
Developmental Origins of Chronic Kidney Disease: Should We Focus on Early Life?



Abstract
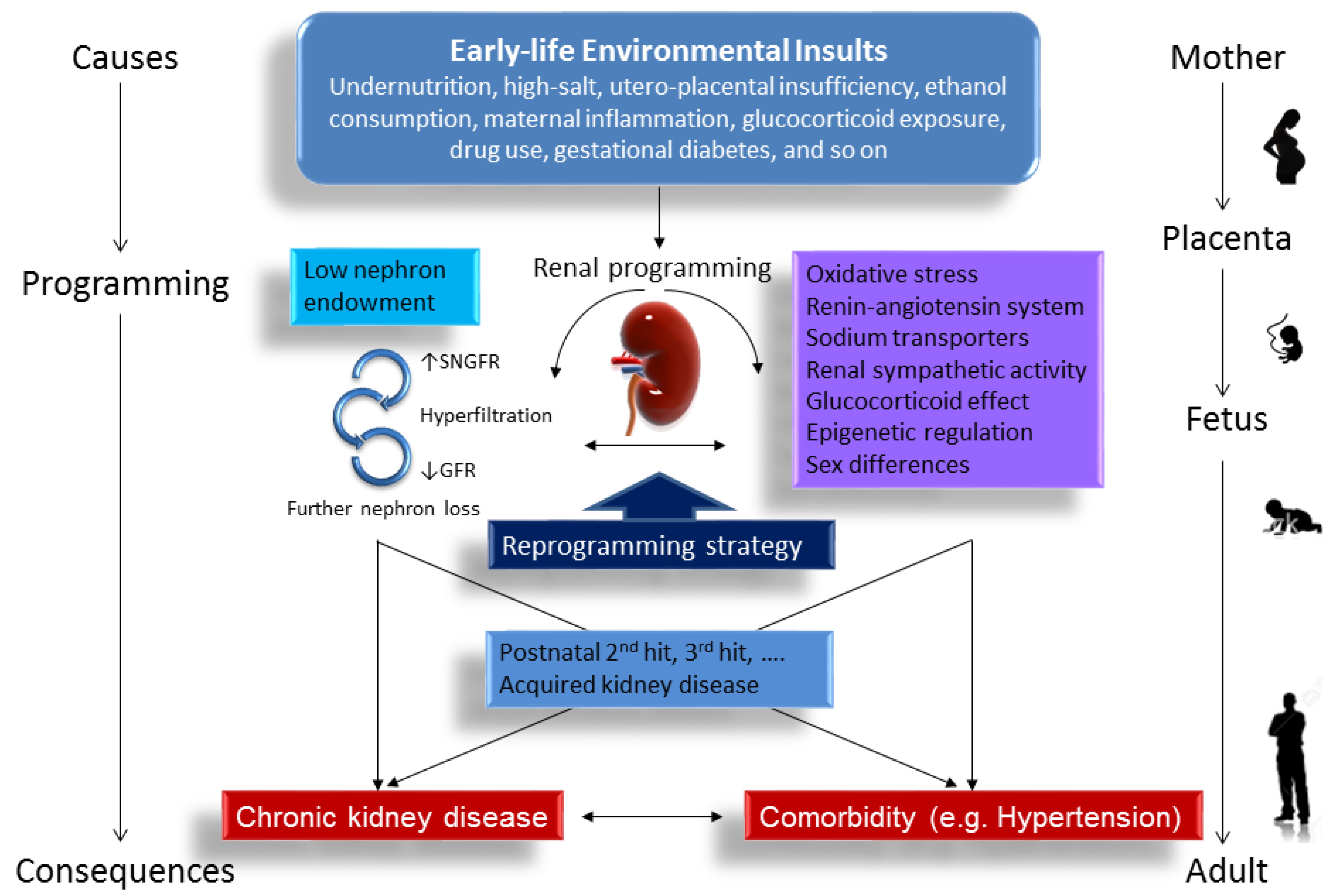
:1. Introduction
2. Evidence for Programming of Kidney Disease in the Human
3. Animal Models of Renal Programming
4. Mechanisms of Renal Programming
4.1. Oxidatice Stress
4.2. Renin-Angiotensin System
4.3. Sodium Transporters
4.4. Renal Sympathetic Activity
4.5. Glucocorticoid Effect
4.6. Epigenetic Regulation
4.7. Sex Differences
5. Changes in Renal Transcriptome in Response to Early-Life Insults
6. Reprogramming Strategy to Prevent the Programming of Kidney Disease
7. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| ACE | Angiotensin converting enzyme |
| ARB | Angiotensin receptor blocker |
| CAKUT | Congenital anomalies of the kidney and urinary tract |
| CKD | Chronic Kidney Disease |
| DEG | Differentially expressed gene |
| DOHaD | Directory of open access journals |
| GSH | Glutathione |
| HDAC | Histone deacetylases |
| KEGG | Kyoto Encyclopedia of Genes and Genomes |
| LBW | Low birth weight |
| miRNA | MicroRNA |
| NaKATPase | Na+/K+ATPase α1 subunit |
| NCD | Non-communicable disease |
| NCC | Na+/Cl− cotransporter |
| NGS | Next-generation sequencing |
| NHE3 | Type 3 sodium hydrogen exchanger |
| NKCC2 | Na-K-2Cl cotransporter |
| PPAR | Peroxisome proliferator-activated receptor |
| RAS | Renin-angiotensin system |
| ROS | Reactive oxygen species |
| SEH | Soluble epoxide hydrolase |
| STZ | Streptozotocin |
| 11β-HSD2 | 11β-hydroxysteroid dehydrogenase type 2 |
References
- Zarocostas, J. Need to increase focus on non-communicable diseases in global health, says WHO. Br. Med. J. 2010, 341, c7065. [Google Scholar] [CrossRef] [PubMed]
- Hanson, M.; Gluckman, P. Developmental origins of noncommunicable disease: Population and public health implications. Am. J. Clin. Nutr. 2011, 94, 1754S–1758S. [Google Scholar] [CrossRef] [PubMed]
- Lucas, A. Programming by early nutrition: An experimental approach. J. Nutr. 1998, 128, 401S–406S. [Google Scholar] [PubMed]
- Couser, W.G.; Remuzzi, G.; Mendis, S.; Tonelli, M. The contribution of chronic kidney disease to the global burden of major noncommunicable diseases. Kidney Int. 2011, 80, 1258–1270. [Google Scholar] [CrossRef] [PubMed]
- Kidney Disease: Improving Global Outcomes (KDIGO) CKD Work Group. KDIGO 2012 clinical practice guideline for the evaluation and management of Chronic Kidney Disease. Kidney Int. Suppl. 2013, 3, S1–S150. [Google Scholar]
- National Institutes of Health; National Institute of Diabetes and Digestive and Kidney Diseases. U.S. Renal Data System, USRDS 2013. Annual Data Report: Atlas of Chronic Kidney Disease and End-Stage Renal Disease in the United States; National Institutes of Health; National Institute of Diabetes and Digestive and Kidney Diseases: Bethesda, MD, USA, 2013.
- Ingelfinger, J.R.; Kalantar-Zadeh, K.; Schaefer, F.; World Kidney Day Steering Committee. World Kidney Day 2016: Averting the legacy of kidney disease-focus on childhood. Pediatr. Nephrol. 2016, 31, 343–348. [Google Scholar] [CrossRef] [PubMed]
- Kett, M.M.; Denton, K.M. Renal programming: Cause for concern? Am. J. Physiol. Regul. Integr. Comp. Physiol. 2011, 300, R791–R803. [Google Scholar] [CrossRef] [PubMed]
- Chong, E.; Yosypiv, I.V. Developmental programming of hypertension and kidney disease. Int. J. Nephrol. 2012, 2012, 760580. [Google Scholar] [CrossRef] [PubMed]
- Paixão, A.D.; Alexander, B.T. How the kidney is impacted by the perinatal maternal environment to develop hypertension. Biol. Reprod. 2013, 89, 144. [Google Scholar] [CrossRef] [PubMed]
- Luyckx, V.A.; Bertram, J.F.; Brenner, B.M.; Fall, C.; Hoy, W.E.; Ozanne, S.E.; Vikse, B.E. Effect of fetal and child health on kidney development and long-term risk of hypertension and kidney disease. Lancet 2013, 382, 273–283. [Google Scholar] [CrossRef] [Green Version]
- Boubred, F.; Saint-Faust, M.; Buffat, C.; Ligi, I.; Grandvuillemin, I.; Simeoni, U. Developmental origins of chronic renal disease: An integrative hypothesis. Int. J. Nephrol. 2013, 2013, 346067. [Google Scholar] [CrossRef] [PubMed]
- Singh, R.R.; Denton, K.M. Role of the kidney in the fetal programming of adult cardiovascular disease: An update. Curr. Opin. Pharmacol. 2015, 21, 53–59. [Google Scholar] [CrossRef] [PubMed]
- Luyckx, V.A.; Brenner, B.M. Birth weight, malnutrition and kidney-associated outcomes—A global concern. Nat. Rev. Nephrol. 2015, 11, 135–149. [Google Scholar] [CrossRef] [PubMed]
- Tain, Y.L.; Joles, J.A. Reprogramming: A preventive strategy in hypertension focusing on the kidney. Int. J. Mol. Sci. 2015. [Google Scholar] [CrossRef] [PubMed]
- Roseboom, T.; de Rooij, S.; Painter, R. The Dutch famine and its long-term consequences for adult health. Early Hum. Dev. 2006, 82, 485–491. [Google Scholar] [CrossRef] [PubMed]
- Painter, R.C.; Roseboom, T.J.; van Montfrans, G.A.; Bossuyt, P.M.; Krediet, R.T.; Osmond, C.; Barker, D.J.; Bleker, O.P. Microalbuminuria in adults after prenatal exposure to the Dutch famine. J. Am. Soc. Nephrol. 2005, 16, 189–194. [Google Scholar] [CrossRef] [PubMed]
- Luyckx, V.A.; Brenner, B.M. The clinical importance of nephron mass. J. Am. Soc. Nephrol. 2010, 21, 898–910. [Google Scholar] [CrossRef] [PubMed]
- Shah, M.M.; Sampogna, R.V.; Sakurai, H.; Bush, K.T.; Nigam, S.K. Branching morphogenesis and kidney disease. Development 2004, 131, 1449–1462. [Google Scholar] [CrossRef] [PubMed]
- Bertram, J.F.; Douglas-Denton, R.N.; Diouf, B.; Hughson, M.D.; Hoy, W.E. Human nephron number: Implications for health and disease. Pediatr. Nephrol. 2011, 26, 1529–1533. [Google Scholar] [CrossRef] [PubMed]
- Brenner, B.M.; Garcia, D.L.; Anderson, S. Glomeruli and blood pressure. Less of one, more the other? Am. J. Hypertens. 1988, 1, 335–347. [Google Scholar] [CrossRef] [PubMed]
- Nenov, V.D.; Taal, M.W.; Sakharova, O.V.; Brenner, B.M. Multi-hit nature of chronic renal disease. Curr. Opin. Nephrol. Hypertens. 2000, 9, 85–97. [Google Scholar] [CrossRef] [PubMed]
- White, S.L.; Perkovic, V.; Cass, A.; Chang, C.L.; Poulter, N.R.; Spector, T.; Haysom, L.; Craig, J.C.; Salmi, I.A.; Chadban, S.J.; et al. Is low birth weight an antecedent of CKD in later life? A systematic review of observational studies. Am. J. Kidney Dis. 2009, 54, 248–261. [Google Scholar] [CrossRef] [PubMed]
- Hsu, C.W.; Yamamoto, K.T.; Henry, R.K.; de Roos, A.J.; Flynn, J.T. Prenatal risk factors for childhood CKD. J. Am. Soc. Nephrol. 2014, 25, 2105–2111. [Google Scholar] [CrossRef] [PubMed]
- Tain, Y.L.; Luh, H.; Lin, C.Y.; Hsu, C.N. Incidence and risks of congenital anomalies of kidney and urinary tract in newborns: A population-based case-control study in Taiwan. Medicine 2016, 95, e2659. [Google Scholar] [CrossRef] [PubMed]
- Beeman, S.C.; Cullen-McEwen, L.A.; Puelles, G.; Zhang, M.; Wu, T.; Baldelomar, E.J.; Dowling, J.; Charlton, J.R.; Forbes, M.S.; Ng, A.; et al. MRI-based glomerular morphology and pathology in whole human kidneys. Am. J. Physiol. Ren. Physiol. 2014, 306, F1381–F1390. [Google Scholar] [CrossRef] [PubMed]
- Hales, C.N.; Barker, D.J. The thrifty phenotype hypothesis. Br. Med. Bull. 2001, 60, 5–20. [Google Scholar] [CrossRef] [PubMed]
- Gluckman, P.D.; Hanson, M.A. Living with the past: Evolution, development, and patterns of disease. Science 2004, 305, 1733–1736. [Google Scholar] [CrossRef] [PubMed]
- Cianfarani, S.; Germani, D.; Branca, F. Low birthweight and adult insulin resistance: The “catch-up growth” hypothesis. Arch. Dis. Child. Fetal Neonatal. 1999, 81, F71–F73. [Google Scholar] [CrossRef]
- Pham, T.D.; MacLennan, N.K.; Chiu, C.T.; Laksana, G.S.; Hsu, J.L.; Lane, R.H. Uteroplacental insufficiency increases apoptosis and alters p53 gene methylation in the full-term IUGR rat kidney. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2003, 285, R962–R970. [Google Scholar] [CrossRef] [PubMed]
- Lelièvre-Pégorier, M.; Vilar, J.; Ferrier, M.L.; Moreau, E.; Freund, N.; Gilbert, T.; Merlet-Bénichou, C. Mild vitamin A deficiency leads to inborn nephron deficit in the rat. Kidney Int. 1998, 54, 1455–1462. [Google Scholar] [CrossRef] [PubMed]
- Koleganova, N.; Piecha, G.; Ritz, E.; Becker, L.E.; Müller, A.; Weckbach, M.; Nyengaard, J.R.; Schirmacher, P.; Gross-Weissmann, M.L. Both high and low maternal salt intake in pregnancy alter kidney development in the offspring. Am. J. Physiol. Ren. Physiol. 2011, 301, F344–F354. [Google Scholar] [CrossRef] [PubMed]
- Merlet-Bénichou, C.; Gilbert, T.; Muffat-Joly, M.; Lelièvre-Pégorier, M.; Leroy, B. Intrauterine growth retardation leads to a permanent nephron deficit in the rat. Pediatr. Nephrol. 1994, 8, 175–180. [Google Scholar] [CrossRef] [PubMed]
- Gray, S.P.; Denton, K.M.; Cullen-McEwen, L.; Bertram, J.F.; Moritz, K.M. Prenatal exposure to alcohol reduces nephron number and raises blood pressure in progeny. J. Am. Soc. Nephrol. 2010, 21, 1891–1902. [Google Scholar] [CrossRef] [PubMed]
- Hao, X.Q.; Zhang, H.G.; Yuan, Z.B.; Yang, D.L.; Hao, L.Y.; Li, X.H. Prenatal exposure to lipopolysaccharide alters the intrarenal renin-angiotensin system and renal damage in offspring rats. Hypertens. Res. 2010, 33, 76–82. [Google Scholar] [CrossRef] [PubMed]
- Celsi, G.; Kistner, A.; Aizman, R.; Eklöf, A.C.; Ceccatelli, S.; de Santiago, A.; Jacobson, S.H. Prenatal dexamethasone causes oligonephronia, sodium retention, and higher blood pressure in the offspring. Pediatr. Res. 1998, 44, 317–322. [Google Scholar] [CrossRef] [PubMed]
- Ortiz, L.A.; Quan, A.; Weinberg, A.; Baum, M. Effect of prenatal dexamethasone on rat renal development. Kidney Int. 2001, 59, 1663–1669. [Google Scholar] [CrossRef] [PubMed]
- Luzardo, R.; Silva, P.A.; Einicker-Lamas, M.; Ortiz-Costa, S.; do Carmo Mda, G.; Vieira-Filho, L.D.; Paixão, A.D.; Lara, L.S.; Vieyra, A. Metabolic programming during lactation stimulates renal Na+ transport in the adult offspring due to an early impact on local angiotensin II pathways. PLoS ONE 2011, 6, e21232. [Google Scholar] [CrossRef] [PubMed]
- Slabiak-Blaz, N.; Adamczak, M.; Gut, N.; Grajoszek, A.; Nyengaard, J.R.; Ritz, E.; Wiecek, A. Administration of cyclosporine a in pregnant rats—The effect on blood pressure and on the glomerular number in their offspring. Kidney Blood Press. Res. 2015, 40, 413–423. [Google Scholar] [PubMed]
- Tain, Y.L.; Hsieh, C.S.; Lin, I.C.; Chen, C.C.; Sheen, J.M.; Huang, L.T. Effects of maternal l-citrulline supplementation on renal function and blood pressure in offspring exposed to maternal caloric restriction: The impact of nitric oxide pathway. Nitric Oxide 2010, 23, 34–41. [Google Scholar] [CrossRef] [PubMed]
- Tain, Y.L.; Lee, W.C.; Hsu, C.N.; Lee, W.C.; Huang, L.T.; Lee, C.T.; Lin, C.Y. Asymmetric dimethylarginine is associated with developmental programming of adult kidney disease and hypertension in offspring of streptozotocin-treated mothers. PLoS ONE 2013, 8, e55420. [Google Scholar] [CrossRef] [PubMed]
- Paixão, A.D.; Maciel, C.R.; Teles, M.B.; Figueiredo-Silva, J. Regional Brazilian diet-induced low birth weight is correlated with changes in renal hemodynamics and glomerular morphometry in adult age. Biol. Neonate 2001, 80, 239–246. [Google Scholar] [CrossRef] [PubMed]
- Woods, L.L.; Morgan, T.K.; Resko, J.A. Castration fails to prevent prenatally programmed hypertension in male rats. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2010, 298, R1111–R1116. [Google Scholar] [CrossRef] [PubMed]
- Tain, Y.L.; Chen, C.C.; Sheen, J.M.; Yu, H.R.; Tiao, M.M.; Kuo, H.C.; Huang, L.T. Melatonin attenuates prenatal dexamethasone-induced blood pressure increase in a rat model. J. Am. Soc. Hypertens. 2014, 8, 216–226. [Google Scholar] [CrossRef] [PubMed]
- Lisle, S.J.; Lewis, R.M.; Petry, C.J.; Ozanne, S.E.; Hales, C.N.; Forhead, A.J. Effect of maternal iron restriction during pregnancy on renal morphology in the adult rat offspring. Br. J. Nutr. 2003, 90, 33–39. [Google Scholar] [CrossRef] [PubMed]
- Hokke, S.; Puelles, V.G.; Armitage, J.A.; Fong, K.; Bertram, J.F.; Cullen-McEwen, L.A. Maternal fat feeding augments offspring nephron endowment in mice. PLoS ONE 2016, 11, e0161578. [Google Scholar] [CrossRef] [PubMed]
- Woods, L.L.; Ingelfinger, J.R.; Rasch, R. Modest maternal protein restriction fails to program adult hypertension in female rats. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2005, 289, R1131–R1136. [Google Scholar] [CrossRef] [PubMed]
- Boubred, F.; Buffat, C.; Feuerstein, J.M.; Daniel, L.; Tsimaratos, M.; Oliver, C.; Lelièvre-Pégorier, M.; Simeoni, U. Effects of early postnatal hypernutrition on nephron number and long-term renal function and structure in rats. Am. J. Physiol. Ren. Physiol. 2007, 293, F1944–F1949. [Google Scholar] [CrossRef] [PubMed]
- Thompson, L.P.; Al-Hasan, Y. Impact of oxidative stress in fetal programming. J. Pregnancy 2012, 2012, 582748. [Google Scholar] [CrossRef] [PubMed]
- Tain, Y.L.; Leu, S.; Wu, K.L.; Lee, W.C.; Chan, J.Y. Melatonin prevents maternal fructose intake-induced programmed hypertension in the offspring: Roles of nitric oxide and arachidonic acid metabolites. J. Pineal Res. 2014, 57, 80–89. [Google Scholar] [CrossRef] [PubMed]
- Tai, I.H.; Sheen, J.M.; Lin, Y.J.; Yu, H.R.; Tiao, M.M.; Chen, C.C.; Huang, L.T.; Tain, Y.L. Maternal N-acetylcysteine therapy regulates hydrogen sulfide-generating pathway and prevents programmed hypertension in male offspring exposed to prenatal dexamethasone and postnatal high-fat diet. Nitric Oxide 2016, 53, 6–12. [Google Scholar] [CrossRef] [PubMed]
- Tain, Y.L.; Hsu, C.N.; Lee, C.T.; Lin, Y.J.; Tsai, C.C. N-Acetylcysteine prevents programmed hypertension in male rat offspring born to suramin-treated mothers. Biol. Reprod. 2016, 95, 8. [Google Scholar] [CrossRef] [PubMed]
- Tain, Y.L.; Lee, C.T.; Chan, J.Y.; Hsu, C.N. Maternal melatonin or N-acetylcysteine therapy regulates hydrogen sulfide-generating pathway and renal transcriptome to prevent prenatal N(G)-Nitro-l-arginine-methyl ester (l-NAME)-induced fetal programming of hypertension in adult male offspring. Am. J. Obstet. Gynecol. 2016, 215, 636. [Google Scholar] [CrossRef] [PubMed]
- Stangenberg, S.; Nguyen, L.T.; Chen, H.; Al-Odat, I.; Killingsworth, M.C.; Gosnell, M.E.; Anwer, A.G.; Goldys, E.M.; Pollock, C.A.; Saad, S. Oxidative stress, mitochondrial perturbations and fetal programming of renal disease induced by maternal smoking. Int. J. Biochem. Cell Biol. 2015, 64, 81–90. [Google Scholar] [CrossRef] [PubMed]
- Cambonie, G.; Comte, B.; Yzydorczyk, C.; Ntimbane, T.; Germain, N.; Lê, N.L.; Pladys, P.; Gauthier, C.; Lahaie, I.; Abran, D.; et al. Antenatal antioxidant prevents adult hypertension, vascular dysfunction, and microvascular rarefaction associated with in utero exposure to a low-protein diet. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2007, 292, R1236–R1245. [Google Scholar] [CrossRef] [PubMed]
- Tain, Y.L.; Huang, L.T. Restoration of asymmetric dimethylarginine-nitric oxide balance to prevent the development of hypertension. Int. J. Mol. Sci. 2014, 15, 11773–11782. [Google Scholar] [CrossRef] [PubMed]
- Tain, Y.L. Targeting redox balance to deprogramme obesity: Are we starting early enough? J. Physiol. 2015, 593, 4689–4690. [Google Scholar] [CrossRef] [PubMed]
- Yosypiv, I.V. Renin-angiotensin system in ureteric bud branching morphogenesis: Insights into the mechanisms. Pediatr. Nephrol. 2011, 26, 1499–1512. [Google Scholar] [CrossRef] [PubMed]
- Te Riet, L.; van Esch, J.H.; Roks, A.J.; van den Meiracker, A.H.; Danser, A.H. Hypertension: Renin-angiotensin-aldosterone system alterations. Circ. Res. 2015, 116, 960–975. [Google Scholar] [CrossRef] [PubMed]
- Bogdarina, I.; Welham, S.; King, P.J.; Burns, S.P.; Clark, A.J. Epigenetic modification of the renin-angiotensin system in the fetal programming of hypertension. Circ. Res. 2007, 100, 520–526. [Google Scholar] [CrossRef] [PubMed]
- Chappell, M.C.; Marshall, A.C.; Alzayadneh, E.M.; Shaltout, H.A.; Diz, D.I. Update on the Angiotensin converting enzyme 2-Angiotensin (1–7)-MAS receptor axis: Fetal programing, sex differences, and intracellular pathways. Front. Endocrinol. 2014, 4, 201. [Google Scholar] [CrossRef] [PubMed]
- Sherman, R.C.; Langley-Evans, S.C. Early administration of angiotensin-converting enzyme inhibitor captopril, prevents the development of hypertension programmed by intrauterine exposure to a maternal low-protein diet in the rat. Clin. Sci. 1998, 94, 373–381. [Google Scholar] [CrossRef] [PubMed]
- Sherman, R.C.; Langley-Evans, S.C. Antihypertensive treatment in early postnatal life modulates prenatal dietary influences upon blood pressure in the rat. Clin. Sci. 2000, 98, 269–275. [Google Scholar] [CrossRef] [PubMed]
- Manning, J.; Vehaskari, V.M. Postnatal modulation of prenatally programmed hypertension by dietary Na and ACE inhibition. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2005, 288, R80–R84. [Google Scholar] [CrossRef] [PubMed]
- Hsu, C.N.; Lee, C.T.; Huang, L.T.; Tain, Y.L. Aliskiren in early postnatal life prevents hypertension and reduces asymmetric dimethylarginine in offspring exposed to maternal caloric restriction. J. Renin Angiotensin Aldosterone Syst. 2015, 16, 506–513. [Google Scholar] [CrossRef] [PubMed]
- Hsu, C.N.; Wu, K.L.; Lee, W.C.; Leu, S.; Chan, J.Y.; Tain, Y.L. Aliskiren administration during early postnatal life sex-specifically alleviates hypertension programmed by maternal high fructose consumption. Front. Physiol. 2016, 7, 299. [Google Scholar] [CrossRef] [PubMed]
- Dagan, A.; Kwon, H.M.; Dwarakanath, V.; Baum, M. Effect of renal denervation on prenatal programming of hypertension and renal tubular transporter abundance. Am. J. Physiol. Ren. Physiol. 2008, 295, F29–F34. [Google Scholar] [CrossRef] [PubMed]
- Tain, Y.L.; Lee, W.C.; Leu, S.; Wu, K.; Chan, J. High salt exacerbates programmed hypertension in maternal fructose-fed male offspring. Nutr. Metab. Cardiovasc. Dis. 2015, 25, 1146–1151. [Google Scholar] [CrossRef] [PubMed]
- Vallon, V.; Lang, F. New insights into the role of serum- and glucocorticoid-inducible kinase SGK1 in the regulation of renal function and blood pressure. Curr. Opin. Nephrol. Hypertens. 2005, 14, 59–66. [Google Scholar] [CrossRef] [PubMed]
- Rexhepaj, R.; Boini, K.M.; Huang, D.Y.; Amann, K.; Artunc, F.; Wang, K.; Brosens, J.J.; Kuhl, D.; Lang, F. Role of maternal glucocorticoid inducible kinase SGK1 in fetal programming of blood pressure in response to prenatal diet. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2008, 294, R2008–R2013. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Barajas, L. The rat renal nerves during development. Anat. Embryol. 1993, 188, 345–361. [Google Scholar] [CrossRef] [PubMed]
- Barajas, L.; Liu, L. The renal nerves in the newborn rat. Pediatr. Nephrol. 1993, 7, 657–666. [Google Scholar] [CrossRef] [PubMed]
- Jansson, T.; Lambert, G.W. Effect of intrauterine growth restriction on blood pressure, glucose tolerance and sympathetic nervous system activity in the rat at 3–4 months of age. J. Hypertens. 1999, 17, 1239–1248. [Google Scholar] [CrossRef] [PubMed]
- Alexander, B.T.; Hendon, A.E.; Ferril, G.; Dwyer, T.M. Renal denervation abolishes hypertension in low-birth-weight offspring from pregnant rats with reduced uterine perfusion. Hypertension 2005, 45, 754–758. [Google Scholar] [CrossRef] [PubMed]
- Sheen, J.M.; Yu, H.R.; Tiao, M.M.; Chen, C.C.; Huang, L.T.; Chang, H.Y.; Tain, Y.L. Prenatal dexamethasone-induced programmed hypertension and renal programming. Life Sci. 2015, 132, 41–48. [Google Scholar] [CrossRef] [PubMed]
- Tain, Y.L.; Wu, K.L.; Lee, W.C.; Leu, S.; Chan, J.Y. Maternal fructose-intake-induced renal programming in adult male offspring. J. Nutr. Biochem. 2015, 26, 642–650. [Google Scholar] [CrossRef] [PubMed]
- Moisiadis, V.G.; Matthews, S.G. Glucocorticoids and fetal programming part 2: Mechanisms. Nat. Rev. Endocrinol. 2014, 10, 403–411. [Google Scholar] [CrossRef] [PubMed]
- Langley-Evans, S.C.; Phillips, G.J.; Benediktsson, R. Protein intake in pregnancy, placental glucocorticoid metabolism and the programming of hypertension in the rat. Placenta 1996, 17, 169–172. [Google Scholar] [CrossRef]
- Mairesse, J.; Lesage, J.; Breton, C.; Breant, B.; Hahn, T.; Darnaudery, M.; Dickson, S.L.; Seckl, J.; Blondeau, B.; Vieau, D.; et al. Maternal stress alters endocrine function of the feto-placental unit in rats. Am. J. Physiol. Endocrinol. Metab. 2007, 292, E1526–E1533. [Google Scholar] [CrossRef] [PubMed]
- Kosicka, K.; Siemiątkowska, A.; Główka, F.K. 11β-Hydroxysteroid dehydrogenase 2 in preeclampsia. Int. J. Endocrinol. 2016, 2016, 5279462. [Google Scholar] [CrossRef] [PubMed]
- Cottrell, E.C.; Seckl, J.R. Prenatal stress, glucocorticoids and the programming of adult disease. Front. Behav. Neurosci. 2009, 3, 19. [Google Scholar] [CrossRef] [PubMed]
- Singh, R.R.; Moritz, K.M.; Bertram, J.F.; Cullen-McEwen, L.A. Effects of dexamethasone exposure on rat metanephric development: In vitro and in vivo studies. Am. J. Physiol. Ren. Physiol. 2007, 293, F548–F554. [Google Scholar] [CrossRef] [PubMed]
- Singh, R.R.; Cuffe, J.S.; Moritz, K.M. Short- and long-term effects of exposure to natural and synthetic glucocorticoids during development. Clin. Exp. Pharmacol. Physiol. 2012, 39, 979–989. [Google Scholar] [CrossRef] [PubMed]
- Marcotte, M.; Sharma, R.; Bouchard, M. Gene regulatory network of renal primordium development. Pediatr. Nephrol. 2014, 29, 637–644. [Google Scholar] [CrossRef] [PubMed]
- Cacalano, G.; Fariñas, I.; Wang, L.C.; Hagler, K.; Forgie, A.; Moore, M.; Armanini, M.; Phillips, H.; Ryan, A.M.; Reichardt, L.F.; et al. GFRα1 is an essential receptor component for GDNF in the developing nervous system and kidney. Neuron 1998, 21, 53–62. [Google Scholar] [CrossRef]
- Bird, A. Perceptions of epigenetics. Nature 2007, 447, 396–398. [Google Scholar] [CrossRef] [PubMed]
- Rees, W.D.; Hay, S.M.; Brown, D.S.; Antipatis, C.; Palmer, R.M. Maternal protein deficiency causes hypermethylation of DNA in the livers of rat fetuses. J. Nutr. 2000, 130, 1821–1826. [Google Scholar] [PubMed]
- Suter, M.; Ma, J.; Harris, A.; Patterson, L.; Brown, K.A.; Shope, C.; Showalter, L.; Abramovici, A.; Aagaard-Tillery, K.M. Maternal tobacco use modestly alters correlated epigenome-wide placental DNA methylation and gene expression. Epigenetics 2011, 6, 1284–1294. [Google Scholar] [CrossRef] [PubMed]
- Sable, P.; Randhir, K.; Kale, A.; Chavan-Gautam, P.; Joshi, S. Maternal micronutrients and brain global methylation patterns in the offspring. Nutr. Neurosci. 2015, 18, 30–36. [Google Scholar] [CrossRef] [PubMed]
- Ly, A.; Ishiguro, L.; Kim, D.; Im, D.; Kim, S.E.; Sohn, K.J.; Croxford, R.; Kim, Y.I. Maternal folic acid supplementation modulates DNA methylation and gene expression in the rat offspring in a gestation period-dependent and organ-specific manner. J. Nutr. Biochem. 2016, 33, 103–110. [Google Scholar] [CrossRef] [PubMed]
- Wu, T.H.; Kuo, H.C.; Lin, I.C.; Chien, S.J.; Huang, L.T.; Tain, Y.L. Melatonin prevents neonatal dexamethasone induced programmed hypertension: Histone deacetylase inhibition. J. Steroid Biochem. Mol. Biol. 2014, 144, 253–259. [Google Scholar] [CrossRef] [PubMed]
- Liu, N.; Zhuang, S. Treatment of chronic kidney diseases with histone deacetylase inhibitors. Front. Physiol. 2015, 6, 121. [Google Scholar] [CrossRef] [PubMed]
- Floris, I.; Kraft, J.D.; Altosaar, I. Roles of microRNA across prenatal and postnatal periods. Int. J. Mol. Sci. 2016, 17. [Google Scholar] [CrossRef] [PubMed]
- Khorram, O.; Han, G.; Bagherpour, R.; Magee, T.R.; Desai, M.; Ross, M.G.; Chaudhri, A.A.; Toloubeydokhti, T.; Pearce, W.J. Effect of maternal undernutrition on vascular expression of micro and messenger RNA in newborn and aging offspring. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2010, 298, R1366–R1374. [Google Scholar] [CrossRef] [PubMed]
- Sene Lde, B.; Mesquita, F.F.; de Moraes, L.N.; Santos, D.C.; Carvalho, R.; Gontijo, J.A.; Boer, P.A. Involvement of renal corpuscle microRNA expression on epithelial-to-mesenchymal transition in maternal low protein diet in adult programmed rats. PLoS ONE 2013, 8, e71310. [Google Scholar] [CrossRef] [PubMed]
- Mouillet, J.F.; Chu, T.; Hubel, C.A.; Nelson, D.M.; Parks, W.T.; Sadovsky, Y. The levels of hypoxia-regulated microRNAs in plasma of pregnant women with fetal growth restriction. Placenta 2010, 31, 781–784. [Google Scholar] [CrossRef] [PubMed]
- Tomat, A.L.; Salazar, F.J. Mechanisms involved in developmental programming of hypertension and renal diseases. Gender differences. Horm. Mol. Biol. Clin. Investig. 2014, 18, 63–77. [Google Scholar] [CrossRef] [PubMed]
- Ojeda, N.B.; Intapad, S.; Alexander, B.T. Sex differences in the developmental programming of hypertension. Acta Physiol. 2014, 210, 307–316. [Google Scholar] [CrossRef] [PubMed]
- Hilliard, L.M.; Sampson, A.K.; Brown, R.D.; Denton, K.M. The “his and hers” of the renin-angiotensin system. Curr. Hypertens. Rep. 2013, 15, 71–79. [Google Scholar] [CrossRef] [PubMed]
- Vina, J.; Gambini, J.; Lopez-Grueso, R.; Abdelaziz, K.M.; Jove, M.; Borras, C. Females live longer than males: Role of oxidative stress. Curr. Pharm. Des. 2011, 17, 3959–3965. [Google Scholar] [CrossRef] [PubMed]
- Kwekel, J.C.; Desai, V.G.; Moland, C.L.; Vijay, V.; Fuscoe, J.C. Sex differences in kidney gene expression during the life cycle of F344 rats. Biol. Sex Differ. 2013, 4, 14. [Google Scholar] [CrossRef] [PubMed]
- Tain, Y.L.; Wu, M.S.; Lin, Y.J. Sex differences in renal transcriptome and programmed hypertension in offspring exposed to prenatal dexamethasone. Steroids 2016, 115, 40–46. [Google Scholar] [CrossRef] [PubMed]
- Mao, J.; Zhang, X.; Sieli, P.T.; Falduto, M.T.; Torres, K.E.; Rosenfeld, C.S. Contrasting effects of different maternal diets on sexually dimorphic gene expression in the murine placenta. Proc. Natl. Acad. Sci. USA 2010, 107, 5557–5562. [Google Scholar] [CrossRef] [PubMed]
- Cox, L.A.; Li, C.; Glenn, J.P.; Lange, K.; Spradling, K.D.; Nathanielsz, P.W.; Jansson, T. Expression of the placental transcriptome in maternal nutrient reduction in baboons is dependent on fetal sex. J. Nutr. 2013, 143, 1698–1708. [Google Scholar] [CrossRef] [PubMed]
- Vaiman, D.; Gascoin-Lachambre, G.; Boubred, F.; Mondon, F.; Feuerstein, J.M.; Ligi, I.; Grandvuillemin, I.; Barbaux, S.; Ghigo, E.; Achard, V. The intensity of IUGR-induced transcriptome deregulations is inversely correlated with the onset of organ function in a rat model. PLoS ONE 2011, 6, e21222. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buffat, C.; Boubred, F.; Mondon, F.; Chelbi, S.T.; Feuerstein, J.M.; Lelièvre-Pégorier, M.; Vaiman, D.; Simeoni, U. Kidney gene expression analysis in a rat model of intrauterine growth restriction revealsmassive alterations of coagulation genes. Endocrinology 2007, 148, 5549–5557. [Google Scholar] [CrossRef] [PubMed]
- Almon, R.R.; Lai, W.; DuBois, D.C.; Jusko, W.J. Corticosteroid-regulated genes in rat kidney: Mining time series array data. Am. J. Physiol. Endocrinol. 2005, 289, E870–E882. [Google Scholar] [CrossRef] [PubMed]
- Tain, Y.L.; Huang, L.T.; Chan, J.Y.; Lee, C.T. Transcriptome analysis in rat kidneys: Importance of genes involved in programmed hypertension. Int. J. Mol. Sci. 2015, 16, 4744–4758. [Google Scholar] [CrossRef] [PubMed]
- Tain, Y.L.; Hsu, C.N.; Chan, J.Y.; Huang, L.T. Renal Transcriptome analysis of programmed hypertension induced by maternal nutritional insults. Int. J. Mol. Sci. 2015, 16, 17826–17837. [Google Scholar] [CrossRef] [PubMed]
- Ruan, X.; Zheng, F.; Guan, Y. PPARs and the kidney in metabolic syndrome. Am. J. Physiol. Ren. Physiol. 2008, 294, F1032–F1047. [Google Scholar] [CrossRef] [PubMed]
- Tain, Y.L.; Hsu, C.N.; Chan, J. PPARs link early life nutritional insults to later programmed hypertension and metabolic syndrome. Int. J. Mol. Sci. 2016. [Google Scholar] [CrossRef] [PubMed]
- Griffith, O.W. Biologic and pharmacologic regulation of mammalian glutathione synthesis. Free Radic. Biol. Med. 1999, 27, 922–935. [Google Scholar] [CrossRef]
- Tain, Y.L.; Lee, W.C.; Wu, K.L.; Leu, S.; Chan, J.Y. Targeting arachidonic acid pathway to prevent programmed hypertension in maternal fructose-fed male adult rat offspring. J. Nutr. Biochem. 2016, 38, 86–92. [Google Scholar] [CrossRef] [PubMed]
- Lu, P.C.; Sheen, J.M.; Yu, H.R.; Lin, Y.J.; Chen, C.C.; Tiao, M.M.; Tsai, C.C.; Huang, L.T.; Tain, Y.L. Early postnatal treatment with soluble epoxide hydrolase inhibitor or 15-deoxy-Δ(12,14)-prostagandin J2 prevents prenatal dexamethasone and postnatal high saturated fat diet induced programmed hypertension in adult rat offspring. Prostaglandins Other Lipid Mediat. 2016, 124, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Costantini, F. GDNF/Ret signaling and renal branching morphogenesis: From mesenchymal signals to epithelial cell behaviors. Organogenesis 2010, 6, 252–262. [Google Scholar] [CrossRef] [PubMed]
- Vieira-Filho, L.D.; Cabral, E.V.; Santos, F.T.; Coimbra, T.M.; Paixão, A.D. α-Tocopherol prevents intrauterine undernutrition-induced oligonephronia in rats. Pediatr. Nephrol. 2011, 26, 2019–2029. [Google Scholar] [CrossRef] [PubMed]
- Khodus, G.R.; Kruusmägi, M.; Li, J.; Liu, X.L.; Aperia, A. Calcium signaling triggered by ouabain protects the embryonic kidney from adverse developmental programming. Pediatr. Nephrol. 2011, 26, 1479–1482. [Google Scholar] [CrossRef] [PubMed]
- Makrakis, J.; Zimanyi, M.A.; Black, M.J. Retinoic acid enhances nephron endowment in rats exposed to maternal protein restriction. Pediatr. Nephrol. 2007, 22, 1861–1867. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| Experimental Model | Renal Phenotype | Age at Evaluation of Nephron Endowment | Ref. |
|---|---|---|---|
| Uteroplacental insufficiency | ↑ Apoptosis | 1 day | [30] |
| Vitamin A-deficient diet from 3 weeks before mating throughout pregnancy | Not evaluated | 1 day | [31] |
| Low sodium diet (0.07%) during pregnancy and lactation | Hypertension at 5 months | 1 week | [32] |
| High sodium diet (3%) during pregnancy and lactation | Glomerular hypertrophy, hypertension at 5 month | 1 week | [32] |
| Partial ligation of uterine ligation | ↓ GFR, glomerular hypertrophy | 2 weeks | [33] |
| Ethanol (1 g/kg/day) at gestational day 13.5 and 14.5 | ↓ GFR at 6 months | 4 weeks | [34] |
| Lipopolysaccharide (0.79 mg/kg/day) i.p. at gestational day 8, 10, and 12 | ↓ GFR | 7 weeks | [35] |
| Dexamethasone (0.1 mg/kg/day) throughout pregnancy | ↓ GFR, glomerular hypertrophy | 2 months | [36] |
| Dexamethasone (0.2 mg/kg/day) at gestational day 15 and 16 or 17 and 18 | ↔ GFR, unchanged glomerular morphology | 2 months | [37] |
| Low protein diet (8% protein) during lactation | Hypertension at 5 months | 2 months | [38] |
| Cyclosporine (3.3 mg/kg/day) from gestational day 10 to postnatal day 7 | ↔ GFR, glomerular hypertrophy | 3 months | [39] |
| 50% caloric restriction during pregnancy and lactation | ↔ GFR, glomerular hypertrophy, hypertension, tubulointerstitial injury | 3 months | [40] |
| Streptozotocin (STZ)-induced diabetes during pregnancy | ↔ GFR, hypertension, tuburointerstitial injury | 3 months | [41] |
| Multideficient diet during pregnancy | ↑ GFR, glomerular hypertrophy | 3 months | [42] |
| Dexamethasone (0.1 mg/kg/day) from gestational day 16 to 22. | Hypertension | 4 months | [43] |
| Low protein diet (8.5% protein) during pregnancy | ↔ GFR, hypertension | 5.5 months | [44] |
| Iron restriction diet (3 mg/kg diet) from 1 week before mating and through pregnancy | Glomerular hypertrophy, hypertension | 18 months | [45] |
| Caloric Restriction | Diabetes |
|---|---|
| Ribosome | Ribosome |
| Cell cycle | ABC transporters |
| Oocyte meiosis | Complement and coagulation cascades |
| DNA replication | Spliceosome |
| Fatty acid metabolism | Antigen processing and presentation |
| Tryptophan metabolism | Prostate cancer |
| Homologous recombination | Drug metabolism |
| Progesterone-mediated oocyte maturation | Histidine metabolism |
| Valine, leucine, and isoleucine degradation | Metabolism of xenobiotics by cytochrome P450 |
| Prostate cancer | ECM-receptor interaction |
| PPAR signaling pathway | Tryptophan metabolism |
| Glutathione metabolism | Glutathione metabolism |
| Arginine and proline metabolism | PPAR signaling pathway |
| High fructose | High salt |
| PPAR signaling pathway | Cell adhesion molecules (CAMs) |
| Butanoate metabolism | Complement and coagulation cascades |
| Arachidonic acid metabolism | Hematopoietic cell lineage |
| Fatty acid metabolism | Systemic lupus erythematosus |
| Glutathione metabolism | Intestinal immune network for IgA production |
| Metabolism of xenobiotics by cytochrome P450 | Graft-versus-host disease |
| Tyrosine metabolism | Allograft rejection |
| Drug metabolism |
| Gene ID | Gene Symbol | CR | STZ | HF | HS |
|---|---|---|---|---|---|
| Expansion and survival of renal stem cells | |||||
| ENSRNOG00000012278 | Fgf10 | 0.52 | 0.38 | 0.55 | 0.87 |
| Formation and extension of the primary nephric duct | |||||
| ENSRNOG00000012819 | Gdnf | 2563 | 1508 | 1836 | 1362 |
| ENSRNOG00000008430 | Spry3 | ND | 123 | 1.04 | 278 |
| ENSRNOG00000022777 | Six1 | 1.58 | 0.4 | 2.64 | 1.55 |
| ENSRNOG00000026053 | Grem1 | 0.57 | 0.47 | 1.21 | 0.83 |
| Initiation of metanephric development | |||||
| ENSRNOG00000003807 | Wnt9b | 0.75 | 1.27 | 0.49 | 0.85 |
| ENSRNOG00000015982 | Wnt11 | 1.18 | 3.37 | 1.29 | 1.3 |
| ENSRNOG00000007002 | Lif | 0.36 | 0.64 | 0.95 | 1.12 |
| ENSRNOG00000017392 | Fgf2 | 2.07 | 2.9 | 1.54 | 0.82 |
| ENSRNOG00000020792 | Etv4 | 0.94 | 2.66 | 1.7 | 1.53 |
| Mesoderm patterning | |||||
| ENSRNOG00000004210 | Osr1 | 0.27 | 0.46 | 0.61 | 0.57 |
| ENSRNOG00000021276 | Bmp2 | 1.72 | 2.39 | 0.86 | 1.05 |
| ENSRNOG00000000556 | Nodal | ND | ND | ND | 242 |
| Nephron development | |||||
| ENSRNOG00000004517 | Igf1 | 0.55 | 0.44 | 0.77 | 0.64 |
| ENSRNOG00000004346 | Notch3 | 1.18 | 2.09 | 0.83 | 1.06 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tain, Y.-L.; Hsu, C.-N. Developmental Origins of Chronic Kidney Disease: Should We Focus on Early Life? Int. J. Mol. Sci. 2017, 18, 381. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms18020381
Tain Y-L, Hsu C-N. Developmental Origins of Chronic Kidney Disease: Should We Focus on Early Life? International Journal of Molecular Sciences. 2017; 18(2):381. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms18020381
Chicago/Turabian StyleTain, You-Lin, and Chien-Ning Hsu. 2017. "Developmental Origins of Chronic Kidney Disease: Should We Focus on Early Life?" International Journal of Molecular Sciences 18, no. 2: 381. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms18020381