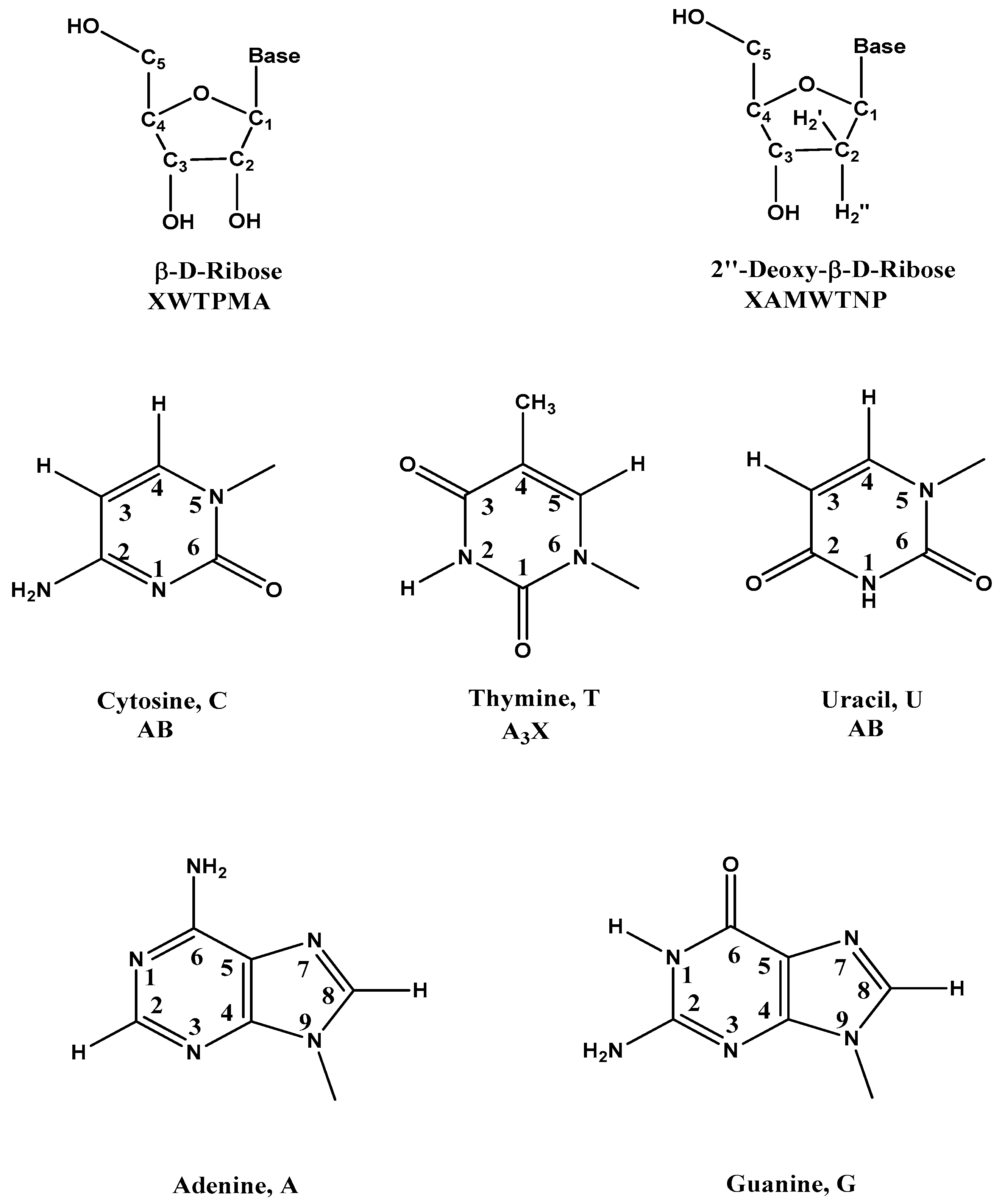
| Code | δ (ppm) | Comments |
|---|---|---|
| β-d-Riboses | ||
| 2′ | 1.8–3.0 | 2′, 2′′ H in DNA |
| 4′ | 3.7–4.5 | 4′, 5′, 5′′ H in DNA |
| 3′ | 4.4–5.2 | 3′ H in DNA |
| 2′, 3′, 4′, 5′, 5′′ | 3.7–5.2 | 2′, 3′, 4′, 5′, 5′′ H in RNA |
| 1′ | 5.3–6.3 | 1′ H in DNA and RNA |
| Five Common Bases | ||
| Me | 1.2–1.6 | Me of T |
| 5 | 5.3–6.0 | 5 H of C and U |
| 6 | 7.1–7.6 | 6 H of C, T, and U |
| 2, 8 | 7.3–8.4 | 8 H of A and G, 2 H of A |
| –NH2 | 6.6–9.0 | NH2 of A, U And G |
 | 10–15 | NH of G, T and U |
| Stage | NMR Experiments | Information | References |
|---|---|---|---|
| Identification (fingerprint) | 1H 31P or 19F 1D; 1H 1D temperature studies | Determine the presence of secondary structure, including the number of base pairs; Determine the secondary structure | [68,69,82,83,84] |
| Impurity or conformational identification | 1H 31P 19F 1D; LC-NMR or DOSY | Identify and/or quantitate the presence of multiple conformations and/or impurities | [85,86,87] |
| Imino assignment | 2D NOESY; 2D TOCSY/COSY | Identify base pairs and show connectivity (imino walk) | [67,68,69,75,88] |
| Base proton assignment of aromatic walk | 13C-HSQC; D2O NOESY; D2O TOCSY | Secondary structure; Aromatic to H1′ sugar sequential assignment (walk); H6 T/C and H8 G/A base assignments | [86,89,90,91] |
| Complete 1H assignment (as possible); 13C assignment (as possible) | Different temperatures and/or mixing times may be necessary (NOESY and TOCSY) | sugar assignment as possible; Additional base protons (e.g., H2 of A bases); Carbon assignments as possible | [78,92,93] |
| Tertiary structure (rough) | Quantitative NOESY; 2D 1H 31P HETCOR; Long-range heteronuclear correlation experiments (HMBC, HSQMBC, J-quantitative J-resolved experiments) | Distance restraints from full assignment of the correlations observed in the two-dimensional NOESY spectra; Angle restraints from quantitative analysis of J coupling constant | [73,90,91,94] |
| Tertiary structure (refined) | 1D and/or 2D NMR spectra in aligning media | residual dipolar couplings utilized to obtain long-range restraints | [83,95] |
| Dynamics | 1D or 2D T1 or T2 experiments such as inversion recovery and CPMG | Sequence dependence dynamics | [88,93,96,97,98] |
| Ligand interaction | Observation of chemical shift perturbation in NMR experiments or cross-correlation experiments; Isotope-filtered NMR methods; STD NMR methods; F-Site-specific-Labeled Nucleotides | Oligonucleotide resonances in contact with ligand (protein) | [71,74,75,84,86,99,100] |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, Q.; Lv, H.; Wang, L.; Chen, M.; Li, F.; Liang, C.; Yu, Y.; Jiang, F.; Lu, A.; Zhang, G. Recent Methods for Purification and Structure Determination of Oligonucleotides. Int. J. Mol. Sci. 2016, 17, 2134. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms17122134
Zhang Q, Lv H, Wang L, Chen M, Li F, Liang C, Yu Y, Jiang F, Lu A, Zhang G. Recent Methods for Purification and Structure Determination of Oligonucleotides. International Journal of Molecular Sciences. 2016; 17(12):2134. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms17122134
Chicago/Turabian StyleZhang, Qiulong, Huanhuan Lv, Lili Wang, Man Chen, Fangfei Li, Chao Liang, Yuanyuan Yu, Feng Jiang, Aiping Lu, and Ge Zhang. 2016. "Recent Methods for Purification and Structure Determination of Oligonucleotides" International Journal of Molecular Sciences 17, no. 12: 2134. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms17122134