
The Temporal Pattern, Flux, and Function of Autophagy in Spinal Cord Injury



Abstract
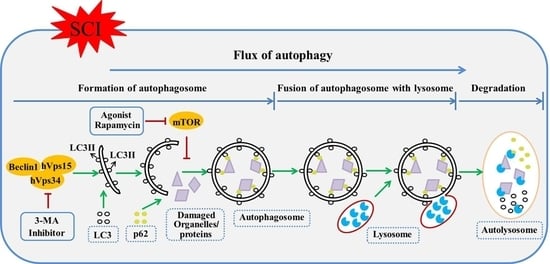
:
1. Introduction
2. Temporal Pattern of Autophagic Activation in SCI
2.1. Temporal Pattern of Autophagy in TSCI
2.1.1. Spinal Cord Hemisection Injury
2.1.2. Spinal Cord Contusion Injury
2.1.3. Spinal Cord Compression Injury
2.2. Temporal Pattern of Autophagy in IRSCI
3. Autophagic Flux Blockade or Enhancement in SCI
4. Autophagic Cell Death in SCI
5. Relationship between Autophagy and Apoptosis after SCI
6. Pharmacological Intervention of Autophagy in SCI
6.1. Therapeutic Agent Effect on Autophagy in SCI
6.2. Agents Directly Modulate Autophagy in SCI
7. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Nowrouzi, B.; Assan-Lebbe, A.; Sharma, B.; Casole, J.; Nowrouzi-Kia, B. Spinal cord injury: A review of the most-cited publications. Eur. Spine J. 2016, 26, 28–39. [Google Scholar] [CrossRef] [PubMed]
- Guilcher, S.J.; Parsons, D.; Craven, B.C.; Jaglal, S.B.; Verrier, M. Developing quality of care indicators for patients with traumatic and non-traumatic spinal cord injury (SCI): A feasibility study using administrative health data. J. Spinal Cord Med. 2015, 38, 765–776. [Google Scholar] [CrossRef] [PubMed]
- Kjell, J.; Olson, L. Rat models of spinal cord injury: From pathology to potential therapies. Dis. Model. Mech. 2016, 9, 1125–1137. [Google Scholar] [CrossRef] [PubMed]
- Saunders, L.L.; Clarke, A.; Tate, D.G.; Forchheimer, M.; Krause, J.S. Lifetime prevalence of chronic health conditions among persons with spinal cord injury. Arch. Phys. Med. Rehabil. 2015, 96, 673–679. [Google Scholar] [CrossRef] [PubMed]
- Coselli, J.S.; LeMaire, S.A.; Miller, C.C.; Schmittling, Z.C.; Koksoy, C.; Pagan, J.; Curling, P.E. Mortality and paraplegia after thoracoabdominal aortic aneurysm repair: A risk factor analysis. Ann. Thorac. Surg. 2000, 69, 409–414. [Google Scholar] [CrossRef]
- Celic, T.; Spanjol, J.; Bobinac, M.; Tovmasyan, A.; Vukelic, I.; Reboucas, J.S.; Batinic-Haberle, I.; Bobinac, D. Mn porphyrin-based SOD mimic, MnTnHex-2-pyp5+, and non-SOD mimic, MnTBAP3−, suppressed rat spinal cord ischemia/reperfusion injury via NF-κB pathways. Free Radic. Res. 2014, 48, 1426–1442. [Google Scholar] [CrossRef] [PubMed]
- Iyer, N.R.; Wilems, T.S.; Sakiyama-Elbert, S.E. Stem cells for spinal cord injury: Strategies to inform differentiation and transplantation. Biotechnol. Bioeng. 2016, 114, 245–259. [Google Scholar] [CrossRef] [PubMed]
- Norenberg, M.D.; Smith, J.; Marcillo, A. The pathology of human spinal cord injury: Defining the problems. J. Neurotrauma 2004, 21, 429–440. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Yao, Y.; He, R.; Meng, Y.; Li, N.; Zhang, D.; Xu, J.; Chen, O.; Cui, J.; Bian, J.; et al. Methane ameliorates spinal cord ischemia-reperfusion injury in rats: Antioxidant, anti-inflammatory and anti-apoptotic activity mediated by Nrf2 activation. Free Radic. Biol. Med. 2016, 103, 69–86. [Google Scholar] [CrossRef] [PubMed]
- Jia, X.; Kowalski, R.G.; Sciubba, D.M.; Geocadin, R.G. Critical care of traumatic spinal cord injury. J. Intensive Care Med. 2013, 28, 12–23. [Google Scholar] [CrossRef] [PubMed]
- Miyamoto, T.A.; Miyamoto, K.J. Retrograde venous perfusion for spinal cord protection. Ann. Thorac. Surg. 2000, 69, 1987–1989. [Google Scholar] [CrossRef]
- Fan, J.; Zhang, Z.; Chao, X.; Gu, J.; Cai, W.; Zhou, W.; Yin, G.; Li, Q. Ischemic preconditioning enhances autophagy but suppresses autophagic cell death in rat spinal neurons following ischemia-reperfusion. Brain Res. 2014, 1562, 76–86. [Google Scholar] [CrossRef] [PubMed]
- Shi, E.; Kazui, T.; Jiang, X.; Washiyama, N.; Suzuki, K.; Yamashita, K.; Terada, H. Ns-7, a novel Na+/Ca2+ channel blocker, prevents neurologic injury after spinal cord ischemia in rabbits. J. Thorac. Cardiovasc. Surg. 2005, 129, 364–371. [Google Scholar] [CrossRef] [PubMed]
- Xiong, M.; Chen, S.; Yu, H.; Liu, Z.; Zeng, Y.; Li, F. Neuroprotection of erythropoietin and methylprednisolone against spinal cord ischemia-reperfusion injury. J. Huazhong Univ. Sci. Technol. Med. Sci. 2011, 31, 652–656. [Google Scholar] [CrossRef] [PubMed]
- Bains, M.; Hall, E.D. Antioxidant therapies in traumatic brain and spinal cord injury. Biochim. Biophys. Acta 2012, 1822, 675–684. [Google Scholar] [CrossRef] [PubMed]
- Kawano, H.; Kimura-Kuroda, J.; Komuta, Y.; Yoshioka, N.; Li, H.P.; Kawamura, K.; Li, Y.; Raisman, G. Role of the lesion scar in the response to damage and repair of the central nervous system. Cell Tissue Res. 2012, 349, 169–180. [Google Scholar] [CrossRef] [PubMed]
- Totoiu, M.O.; Keirstead, H.S. Spinal cord injury is accompanied by chronic progressive demyelination. J. Comp. Neurol. 2005, 486, 373–383. [Google Scholar] [CrossRef] [PubMed]
- Alblas, C.L.; Bouvy, W.H.; Lycklama, A.N.G.J.; Boiten, J. Acute spinal-cord ischemia: Evolution of MRI findings. J. Clin. Neurol. 2012, 8, 218–223. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Li, W.; Kang, Z.; Liu, Y.; Deng, X.; Tao, H.; Xu, W.; Li, R.; Sun, X.; Zhang, J.H. Hyperbaric oxygen preconditioning attenuates early apoptosis after spinal cord ischemia in rats. J. Neurotrauma 2009, 26, 55–66. [Google Scholar] [CrossRef] [PubMed]
- Li, X.Q.; Wang, J.; Fang, B.; Tan, W.F.; Ma, H. Intrathecal antagonism of microglial TLR4 reduces inflammatory damage to blood-spinal cord barrier following ischemia/reperfusion injury in rats. Mol. Brain 2014, 7, 28. [Google Scholar] [CrossRef] [PubMed]
- Shin, H.J.; Kim, H.; Oh, S.; Lee, J.G.; Kee, M.; Ko, H.J.; Kweon, M.N.; Won, K.J.; Baek, S.H. AMPK-SKP2-CARM1 signalling cascade in transcriptional regulation of autophagy. Nature 2016, 534, 553–557. [Google Scholar] [CrossRef] [PubMed]
- Glick, D.; Barth, S.; Macleod, K.F. Autophagy: Cellular and molecular mechanisms. J. Pathol. 2010, 221, 3–12. [Google Scholar] [CrossRef] [PubMed]
- Nikoletopoulou, V.; Papandreou, M.E.; Tavernarakis, N. Autophagy in the physiology and pathology of the central nervous system. Cell Death Differ. 2015, 22, 398–407. [Google Scholar] [CrossRef] [PubMed]
- Galluzzi, L.; Bravo-San Pedro, J.M.; Blomgren, K.; Kroemer, G. Autophagy in acute brain injury. Nat. Rev. Neurosci. 2016, 17, 467–484. [Google Scholar] [CrossRef] [PubMed]
- Lipinski, M.M.; Wu, J.; Faden, A.I.; Sarkar, C. Function and mechanisms of autophagy in brain and spinal cord trauma. Antioxid. Redox Signal. 2015, 23, 565–577. [Google Scholar] [CrossRef] [PubMed]
- Zhou, K.L.; Zhou, Y.F.; Wu, K.; Tian, N.F.; Wu, Y.S.; Wang, Y.L.; Chen, D.H.; Zhou, B.; Wang, X.Y.; Xu, H.Z.; et al. Stimulation of autophagy promotes functional recovery in diabetic rats with spinal cord injury. Scient. Rep. 2015, 5, 17130. [Google Scholar] [CrossRef] [PubMed]
- Tang, P.; Hou, H.; Zhang, L.; Lan, X.; Mao, Z.; Liu, D.; He, C.; Du, H.; Zhang, L. Autophagy reduces neuronal damage and promotes locomotor recovery via inhibition of apoptosis after spinal cord injury in rats. Mol. Neurobiol. 2014, 49, 276–287. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.Y.; Wang, Z.G.; Wu, F.Z.; Kong, X.X.; Yang, J.; Lin, B.B.; Zhu, S.P.; Lin, L.; Gan, C.S.; Fu, X.B.; et al. Regulation of autophagy and ubiquitinated protein accumulation by BFGF promotes functional recovery and neural protection in a rat model of spinal cord injury. Mol. Neurobiol. 2013, 48, 452–464. [Google Scholar] [CrossRef] [PubMed]
- Yu, D.; Li, M.; Ni, B.; Kong, J.; Zhang, Z. Induction of neuronal mitophagy in acute spinal cord injury in rats. Neurotox. Res. 2013, 24, 512–522. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.C.; Zhang, S.; Du, T.Y.; Wang, B.; Sun, X.Q. Hyperbaric oxygen preconditioning reduces ischemia-reperfusion injury by stimulating autophagy in neurocyte. Brain Res. 2010, 1323, 149–151. [Google Scholar] [CrossRef] [PubMed]
- Kanno, H.; Ozawa, H.; Sekiguchi, A.; Itoi, E. Spinal cord injury induces upregulation of beclin 1 and promotes autophagic cell death. Neurobiol. Dis. 2009, 33, 143–148. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Sarkar, C.; Dinizo, M.; Faden, A.I.; Koh, E.Y.; Lipinski, M.M.; Wu, J. Disrupted autophagy after spinal cord injury is associated with er stress and neuronal cell death. Cell Death Dis. 2015, 6, e1582. [Google Scholar] [CrossRef] [PubMed]
- Kabeya, Y.; Mizushima, N.; Ueno, T.; Yamamoto, A.; Kirisako, T.; Noda, T.; Kominami, E.; Ohsumi, Y.; Yoshimori, T. LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J. 2000, 19, 5720–5728. [Google Scholar] [CrossRef] [PubMed]
- Tanida, I.; Ueno, T.; Kominami, E. Lc3 conjugation system in mammalian autophagy. Int. J. Biochem. Cell Biol. 2004, 36, 2503–2518. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Liu, D.; Su, P.; Lin, F.; Tang, Q. Changes in autophagy in rats after spinal cord injury and the effect of hyperbaric oxygen on autophagy. Neurosci. Lett. 2016, 618, 139–145. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.Y.; Lin, J.H.; Muharram, A.; Liu, W.G. Beclin-1-mediated autophagy protects spinal cord neurons against mechanical injury-induced apoptosis. Apoptosis 2014, 19, 933–945. [Google Scholar] [CrossRef] [PubMed]
- Bjorkoy, G.; Lamark, T.; Brech, A.; Outzen, H.; Perander, M.; Overvatn, A.; Stenmark, H.; Johansen, T. P62/SQSTM1 forms protein aggregates degraded by autophagy and has a protective effect on huntingtin-induced cell death. J. Cell Biol. 2005, 171, 603–614. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Xuan, J.; Zheng, B.B.; Zhou, Y.L.; Lin, Y.; Wu, Y.S.; Zhou, Y.F.; Huang, Y.X.; Wang, Q.; Shen, L.Y.; et al. Metformin improves functional recovery after spinal cord injury via autophagy flux stimulation. Mol. Neurobiol. 2016. [Google Scholar] [CrossRef] [PubMed]
- Kanno, H.; Ozawa, H.; Sekiguchi, A.; Yamaya, S.; Itoi, E. Induction of autophagy and autophagic cell death in damaged neural tissue after acute spinal cord injury in mice. Spine 2011, 36, E1427–E1434. [Google Scholar] [CrossRef] [PubMed]
- Baba, H.; Sakurai, M.; Abe, K.; Tominaga, R. Autophagy-mediated stress response in motor neuron after transient ischemia in rabbits. J. Vasc. Surg. 2009, 50, 381–387. [Google Scholar] [CrossRef] [PubMed]
- Kanno, H.; Ozawa, H.; Sekiguchi, A.; Itoi, E. The role of autophagy in spinal cord injury. Autophagy 2009, 5, 390–392. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Jiang, H.K.; Li, Y.P.; Guo, Y.P. Hydrogen sulfide protects spinal cord and induces autophagy via miR-30c in a rat model of spinal cord ischemia-reperfusion injury. J. Biomed. Sci. 2015, 22, 50. [Google Scholar] [CrossRef] [PubMed]
- Anwar, M.A.; Al Shehabi, T.S.; Eid, A.H. Inflammogenesis of secondary spinal cord injury. Front. Cell. Neurosci. 2016, 10, 98. [Google Scholar] [CrossRef] [PubMed]
- Sekiguchi, A.; Kanno, H.; Ozawa, H.; Yamaya, S.; Itoi, E. Rapamycin promotes autophagy and reduces neural tissue damage and locomotor impairment after spinal cord injury in mice. J. Neurotrauma 2012, 29, 946–956. [Google Scholar] [CrossRef] [PubMed]
- Hou, H.; Zhang, L.; Zhang, L.; Tang, P. Acute spinal cord injury in rats should target activated autophagy. J. Neurosurg. Spine 2014, 20, 568–577. [Google Scholar] [CrossRef] [PubMed]
- Okada, S.; Nakamura, M.; Mikami, Y.; Shimazaki, T.; Mihara, M.; Ohsugi, Y.; Iwamoto, Y.; Yoshizaki, K.; Kishimoto, T.; Toyama, Y.; et al. Blockade of interleukin-6 receptor suppresses reactive astrogliosis and ameliorates functional recovery in experimental spinal cord injury. J. Neurosci. Res. 2004, 76, 265–276. [Google Scholar] [CrossRef] [PubMed]
- Cui, S.S.; Yang, C.P.; Bowen, R.C.; Bai, O.; Li, X.M.; Jiang, W.; Zhang, X. Valproic acid enhances axonal regeneration and recovery of motor function after sciatic nerve axotomy in adult rats. Brain Res. 2003, 975, 229–236. [Google Scholar] [CrossRef]
- Gruner, J.A. A monitored contusion model of spinal cord injury in the rat. J. Neurotrauma 1992, 9, 123–128. [Google Scholar] [CrossRef] [PubMed]
- Agrawal, G.; Kerr, C.; Thakor, N.V.; All, A.H. Characterization of graded multicenter animal spinal cord injury study contusion spinal cord injury using somatosensory-evoked potentials. Spine 2010, 35, 1122–1127. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.C.; Fong, T.H.; Lee, A.W.; Chiu, W.T. Autophagy is activated in injured neurons and inhibited by methylprednisolone after experimental spinal cord injury. Spine 2012, 37, 470–475. [Google Scholar] [CrossRef] [PubMed]
- Hao, H.H.; Wang, L.; Guo, Z.J.; Bai, L.; Zhang, R.P.; Shuang, W.B.; Jia, Y.J.; Wang, J.; Li, X.Y.; Liu, Q. Valproic acid reduces autophagy and promotes functional recovery after spinal cord injury in rats. Neurosci. Bull. 2013, 29, 484–492. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Guo, Z.; Xu, Z.; Meng, Q.; Chen, C.; Zhang, Y.; Cao, X. Effect of pollen typhae on inhibiting autophagy in spinal cord injury of rats and its mechanisms. Int. J. Clin. Exp. Pathol. 2015, 8, 2375–2383. [Google Scholar]
- Gao, K.; Wang, G.; Wang, Y.; Han, D.; Bi, J.; Yuan, Y.; Yao, T.; Wan, Z.; Li, H.; Mei, X. Neuroprotective effect of simvastatin via inducing the autophagy on spinal cord injury in the rat model. BioMed Res. Int. 2015, 2015, 260161. [Google Scholar] [CrossRef] [PubMed]
- Walker, C.L.; Walker, M.J.; Liu, N.K.; Risberg, E.C.; Gao, X.; Chen, J.; Xu, X.M. Systemic bisperoxovanadium activates Akt/mTOR, reduces autophagy, and enhances recovery following cervical spinal cord injury. PLoS ONE 2012, 7, e30012. [Google Scholar] [CrossRef] [PubMed]
- Li, H.T.; Zhao, X.Z.; Zhang, X.R.; Li, G.; Jia, Z.Q.; Sun, P.; Wang, J.Q.; Fan, Z.K.; Lv, G. Exendin-4 enhances motor function recovery via promotion of autophagy and inhibition of neuronal apoptosis after spinal cord injury in rats. Mol. Neurobiol. 2016, 53, 4073–4082. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.C.; Fong, T.H.; Hsu, P.W.; Chiu, W.T. Multifaceted effects of rapamycin on functional recovery after spinal cord injury in rats through autophagy promotion, anti-inflammation, and neuroprotection. J. Surg. Res. 2013, 179, e203–e210. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.C.; Hsu, P.W.; Tzaan, W.C.; Lee, A.W. Effects of the combined administration of vitamins c and e on the oxidative stress status and programmed cell death pathways after experimental spinal cord injury. Spinal Cord 2014, 52, 24–28. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.Y.; Liu, W.G.; Muharram, A.; Wu, Z.Y.; Lin, J.H. Neuroprotective effects of autophagy induced by rapamycin in rat acute spinal cord injury model. Neuroimmunomodulation 2014, 21, 257–267. [Google Scholar] [CrossRef] [PubMed]
- Abdullahi, D.; Annuar, A.A.; Mohamad, M.; Aziz, I.; Sanusi, J. Experimental spinal cord trauma: A review of mechanically induced spinal cord injury in rat models. Rev. Neurosci. 2016, 28, 15–20. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Zheng, B.; Ye, L.; Zhang, H.; Zhu, S.; Zheng, X.; Xia, Q.; He, Z.; Wang, Q.; Xiao, J.; et al. Retinoic acid prevents disruption of blood-spinal cord barrier by inducing autophagic flux after spinal cord injury. Neurochem. Res. 2016, 41, 813–825. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Zhu, Q.; Man, X.; Guo, L.; Hao, L. Ginsenoside RD inhibits apoptosis following spinal cord ischemia/reperfusion injury. Neural Regen. Res. 2014, 9, 1678–1687. [Google Scholar]
- Gokce, E.C.; Kahveci, R.; Gokce, A.; Sargon, M.F.; Kisa, U.; Aksoy, N.; Cemil, B.; Erdogan, B. Curcumin attenuates inflammation, oxidative stress, and ultrastructural damage induced by spinal cord ischemia-reperfusion injury in rats. J. Stroke Cerebrovasc. Dis. 2016, 25, 1196–1207. [Google Scholar] [CrossRef] [PubMed]
- Fang, B.; Li, X.Q.; Bao, N.R.; Tan, W.F.; Chen, F.S.; Pi, X.L.; Zhang, Y.; Ma, H. Role of autophagy in the bimodal stage after spinal cord ischemia reperfusion injury in rats. Neuroscience 2016, 328, 107–116. [Google Scholar] [CrossRef] [PubMed]
- Wei, X.; Zhou, Z.; Li, L.; Gu, J.; Wang, C.; Xu, F.; Dong, Q.; Zhou, X. Intrathecal injection of 3-methyladenine reduces neuronal damage and promotes functional recovery via autophagy attenuation after spinal cord ischemia/reperfusion injury in rats. Biol. Pharm. Bull. 2016, 39, 665–673. [Google Scholar] [CrossRef] [PubMed]
- Sakai, K.; Fukuda, T.; Iwadate, K. Immunohistochemical analysis of the ubiquitin proteasome system and autophagy lysosome system induced after traumatic intracranial injury: Association with time between the injury and death. Am. J. Forensic Med. Pathol. 2014, 35, 38–44. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.J.; Chen, S.; Huang, K.X.; Le, W.D. Why should autophagic flux be assessed? Acta Pharm. Sin. 2013, 34, 595–599. [Google Scholar] [CrossRef] [PubMed]
- Mizushima, N.; Levine, B. Autophagy in mammalian development and differentiation. Nat. Cell Biol. 2010, 12, 823–830. [Google Scholar] [CrossRef] [PubMed]
- Lipinski, M.M.; Wu, J. Modification of autophagy-lysosomal pathway as a neuroprotective treatment for spinal cord injury. Neural Regen. Res. 2015, 10, 892–893. [Google Scholar] [CrossRef] [PubMed]
- Castillo, K.; Valenzuela, V.; Matus, S.; Nassif, M.; Onate, M.; Fuentealba, Y.; Encina, G.; Irrazabal, T.; Parsons, G.; Court, F.A.; et al. Measurement of autophagy flux in the nervous system in vivo. Cell Death Dis. 2013, 4, e917. [Google Scholar] [CrossRef] [PubMed]
- El-Horany, H.E.; El-Latif, R.N.; ElBatsh, M.M.; Emam, M.N. Ameliorative effect of quercetin on neurochemical and behavioral deficits in rotenone rat model of parkinson’s disease: Modulating autophagy (quercetin on experimental parkinson’s disease). J. Biochem. Mol. Toxicol. 2016, 30, 360–369. [Google Scholar] [CrossRef] [PubMed]
- Son, J.H.; Shim, J.H.; Kim, K.H.; Ha, J.Y.; Han, J.Y. Neuronal autophagy and neurodegenerative diseases. Exp. Mol. Med. 2012, 44, 89–98. [Google Scholar] [CrossRef] [PubMed]
- Ventruti, A.; Cuervo, A.M. Autophagy and neurodegeneration. Curr. Neurol. Neurosci. Rep. 2007, 7, 443–451. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.W.; Chen, B.; Huang, K.L.; Dai, Y.S.; Teng, H.L. Inhibition of autophagy by estradiol promotes locomotor recovery after spinal cord injury in rats. Neurosci. Bull. 2016, 32, 137–144. [Google Scholar] [CrossRef] [PubMed]
- Mizushima, N.; Yoshimori, T.; Levine, B. Methods in mammalian autophagy research. Cell 2010, 140, 313–326. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Zhang, H.; Zheng, B.; Ye, L.; Zhu, S.; Johnson, N.R.; Wang, Z.; Wei, X.; Chen, D.; Cao, G.; et al. Retinoic acid induced-autophagic flux inhibits ER-stress dependent apoptosis and prevents disruption of blood-spinal cord barrier after spinal cord injury. Int. J. Biol. Sci. 2016, 12, 87–99. [Google Scholar] [CrossRef] [PubMed]
- Joshi, M.; Fehlings, M.G. Development and characterization of a novel, graded model of clip compressive spinal cord injury in the mouse: Part 2. Quantitative neuroanatomical assessment and analysis of the relationships between axonal tracts, residual tissue, and locomotor recovery. J. Neurotrauma 2002, 19, 191–203. [Google Scholar] [CrossRef] [PubMed]
- Zhou, K.L.; Chen, D.H.; Jin, H.M.; Wu, K.; Wang, X.Y.; Xu, H.Z.; Zhang, X.L. Effects of calcitriol on experimental spinal cord injury in rats. Spinal Cord 2016, 54, 510–516. [Google Scholar] [CrossRef] [PubMed]
- Fuchs, Y.; Steller, H. Programmed cell death in animal development and disease. Cell 2011, 147, 742–758. [Google Scholar] [CrossRef] [PubMed]
- Yonekawa, T.; Thorburn, A. Autophagy and cell death. Essays Biochem. 2013, 55, 105–117. [Google Scholar] [CrossRef] [PubMed]
- Clarke, P.G. Developmental cell death: Morphological diversity and multiple mechanisms. Anat. Embryol. 1990, 181, 195–213. [Google Scholar] [CrossRef] [PubMed]
- Kroemer, G.; Levine, B. Autophagic cell death: The story of a misnomer. Nat. Rev. Mol. Cell Biol. 2008, 9, 1004–1010. [Google Scholar] [CrossRef] [PubMed]
- Larsen, K.E.; Sulzer, D. Autophagy in neurons: A review. Histol. Histopathol. 2002, 17, 897–908. [Google Scholar] [PubMed]
- Rami, A.; Langhagen, A.; Steiger, S. Focal cerebral ischemia induces upregulation of beclin 1 and autophagy-like cell death. Neurobiol. Dis. 2008, 29, 132–141. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, C.; Isaka, Y.; Takabatake, Y.; Tanaka, H.; Koike, M.; Shibata, M.; Uchiyama, Y.; Takahara, S.; Imai, E. Participation of autophagy in renal ischemia/reperfusion injury. Biochem. Biophys. Res. Commun. 2008, 368, 100–106. [Google Scholar] [CrossRef] [PubMed]
- Krysko, D.V.; Vanden Berghe, T.; D’Herde, K.; Vandenabeele, P. Apoptosis and necrosis: Detection, discrimination and phagocytosis. Methods 2008, 44, 205–221. [Google Scholar] [CrossRef] [PubMed]
- Pattingre, S.; Tassa, A.; Qu, X.; Garuti, R.; Liang, X.H.; Mizushima, N.; Packer, M.; Schneider, M.D.; Levine, B. Bcl-2 antiapoptotic proteins inhibit beclin 1-dependent autophagy. Cell 2005, 122, 927–939. [Google Scholar] [CrossRef] [PubMed]
- Marino, G.; Niso-Santano, M.; Baehrecke, E.H.; Kroemer, G. Self-consumption: The interplay of autophagy and apoptosis. Nat. Rev. Mol. Cell Biol. 2014, 15, 81–94. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Gao, C.; Yao, S.; Xie, B. Blocking autophagic flux enhances matrine-induced apoptosis in human hepatoma cells. Int. J. Mol. Sci. 2013, 14, 23212–23230. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Rodriguez, A.; Mayoral, R.; Agra, N.; Valdecantos, M.P.; Pardo, V.; Miquilena-Colina, M.E.; Vargas-Castrillon, J.; Lo Iacono, O.; Corazzari, M.; Fimia, G.M.; et al. Impaired autophagic flux is associated with increased endoplasmic reticulum stress during the development of nafld. Cell Death Dis. 2014, 5, e1179. [Google Scholar] [CrossRef] [PubMed]
- Shaerzadeh, F.; Motamedi, F.; Minai-Tehrani, D.; Khodagholi, F. Monitoring of neuronal loss in the hippocampus of abeta-injected rat: Autophagy, mitophagy, and mitochondrial biogenesis stand against apoptosis. Neuromol. Med. 2014, 16, 175–190. [Google Scholar] [CrossRef] [PubMed]
- Meira Martins, L.A.; Vieira, M.Q.; Ilha, M.; de Vasconcelos, M.; Biehl, H.B.; Lima, D.B.; Schein, V.; Barbe-Tuana, F.; Borojevic, R.; Guma, F.C. The interplay between apoptosis, mitophagy and mitochondrial biogenesis induced by resveratrol can determine activated hepatic stellate cells death or survival. Cell Biochem. Biophys. 2015, 71, 657–672. [Google Scholar] [CrossRef] [PubMed]
- Green, D.R.; Reed, J.C. Mitochondria and apoptosis. Science 1998, 281, 1309–1312. [Google Scholar] [CrossRef] [PubMed]
- Carloni, S.; Buonocore, G.; Balduini, W. Protective role of autophagy in neonatal hypoxia-ischemia induced brain injury. Neurobiol. Dis. 2008, 32, 329–339. [Google Scholar] [CrossRef] [PubMed]
- Diskin, T.; Tal-Or, P.; Erlich, S.; Mizrachy, L.; Alexandrovich, A.; Shohami, E.; Pinkas-Kramarski, R. Closed head injury induces upregulation of beclin 1 at the cortical site of injury. J. Neurotrauma 2005, 22, 750–762. [Google Scholar] [CrossRef] [PubMed]
- Gao, L.; Jiang, T.; Guo, J.; Liu, Y.; Cui, G.; Gu, L.; Su, L.; Zhang, Y. Inhibition of autophagy contributes to ischemic postconditioning-induced neuroprotection against focal cerebral ischemia in rats. PLoS ONE 2012, 7, e46092. [Google Scholar] [CrossRef] [PubMed]
- Uchiyama, Y.; Koike, M.; Shibata, M. Autophagic neuron death in neonatal brain ischemia/hypoxia. Autophagy 2008, 4, 404–408. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.; Liu, S.; Zhang, X.; Wang, L.; Gao, J.; Han, A.; Hao, A. G-csf promotes autophagy and reduces neural tissue damage after spinal cord injury in mice. Lab. Investig. 2015, 95, 1439–1449. [Google Scholar] [CrossRef] [PubMed]
- Gao, S.; Zhang, Z.M.; Shen, Z.L.; Gao, K.; Chang, L.; Guo, Y.; Li, Z.; Wang, W.; Wang, A.M. Atorvastatin activates autophagy and promotes neurological function recovery after spinal cord injury. Neural Regen. Res. 2016, 11, 977–982. [Google Scholar] [CrossRef] [PubMed]
- Seo, J.Y.; Kim, Y.H.; Kim, J.W.; Kim, S.I.; Ha, K.Y. Effects of therapeutic hypothermia on apoptosis and autophagy after spinal cord injury in rats. Spine 2015, 40, 883–890. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Liu, C.; Gao, K.; Zhao, H.; Zhou, Z.; Shen, Z.; Guo, Y.; Li, Z.; Yao, T.; Mei, X. Metformin preconditioning provide neuroprotection through enhancement of autophagy and suppression of inflammation and apoptosis after spinal cord injury. Biochem. Biophys. Res. Commun. 2016, 477, 534–540. [Google Scholar] [CrossRef] [PubMed]
- Goldshmit, Y.; Kanner, S.; Zacs, M.; Frisca, F.; Pinto, A.R.; Currie, P.D.; Pinkas-Kramarski, R. Rapamycin increases neuronal survival, reduces inflammation and astrocyte proliferation after spinal cord injury. Mol. Cell. Neurosci. 2015, 68, 82–91. [Google Scholar] [CrossRef] [PubMed]
- Fu, J.; Xue, R.; Gu, J.; Xiao, Y.; Zhong, H.; Pan, X.; Ran, R. Neuroprotective effect of calcitriol on ischemic/reperfusion injury through the NR3A/CREB pathways in the rat hippocampus. Mol. Med. Rep. 2013, 8, 1708–1714. [Google Scholar] [PubMed]
- Calvert, J.W.; Gundewar, S.; Jha, S.; Greer, J.J.; Bestermann, W.H.; Tian, R.; Lefer, D.J. Acute metformin therapy confers cardioprotection against myocardial infarction via AMPK-eNOS-mediated signaling. Diabetes 2008, 57, 696–705. [Google Scholar] [CrossRef] [PubMed]
- Tran, A.T.; Ramalinga, M.; Kedir, H.; Clarke, R.; Kumar, D. Autophagy inhibitor 3-methyladenine potentiates apoptosis induced by dietary tocotrienols in breast cancer cells. Eur. J. Nutr. 2015, 54, 265–272. [Google Scholar] [CrossRef] [PubMed]
- He, M.; Ding, Y.; Chu, C.; Tang, J.; Xiao, Q.; Luo, Z.G. Autophagy induction stabilizes microtubules and promotes axon regeneration after spinal cord injury. Proc. Nat. Acad. Sci. USA 2016, 113, 11324–11329. [Google Scholar] [CrossRef] [PubMed]
- Barth, S.; Glick, D.; Macleod, K.F. Autophagy: Assays and artifacts. J. Pathol. 2010, 221, 117–124. [Google Scholar] [CrossRef] [PubMed]
- Yoon, Y.H.; Cho, K.S.; Hwang, J.J.; Lee, S.J.; Choi, J.A.; Koh, J.Y. Induction of lysosomal dilatation, arrested autophagy, and cell death by chloroquine in cultured ARPE-19 cells. Investig. Ophthalmol. Vis. Sci. 2010, 51, 6030–6037. [Google Scholar] [CrossRef] [PubMed]
- Blight, A.R. Macrophages and inflammatory damage in spinal cord injury. J. Neurotrauma 1992, 9 (Suppl. 1), S83–S91. [Google Scholar] [PubMed]
- Alirezaei, M.; Kemball, C.C.; Whitton, J.L. Autophagy, inflammation and neurodegenerative disease. Eur. J. Neurosci. 2011, 33, 197–204. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
| Agents | Classification | Autophagic Mechanism | Autophagic Regulation | Autophagosomes | Flux | Pathologic Mechanism | Behavior Test | Models |
|---|---|---|---|---|---|---|---|---|
| Vitamins C and E [57] | Antioxidants | Not referred | Upregulation | LC3II↑ | Not referred | Oxidative stress↓ Apoptosis↓ | Hindlimb function↑ | Contusion injury (T9–T10) |
| Exendin-4 [55] | Glucagon-like peptide-1 agonist | Not referred | Upregulation | LC3II/I↑ Beclin1↑ | Not referred | Neuron loss↓ Cavity formation↓ Apoptosis↓ | Hindlimb function↑ | Contusion injury (T9–T11) |
| Simvastation [53] | Inhibitor of 3-hydroxy-3-methylglutaryl-coenzyme A reductase | mammalian target of rapamycin (mTOR) inhibition | Upregulation | LC3II↑ Beclin1↑ | Not referred | Brain-derived neurotrophic factor (BDNF)↓ Glial cell line-derived neurotrophic factor (GDNF)↓ Apoptosis↓ | Hindlimb function↑ | Contusion injury (T9–T10) |
| Atorvastatin [98] | Inhibitor of 3-hydroxy-3-methylglutaryl-coenzyme A reductase | Not referred | Upregulation | LC3II↑ Beclin1↑ | Not referred | Apoptosis↓ | Hindlimb function↑ | Contusion injury (T9–T10) |
| Systemic bisperoxovanadium [54] | Small-molecule protein tyrosine phosphatase (PTP) inhibitor | mTOR activation | Downregulation | LC3II/I↓ | Not referred | Neuron loss↓Cavity formation↓ | Forelimb function↑ | Contusion injury (C5) |
| Valproic acid [51] | Histone deacetylase (HDA) inhibitor | Not referred | Downregulation | LC3II↓ Beclin1↓ | Not referred | Neuron loss↓ Demyelination↓ | Hindlimb function↑ | Contusion injury (T10) |
| Methylprednisolone [99] | Synthetic glucocorticoid hormone | Not referred | Downregulation | LC3II↓ | Not referred | Apoptosis↓ | Hindlimb function↑ | Contusion injury (T9) |
| Calcitriol [77] | Biologically active metabolite of vitamin D | Not referred | Upregulation | LC3II↑ Beclin1↑ | Enhancement p62↓ | Neuron loss↓ Cavity formation↓ Apoptosis↓ | Hindlimb function↑ | Compression injury (T9) |
| Metformin [38,100] | Hypoglycemic agent for the therapy of type 2 diabetes mellitus | Adenosine monophosphate-activated protein kinase (AMPK) actiavation | Upregulation | LC3II↑ Beclin1↑ | Enhancement p62↓ | Neuron loss↓ Cavity formation↓ Apoptosis↓ | Hindlimb function↑ | Compression injury (T9); Contusion injury (T9–T10) |
| Basic fibroblast growth factor (bFGF) [28] | Member of the fibroblast growth factors | mTOR activation | Downregulation | LC3II/I↓ | Inhibition p62↑ | Neuron loss↓ Cavity formation↓ Ubiquitinated protein↓ | Hindlimb function↑ | Compression injury (T9) |
| Estradiol [73] | a17β-estradiol, E2 | Not referred | Downregulation | LC3II/I↓ Beclin1↓ Atg5↓ Atg7↓ | Inhibition p62↑ | Neuron loss↓ Cavity formation↓ | Hindlimb function↑ | Compression injury (T10) |
| Granulocyte colony-stimulating factor (G-CSF) [97] | Member of the CSF family of hormone-like glycoproteins | Not referred | Upregulation | LC3II↑ | Not referred | Apoptosis↓ | Hindlimb function↑ | Hemisection injury (T10) |
| Retinoic acid [75] | Biologically active metabolite of vitamin A | Not referred | Upregulation | LC3II↑ | Enhancement p62↓ | Apoptosis↓ Disruption of blood-spinal cord barrier↓ | Hindlimb function↑ | Hemisection injury (T9) |
| Hydrogen sulfide [42] | Novel gaseous mediator | Micro-RNA-30c inhibition | Upregulation | LC3II↑ Beclin1↑ | Not referred | Spinal cord infarction zone↓ | Hindlimb function↑ | I/R injury (thoracic aorta blocking) |
| Rapamycin [26,27,56,58,101] | Specifical mTOR agonist | mTOR activation | Upregulation | LC3II↑ Beclin1↑ | Enhancement p62↓ | Apoptosis↓ Inflammation↓ Neuron loss↓ Cavity formation↓ | Hindlimb function↑ | Contusion injury (T9–T10); Hemisection injury (T9–T10; T12); Compression injury (T9) |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhou, K.; Sansur, C.A.; Xu, H.; Jia, X. The Temporal Pattern, Flux, and Function of Autophagy in Spinal Cord Injury. Int. J. Mol. Sci. 2017, 18, 466. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms18020466
Zhou K, Sansur CA, Xu H, Jia X. The Temporal Pattern, Flux, and Function of Autophagy in Spinal Cord Injury. International Journal of Molecular Sciences. 2017; 18(2):466. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms18020466
Chicago/Turabian StyleZhou, Kailiang, Charles A. Sansur, Huazi Xu, and Xiaofeng Jia. 2017. "The Temporal Pattern, Flux, and Function of Autophagy in Spinal Cord Injury" International Journal of Molecular Sciences 18, no. 2: 466. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms18020466