
Dermal Delivery of Constructs Encoding Cre Recombinase to Induce Skin Tumors in PtenLoxP/LoxP;BrafCA/+ Mice

Abstract
:1. Introduction
2. Results
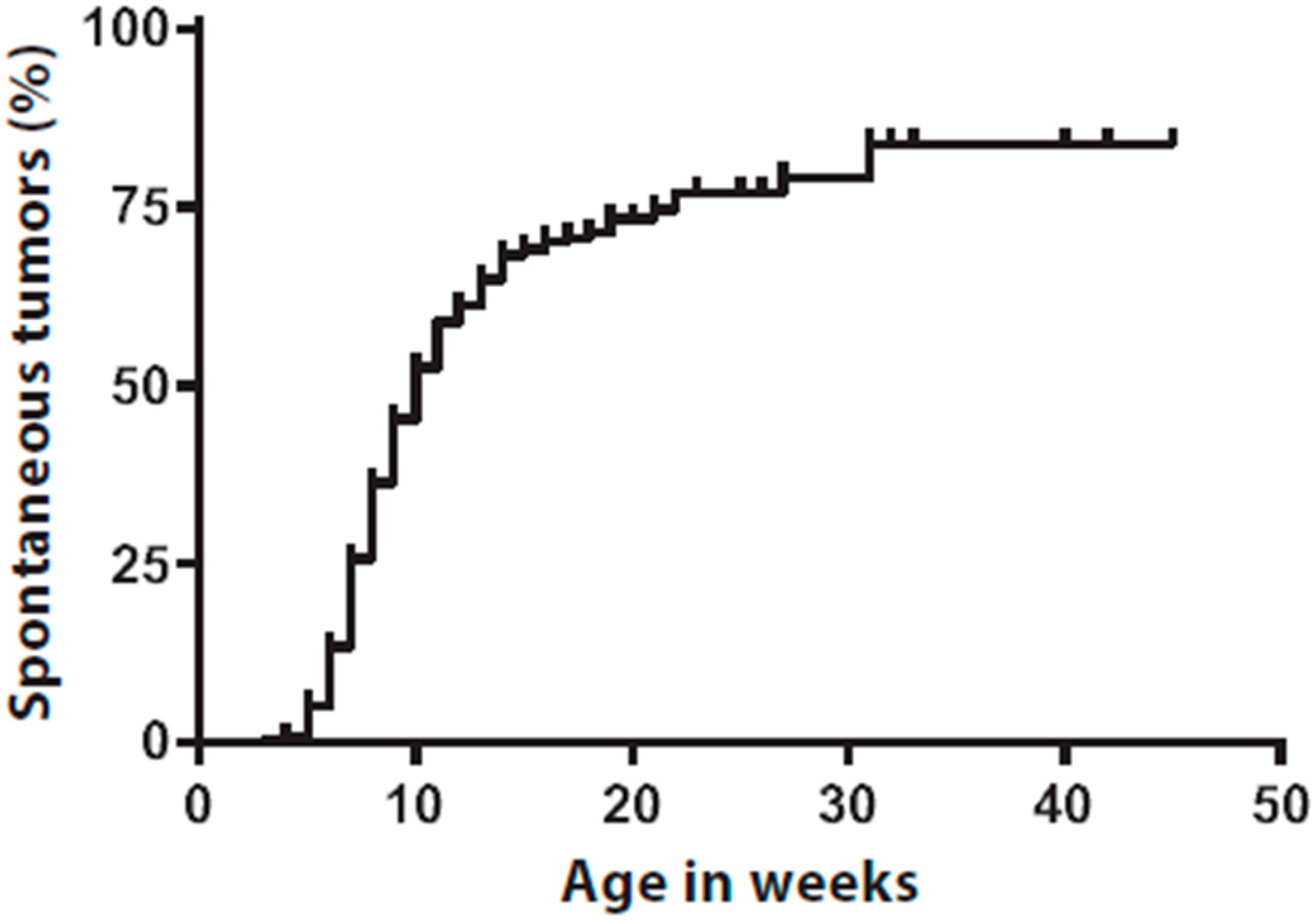
2.1. Spontaneous Tumors Occur in Tyr::CreERT2;PtenLoxP/LoxP;BrafCA/+ Mice
2.2. Dermal Delivery of CAG-Cre Recombinase DNA Led to Tumor Formation
2.3. Dermal Delivery of Cre under the Control of Melanocyte Specific Promoters Results in Melanoma Induction
2.4. Tumors Induced by Dermal Delivery Were Sensitive to Selective BRAF Inhibition
3. Discussion
4. Materials and Methods
4.1. Mouse Model and Tumor Induction
4.2. Viral Production and Plasmid Construction
4.3. Tumor Induction
4.4. Treatment of Tumor-Bearing Mice
4.5. Histological and Immunohistochemical Analysis
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| Tyr | Tyrosinase promoter (6.1-kb version) |
| mTyr | Tyrosinase promoter (577-bp version) |
References
- Hodi, F.S.; O’Day, S.J.; McDermott, D.F.; Weber, R.W.; Sosman, J.A.; Haanen, J.B.; Gonzalez, R.; Robert, C.; Schadendorf, D.; Hassel, J.C.; et al. Improved survival with ipilimumab in patients with metastatic melanoma. N. Engl. J. Med. 2010, 363, 711–723. [Google Scholar] [CrossRef] [PubMed]
- Topalian, S.L.; Hodi, F.S.; Brahmer, J.R.; Gettinger, S.N.; Smith, D.C.; McDermott, D.F.; Powderly, J.D.; Carvajal, R.D.; Sosman, J.A.; Atkins, M.B.; et al. Safety, activity, and immune correlates of anti-pd-1 antibody in cancer. N. Engl. J. Med. 2012, 366, 2443–2454. [Google Scholar] [CrossRef] [PubMed]
- Brahmer, J.R.; Tykodi, S.S.; Chow, L.Q.; Hwu, W.J.; Topalian, S.L.; Hwu, P.; Drake, C.G.; Camacho, L.H.; Kauh, J.; Odunsi, K.; et al. Safety and activity of anti-pd-l1 antibody in patients with advanced cancer. N. Engl. J. Med. 2012, 366, 2455–2465. [Google Scholar] [CrossRef] [PubMed]
- Chapman, P.B.; Hauschild, A.; Robert, C.; Haanen, J.B.; Ascierto, P.; Larkin, J.; Dummer, R.; Garbe, C.; Testori, A.; Maio, M.; et al. Improved survival with vemurafenib in melanoma with braf v600e mutation. N. Engl. J. Med. 2011, 364, 2507–2516. [Google Scholar] [CrossRef] [PubMed]
- Larkin, J.; Ascierto, P.A.; Dreno, B.; Atkinson, V.; Liszkay, G.; Maio, M.; Mandala, M.; Demidov, L.; Stroyakovskiy, D.; Thomas, L.; et al. Combined vemurafenib and cobimetinib in braf-mutated melanoma. N. Engl. J. Med. 2014, 371, 1867–1876. [Google Scholar] [CrossRef] [PubMed]
- Larkin, J.; Chiarion-Sileni, V.; Gonzalez, R.; Grob, J.J.; Cowey, C.L.; Lao, C.D.; Schadendorf, D.; Dummer, R.; Smylie, M.; Rutkowski, P.; et al. Combined nivolumab and ipilimumab or monotherapy in untreated melanoma. N. Engl. J. Med. 2015, 373, 23–34. [Google Scholar] [CrossRef] [PubMed]
- Postow, M.A.; Chesney, J.; Pavlick, A.C.; Robert, C.; Grossmann, K.; McDermott, D.; Linette, G.P.; Meyer, N.; Giguere, J.K.; Agarwala, S.S.; et al. Nivolumab and ipilimumab versus ipilimumab in untreated melanoma. N. Engl. J. Med. 2015, 372, 2006–2017. [Google Scholar] [CrossRef] [PubMed]
- Schadendorf, D.; Hodi, F.S.; Robert, C.; Weber, J.S.; Margolin, K.; Hamid, O.; Patt, D.; Chen, T.T.; Berman, D.M.; Wolchok, J.D. Pooled analysis of long-term survival data from phase ii and phase iii trials of ipilimumab in unresectable or metastatic melanoma. J. Clin. Oncol. 2015, 33, 1889–1894. [Google Scholar] [CrossRef] [PubMed]
- Daud, A.; Ribas, A.; Robert, C.; Hodi, F.S.; Wolchok, J.D.; Joshua, A.M.; Hwu, W.-J.; Weber, J.S.; Gangadhar, T.C.; Joseph, R.W.; et al. Long-term efficacy of pembrolizumab (pembro; mk-3475) in a pooled analysis of 655 patients (pts) with advanced melanoma (mel) enrolled in keynote-001. ASCO Meet. Abstr. 2015, 33, 9005. [Google Scholar]
- Blank, C.U.; Hooijkaas, A.I.; Haanen, J.B.; Schumacher, T.N. Combination of targeted therapy and immunotherapy in melanoma. Cancer Immunol. Immunother. 2011, 60, 1359–1371. [Google Scholar] [CrossRef] [PubMed]
- Ribas, A.; Hodi, F.S.; Callahan, M.; Konto, C.; Wolchok, J. Hepatotoxicity with combination of vemurafenib and ipilimumab. N. Engl. J. Med. 2013, 368, 1365–1366. [Google Scholar] [CrossRef] [PubMed]
- Minor, D.R.; Puzanov, I.; Callahan, M.K.; Hug, B.A.; Hoos, A. Severe gastrointestinal toxicity with administration of trametinib in combination with dabrafenib and ipilimumab. Pigment Cell Melanoma Res. 2015, 28, 611–612. [Google Scholar] [CrossRef] [PubMed]
- Ribas, A.; Butler, M.; Lutzky, J.; Lawrence, D.P.; Robert, C.; Miller, W.; Linette, G.P.; Ascierto, P.A.; Kuzel, T.; Algazi, A.P.; et al. Phase i study combining anti-pd-l1 (medi4736) with braf (dabrafenib) and/or mek (trametinib) inhibitors in advanced melanoma. ASCO Meet. Abstr. 2015, 33, 3003. [Google Scholar]
- Hwu, P.; Hamid, O.; Gonzalez, R.; Infante, J.R.; Patel, M.; Hodi, F.S.; Lewis, K.; Wallin, J.; Pitcher, B.; Cha, E.; et al. Preliminary safety and clinical activity of atezolizumab combined with cobimetinib and vemurafenib in braf v600–mutant metastatic melanoma. Ann. Oncol. 2016, 27. [Google Scholar] [CrossRef]
- Ribas, A.; Wolchok, J.D. Combining cancer immunotherapy and targeted therapy. Curr. Opin. Immunol. 2013, 25, 291–296. [Google Scholar] [CrossRef] [PubMed]
- Hu-Lieskovan, S.; Robert, L.; Homet Moreno, B.; Ribas, A. Combining targeted therapy with immunotherapy in braf-mutant melanoma: Promise and challenges. J. Clin. Oncol. 2014, 32, 2248–2254. [Google Scholar] [CrossRef] [PubMed]
- Kuzu, O.F.; Nguyen, F.D.; Noory, M.A.; Sharma, A. Current state of animal (mouse) modeling in melanoma research. Cancer Growth Metastasis 2015, 8, 81–94. [Google Scholar] [PubMed]
- Hooijkaas, A.I.; Gadiot, J.; van der Valk, M.; Mooi, W.J.; Blank, C.U. Targeting brafv600e in an inducible murine model of melanoma. Am. J. Pathol. 2012, 181, 785–794. [Google Scholar] [CrossRef] [PubMed]
- Dankort, D.; Curley, D.P.; Cartlidge, R.A.; Nelson, B.; Karnezis, A.N.; Damsky, W.E., Jr.; You, M.J.; DePinho, R.A.; McMahon, M.; Bosenberg, M. Braf(v600e) cooperates with PTEN loss to induce metastatic melanoma. Nat. Genet. 2009, 41, 544–552. [Google Scholar] [CrossRef] [PubMed]
- Hans, S.; Freudenreich, D.; Geffarth, M.; Kaslin, J.; Machate, A.; Brand, M. Generation of a non-leaky heat shock-inducible cre line for conditional cre/lox strategies in zebrafish. Dev. Dyn. 2011, 240, 108–115. [Google Scholar] [CrossRef] [PubMed]
- Bosenberg, M.; Muthusamy, V.; Curley, D.P.; Wang, Z.; Hobbs, C.; Nelson, B.; Nogueira, C.; Horner, J.W., 2nd; Depinho, R.; Chin, L. Characterization of melanocyte-specific inducible cre recombinase transgenic mice. Genesis 2006, 44, 262–267. [Google Scholar] [CrossRef] [PubMed]
- Bins, A.D.; Jorritsma, A.; Wolkers, M.C.; Hung, C.F.; Wu, T.C.; Schumacher, T.N.; Haanen, J.B. A rapid and potent DNA vaccination strategy defined by in vivo monitoring of antigen expression. Nat. Med. 2005, 11, 899–904. [Google Scholar] [CrossRef] [PubMed]
- Boissy, R.E. Extracutaneous melanocytes. In The Pigmentary System; Nordlund, J.J., Boissy, R.E., Hearing, V.J., King, R.A., Ortone, J.P., Eds.; Blackwell Publishing: Hoboken, NJ, USA, 1999; pp. 59–72. [Google Scholar]
- Melero, I.; Berman, D.M.; Aznar, M.A.; Korman, A.J.; Gracia, J.L.; Haanen, J. Evolving synergistic combinations of targeted immunotherapies to combat cancer. Nat. Rev. Cancer 2015, 15, 457–472. [Google Scholar] [CrossRef] [PubMed]
- Su, F.; Viros, A.; Milagre, C.; Trunzer, K.; Bollag, G.; Spleiss, O.; Reis-Filho, J.S.; Kong, X.; Koya, R.C.; Flaherty, K.T.; et al. Ras mutations in cutaneous squamous-cell carcinomas in patients treated with braf inhibitors. N. Engl. J. Med. 2012, 366, 207–215. [Google Scholar] [CrossRef] [PubMed]
- Byrne, C.; Fuchs, E. Probing keratinocyte and differentiation specificity of the human k5 promoter in vitro and in transgenic mice. Mol. Cell. Biol. 1993, 13, 3176–3190. [Google Scholar] [CrossRef] [PubMed]
- Carroll, J.M.; Albers, K.M.; Garlick, J.A.; Harrington, R.; Taichman, L.B. Tissue- and stratum-specific expression of the human involucrin promoter in transgenic mice. Proc. Natl. Acad. Sci. USA 1993, 90, 10270–10274. [Google Scholar] [CrossRef] [PubMed]
- Larue, L. Origin of mouse melanomas. J. Investig. Dermatol. 2012, 132, 2135–2136. [Google Scholar] [CrossRef] [PubMed]
- McLenachan, S.; Sarsero, J.P.; Ioannou, P.A. Flow-cytometric analysis of mouse embryonic stem cell lipofection using small and large DNA constructs. Genomics 2007, 89, 708–720. [Google Scholar] [CrossRef] [PubMed]
- Yin, W.; Xiang, P.; Li, Q. Investigations of the effect of DNA size in transient transfection assay using dual luciferase system. Anal. Biochem. 2005, 346, 289–294. [Google Scholar] [CrossRef] [PubMed]
- Slominski, A.; Paus, R. Melanogenesis is coupled to murine anagen: Toward new concepts for the role of melanocytes and the regulation of melanogenesis in hair growth. J. Investig. Dermatol. 1993, 101, 90–97. [Google Scholar] [CrossRef]
- Slominski, A.; Wortsman, J.; Plonka, P.M.; Schallreuter, K.U.; Paus, R.; Tobin, D.J. Hair follicle pigmentation. J. Investig. Dermatol. 2005, 124, 13–21. [Google Scholar] [CrossRef] [PubMed]
- Cho, J.H.; Robinson, J.P.; Arave, R.A.; Burnett, W.J.; Kircher, D.A.; Chen, G.; Davies, M.A.; Grossmann, A.H.; VanBrocklin, M.W.; McMahon, M.; et al. Akt1 activation promotes development of melanoma metastases. Cell Rep. 2015, 13, 898–905. [Google Scholar] [CrossRef] [PubMed]
- VanBrocklin, M.W.; Robinson, J.P.; Lastwika, K.J.; Khoury, J.D.; Holmen, S.L. Targeted delivery of nrasq61r and cre-recombinase to post-natal melanocytes induces melanoma in ink4a/arflox/lox mice. Pigment Cell Melanoma Res. 2010, 23, 531–541. [Google Scholar] [CrossRef] [PubMed]
- Matsuda, T.; Cepko, C.L. Controlled expression of transgenes introduced by in vivo electroporation. Proc. Natl. Acad. Sci. USA 2007, 104, 1027–1032. [Google Scholar] [CrossRef] [PubMed]
- Porter, S.D.; Meyer, C.J. A distal tyrosinase upstream element stimulates gene expression in neural-crest-derived melanocytes of transgenic mice: Position-independent and mosaic expression. Development 1994, 120, 2103–2111. [Google Scholar] [PubMed]
- Yajima, I.; Belloir, E.; Bourgeois, Y.; Kumasaka, M.; Delmas, V.; Larue, L. Spatiotemporal gene control by the cre-ert2 system in melanocytes. Genesis 2006, 44, 34–43. [Google Scholar] [CrossRef] [PubMed]
- Vile, R.; Miller, N.; Chernajovsky, Y.; Hart, I. A comparison of the properties of different retroviral vectors containing the murine tyrosinase promoter to achieve transcriptionally targeted expression of the hsvtk or il-2 genes. Gene Ther. 1994, 1, 307–316. [Google Scholar] [PubMed]
- Bins, A.D.; van Rheenen, J.; Jalink, K.; Halstead, J.R.; Divecha, N.; Spencer, D.M.; Haanen, J.B.; Schumacher, T.N. Intravital imaging of fluorescent markers and fret probes by DNA tattooing. BMC Biotechnol. 2007, 7, 2. [Google Scholar] [CrossRef] [PubMed]
- Bins, A.D.; Wolkers, M.C.; van den Boom, M.D.; Haanen, J.B.; Schumacher, T.N. In vivo antigen stability affects DNA vaccine immunogenicity. J. Immunol. 2007, 179, 2126–2133. [Google Scholar] [CrossRef] [PubMed]
- Hooijkaas, A.; Gadiot, J.; Morrow, M.; Stewart, R.; Schumacher, T.; Blank, C.U. Selective braf inhibition decreases tumor-resident lymphocyte frequencies in a mouse model of human melanoma. Oncoimmunology 2012, 1, 609–617. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Delivery Method | Construct | n | % | Mean Latency | Tumor Type |
|---|---|---|---|---|---|
| Intradermal injection | pBOB-CAG-iCRE-SD | 3/6 | 50 | 73d | melanoma * |
| Tattoo flank | pVAX1/noCMV-Cre | 0/6 | 0 | - | |
| pVAX1/CAG-Cre | 12/12 | 100 | 27d | melanoma * and secondary lesions | |
| pVAX1/Tyr-Cre | 2/8 | 25 | 29d | melanoma * | |
| pVAX1/mTyr-Cre | 6/8 | 75 | 42d | melanoma * | |
| Gene gun flank | pVAX1/CAG-Cre | 8/8 | 100 | 33d | epidermal lesions |
| pVAX1/Tyr-Cre | 2/8 | 25 | 50d | melanoma * | |
| pVAX1/mTyr-Cre | 2/8 | 25 | 42d | melanoma * | |
| Gene gun ear | pVAX1/CAG-Cre | 4/4 | 100 | 56d | melanoma * and secondary lesions |
| pVAX1/Tyr-Cre | 2/4 | 50 | 59d | melanoma * | |
| pVAX1/mTyr-Cre | 4/4 | 100 | 56d | melanoma * |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Deken, M.A.; Song, J.-Y.; Gadiot, J.; Bins, A.D.; Kroon, P.; Verbrugge, I.; Blank, C.U. Dermal Delivery of Constructs Encoding Cre Recombinase to Induce Skin Tumors in PtenLoxP/LoxP;BrafCA/+ Mice. Int. J. Mol. Sci. 2016, 17, 2149. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms17122149
Deken MA, Song J-Y, Gadiot J, Bins AD, Kroon P, Verbrugge I, Blank CU. Dermal Delivery of Constructs Encoding Cre Recombinase to Induce Skin Tumors in PtenLoxP/LoxP;BrafCA/+ Mice. International Journal of Molecular Sciences. 2016; 17(12):2149. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms17122149
Chicago/Turabian StyleDeken, Marcel A., Ji-Ying Song, Jules Gadiot, Adriaan D. Bins, Paula Kroon, Inge Verbrugge, and Christian U. Blank. 2016. "Dermal Delivery of Constructs Encoding Cre Recombinase to Induce Skin Tumors in PtenLoxP/LoxP;BrafCA/+ Mice" International Journal of Molecular Sciences 17, no. 12: 2149. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms17122149