
L1198F Mutation Resensitizes Crizotinib to ALK by Altering the Conformation of Inhibitor and ATP Binding Sites

Abstract
:1. Introduction
2. Results and Discussion
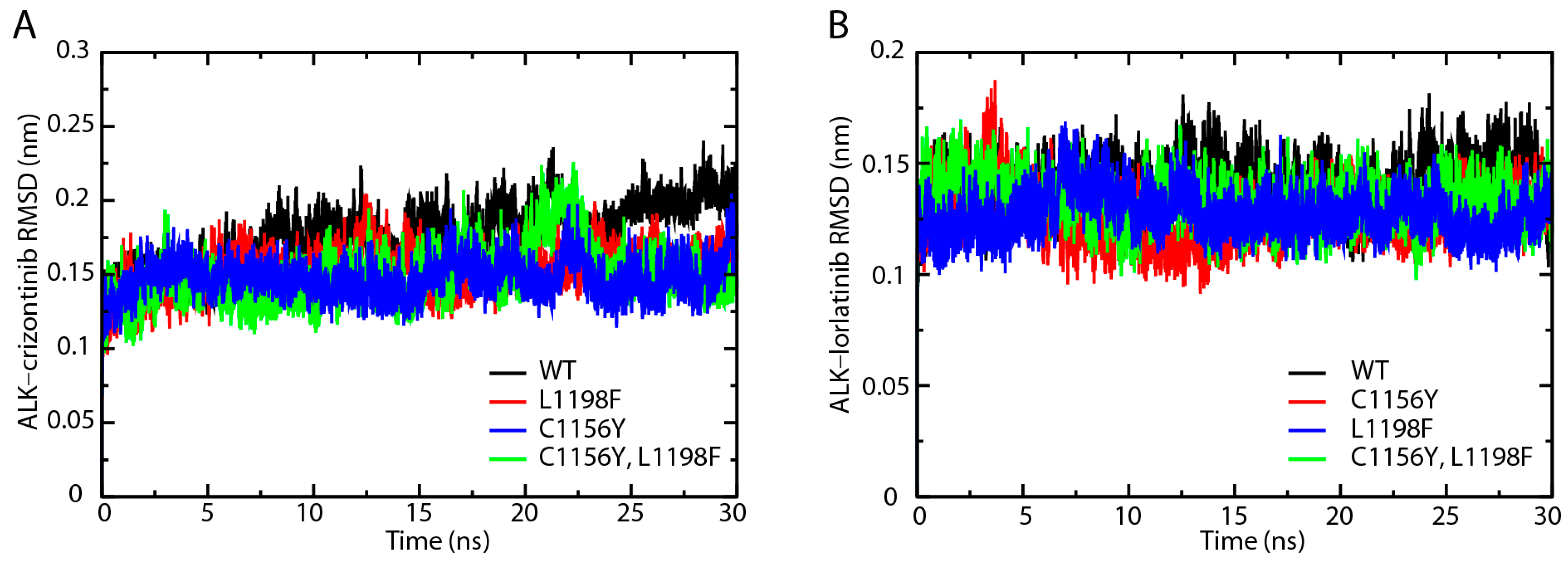
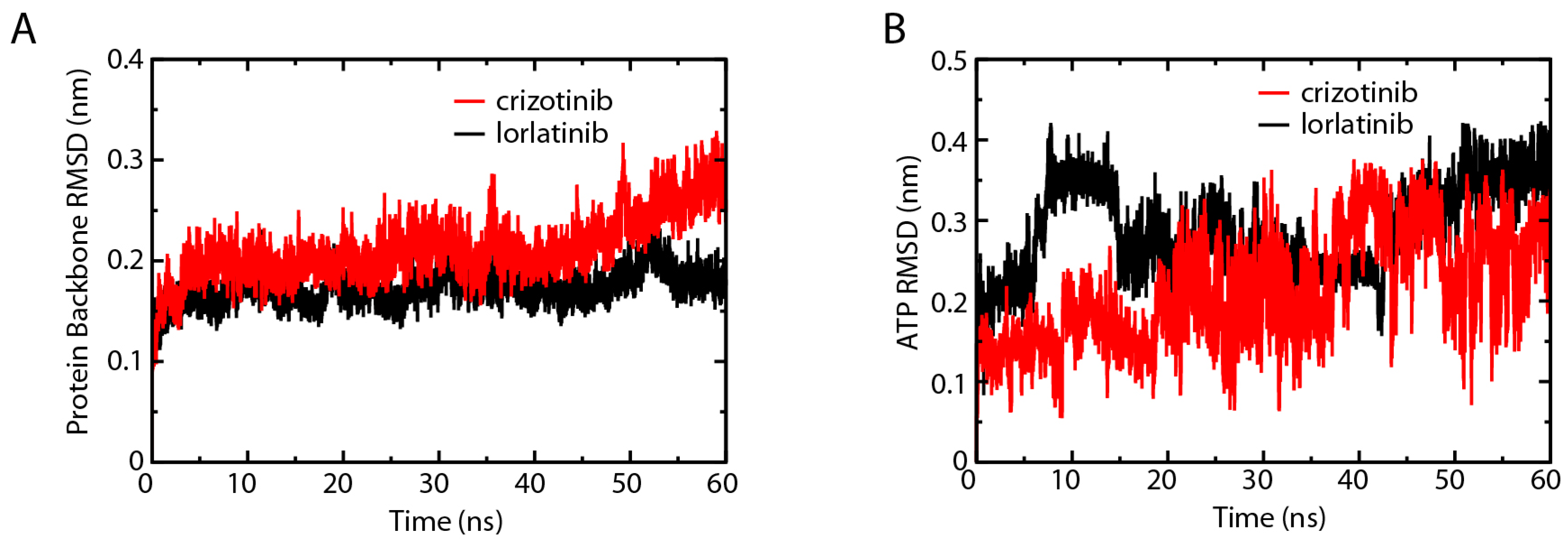
2.1. Root-Mean-Square Deviation Analysis of the Protein Backbones in Crizotinib/Lorlatinib Associated ALKs
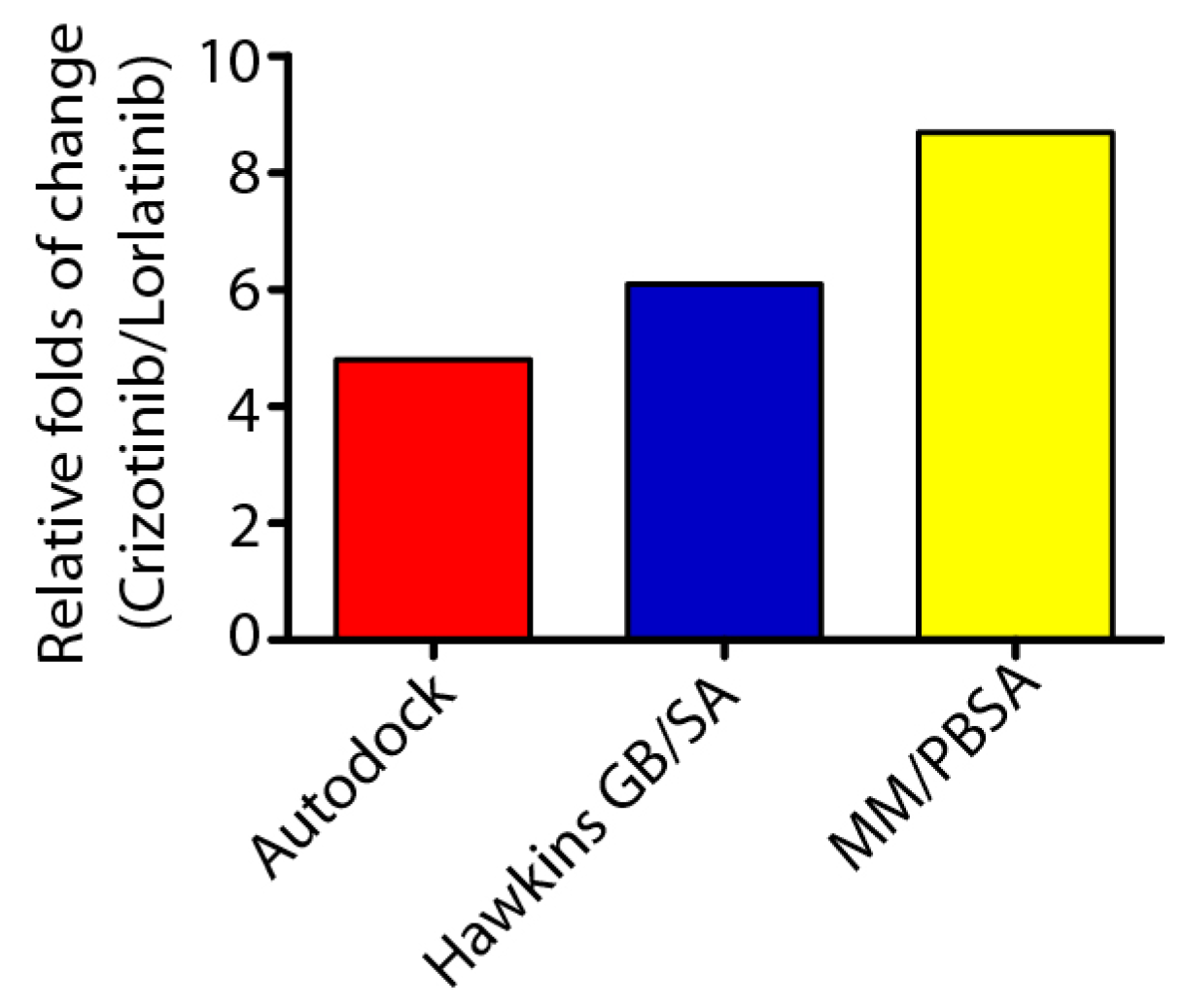
2.2. Crizontinib and Lorlatinib Binds to ALK (C1156Y-L1198F) with Different Affinity
2.3. Root-Mean-Square Deviation Comparison of ALK–Crizotinib and ALK–Lorlatinib Complexes
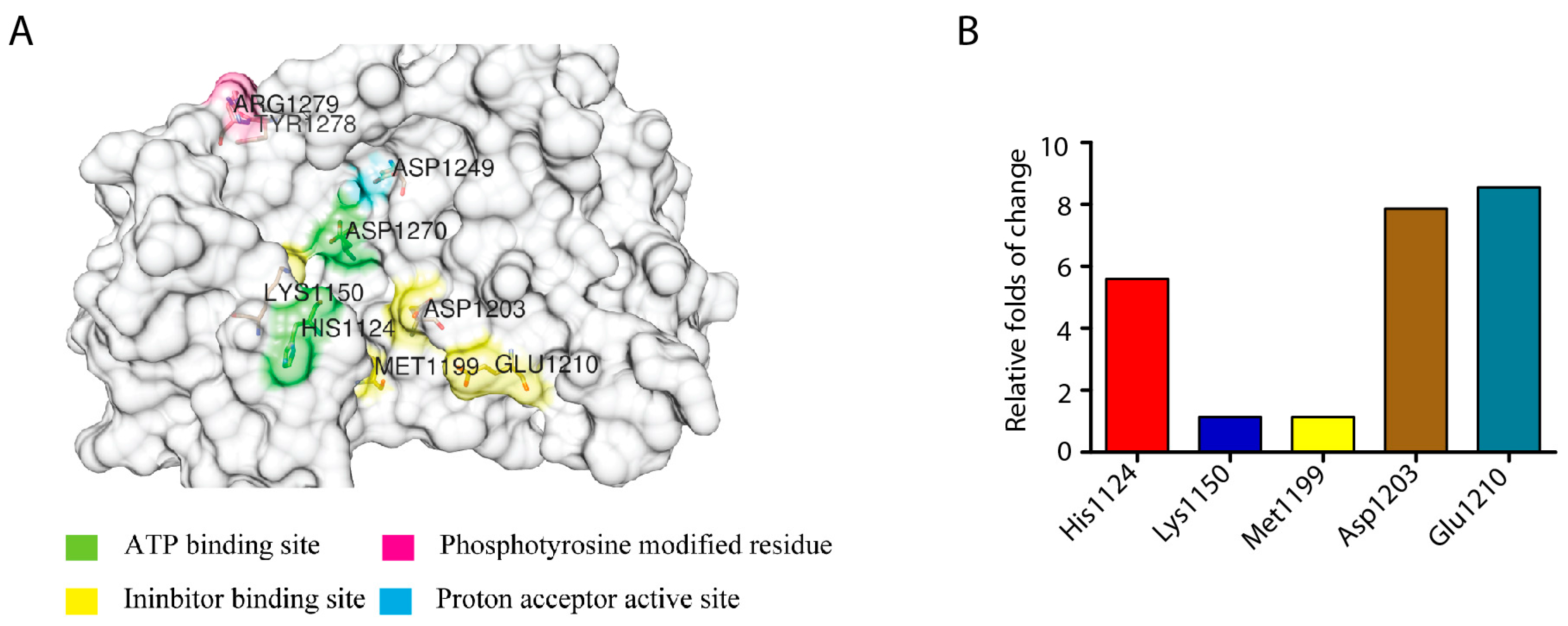
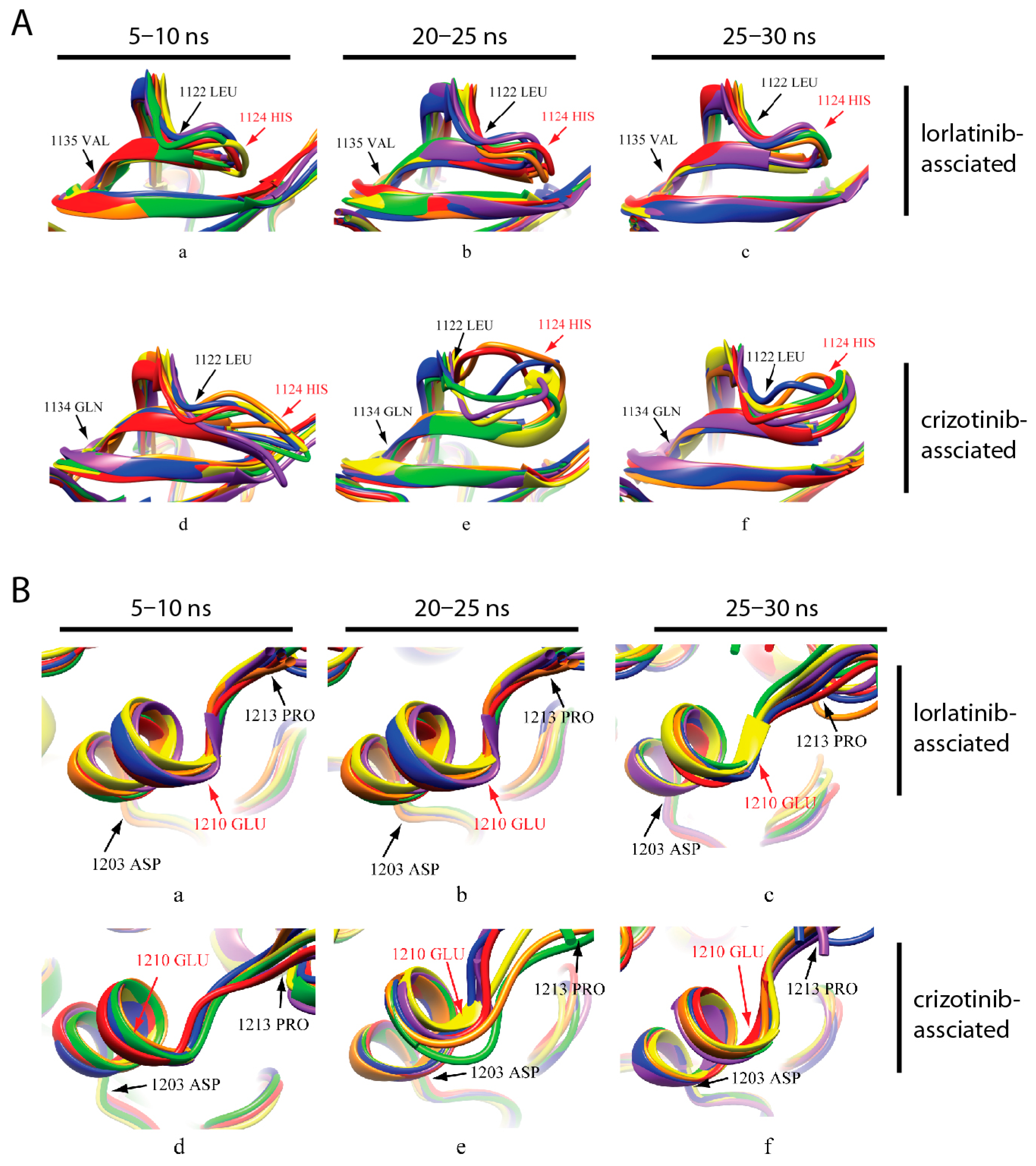
2.4. Identification of the Key Amino Acid Residues Affecting ALK Activity
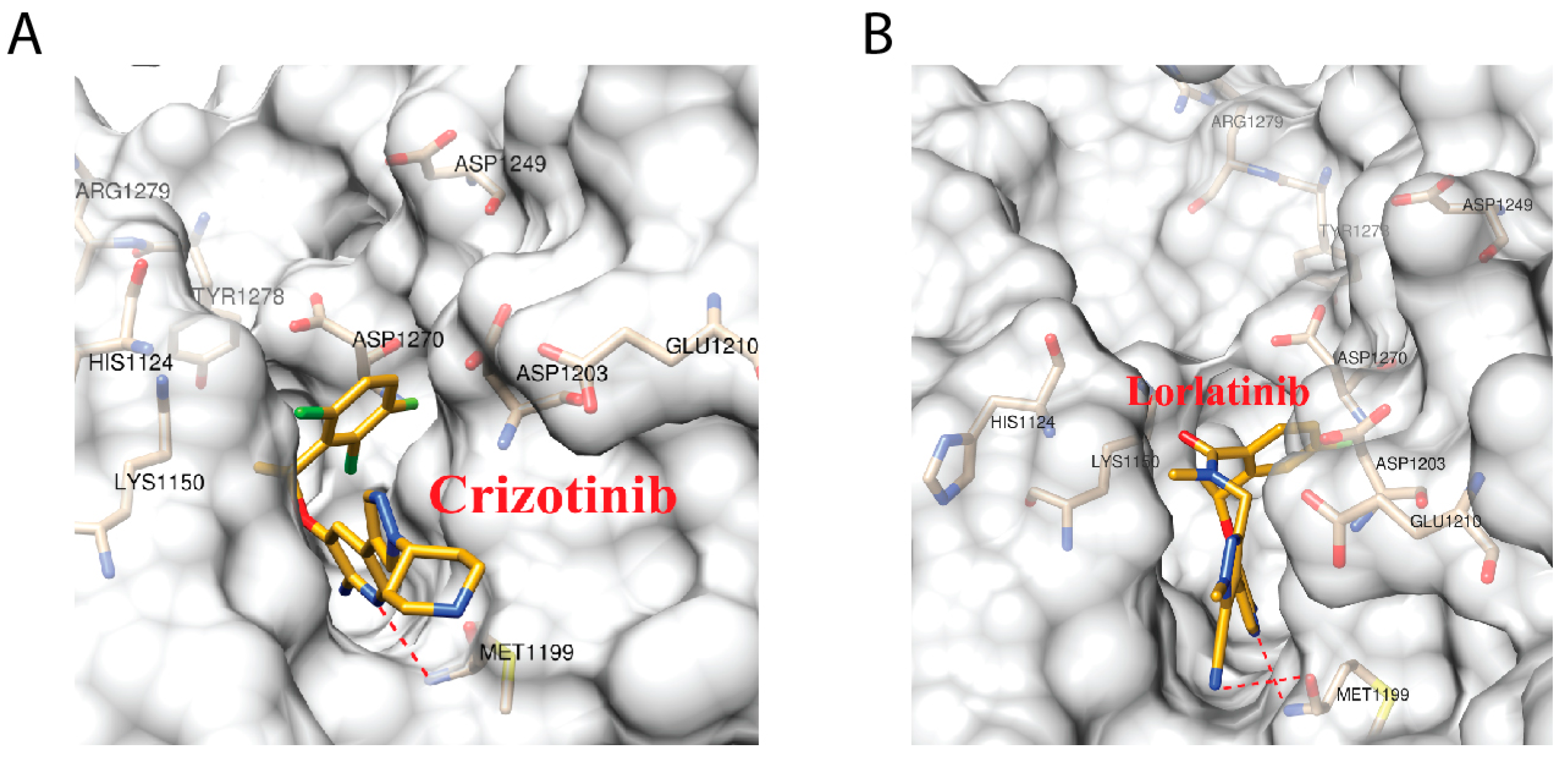
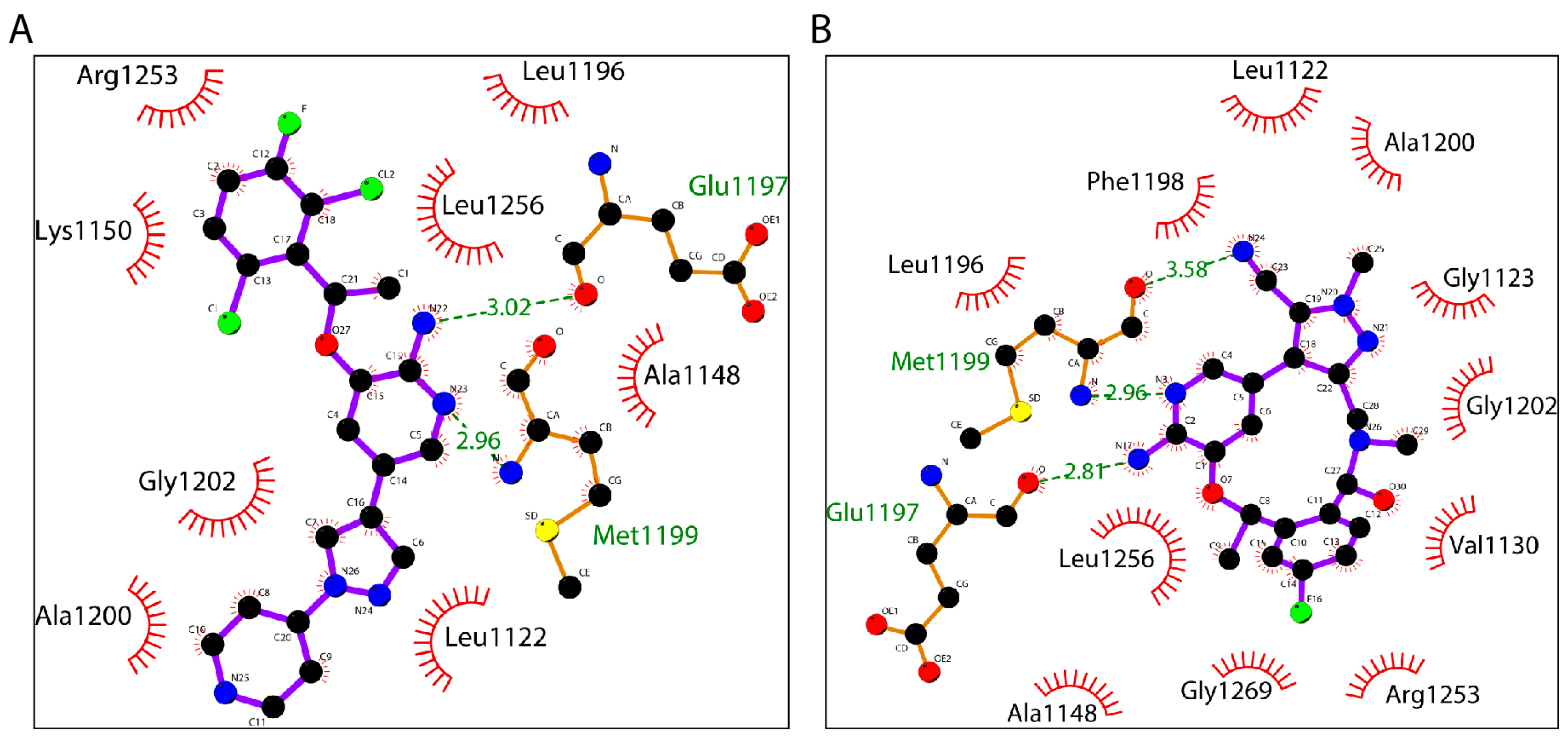
2.5. Crizotinib and Lorlatinib Interacts with ALK in Different Modes
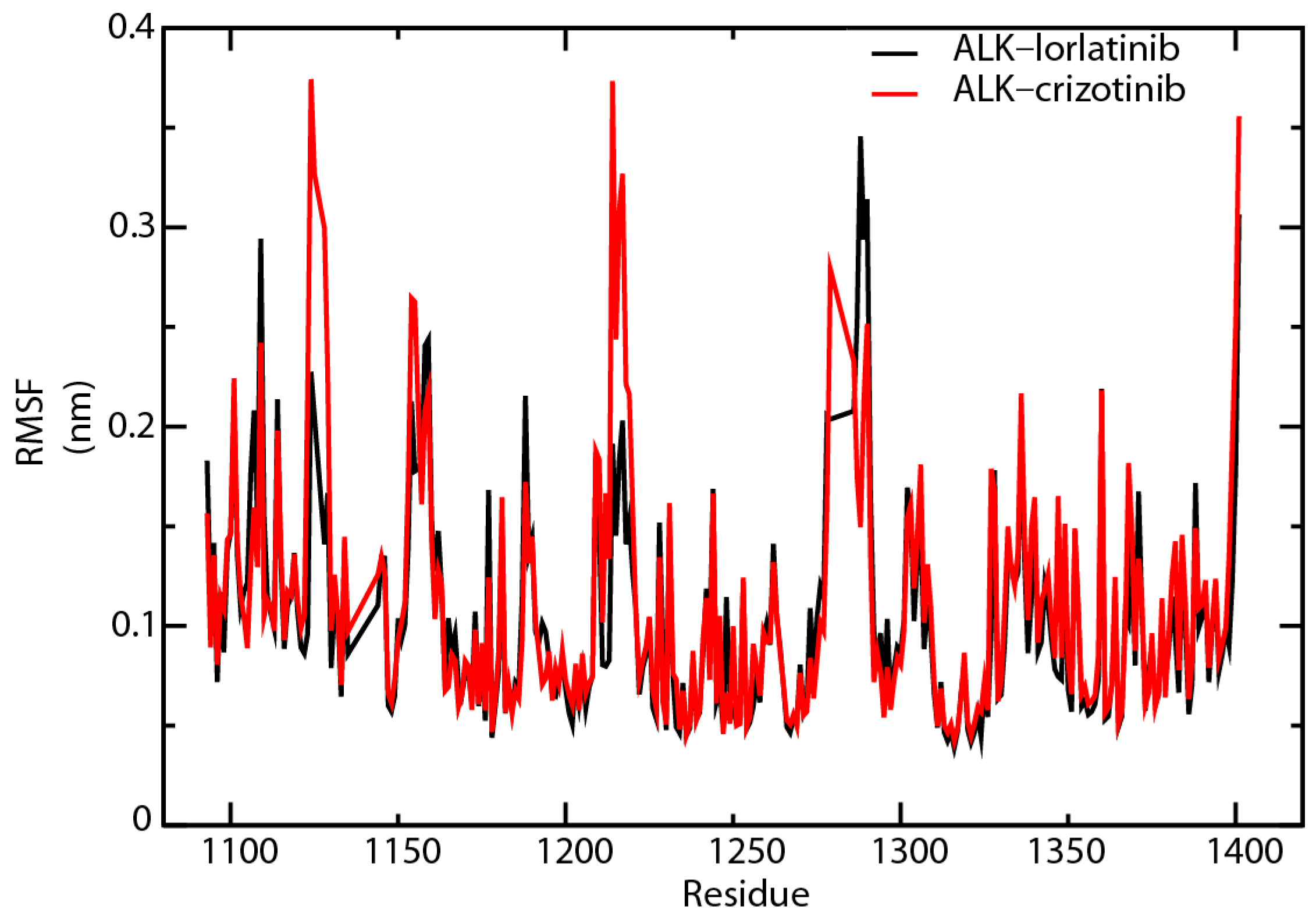
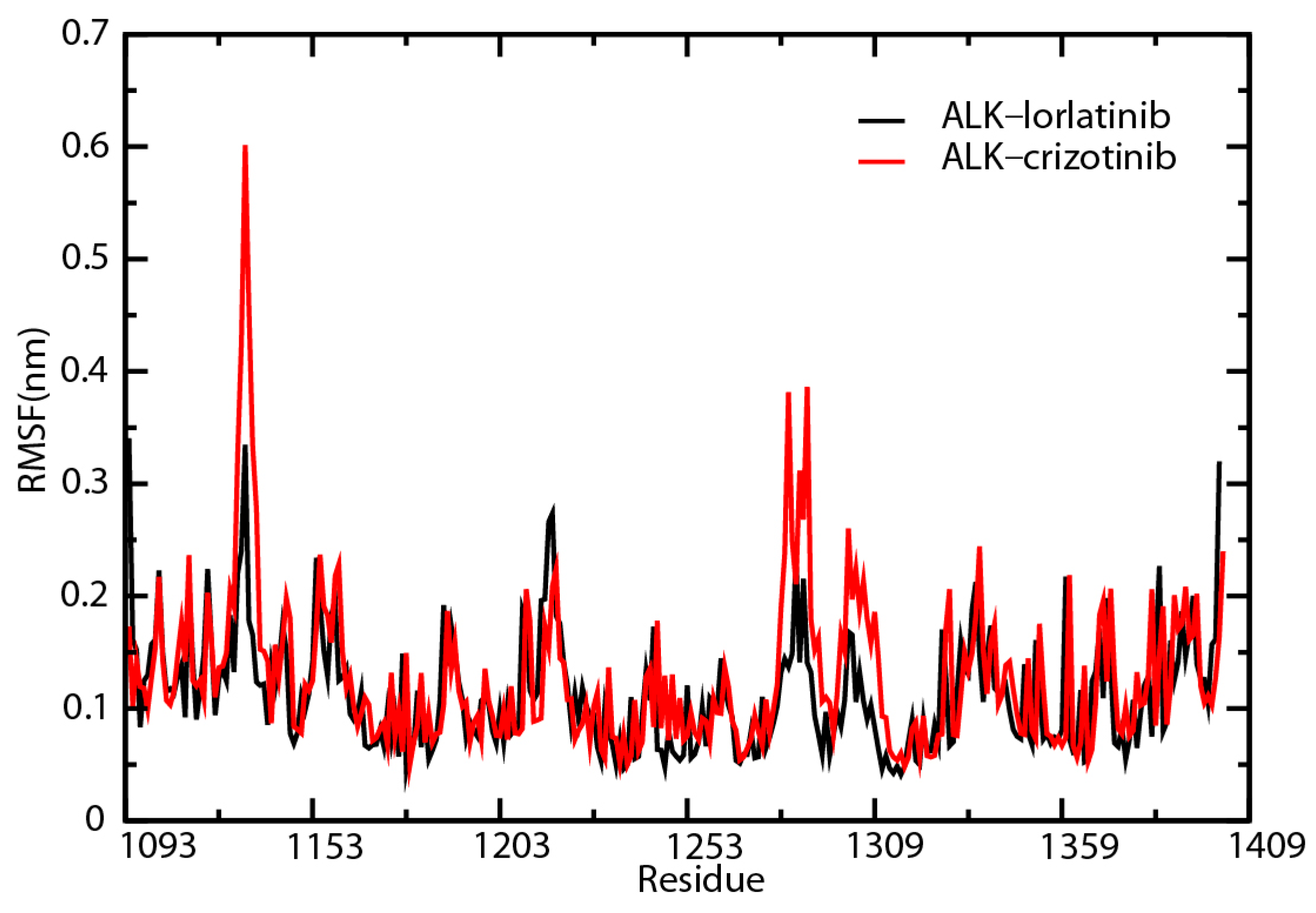
2.6. Root-Mean-Square Fluctuation Analysis of the Key Amino Acid Residues
2.7. L1198F Mutation Affects the ATP Association of ALK
2.8. Secondary Structure Analysis of the ATP and Inhibitor Binding Sites
3. Materials and Method
3.1. Retrieving the Protein Data Bank Files
3.2. Molecular Docking
3.3. Molecular Dynamics Simulation
3.4. Root-Mean-Square Fluctuation and Root-Mean-Square Deviation Analysis
3.5. Binding Energy Calculation and Energy Decomposition
3.6. Calculation of the Electrostatic Energy and Root-Mean-Square Fluctuation Change Trends
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Bray, F.; Ren, J.S.; Masuyer, E.; Ferlay, J. Global estimates of cancer prevalence for 27 sites in the adult population in 2008. Int. J. Cancer 2013, 132, 1133–1145. [Google Scholar] [CrossRef] [PubMed]
- Lemmon, M.A.; Schlessinger, J. Cell signaling by receptor tyrosine kinases. Cell 2010, 141, 1117–1134. [Google Scholar] [CrossRef] [PubMed]
- Katayama, R.; Lovly, C.M.; Shaw, A.T. Therapeutic targeting of anaplastic lymphoma kinase in lung cancer: A paradigm for precision cancer medicine. Clin. Cancer Res. 2015, 21, 2227–2235. [Google Scholar] [CrossRef] [PubMed]
- Vijayvergia, N.; Mehra, R. Clinical challenges in targeting anaplastic lymphoma kinase in advanced non-small cell lung cancer. Cancer Chemother. Pharmacol. 2014, 74, 437–446. [Google Scholar] [CrossRef] [PubMed]
- Jemal, A.; Siegel, R.; Ward, E.; Murray, T.; Xu, J.; Smigal, C.; Thun, M.J. Cancer statistics, 2006. CA Cancer J. Clin. 2006, 56, 106–130. [Google Scholar] [CrossRef] [PubMed]
- Druker, B.J.; Talpaz, M.; Resta, D.J.; Peng, B.; Buchdunger, E.; Ford, J.M.; Lydon, N.B.; Kantarjian, H.; Capdeville, R.; Ohno-Jones, S.; et al. Efficacy and safety of a specific inhibitor of the BCR-ABL tyrosine kinase in chronic myeloid leukemia. N. Engl. J. Med. 2001, 344, 1031–1037. [Google Scholar] [CrossRef] [PubMed]
- Lynch, T.J.; Bell, D.W.; Sordella, R.; Gurubhagavatula, S.; Okimoto, R.A.; Brannigan, B.W.; Harris, P.L.; Haserlat, S.M.; Supko, J.G.; Haluska, F.G.; et al. Activating mutations in the epidermal growth factor receptor underlying responsiveness of non-small-cell lung cancer to gefitinib. N. Engl. J. Med. 2004, 350, 2129–2139. [Google Scholar] [CrossRef] [PubMed]
- Paez, J.G.; Janne, P.A.; Lee, J.C.; Tracy, S.; Greulich, H.; Gabriel, S.; Herman, P.; Kaye, F.J.; Lindeman, N.; Boggon, T.J.; et al. EGFR mutations in lung cancer: Correlation with clinical response to gefitinib therapy. Science 2004, 304, 1497–1500. [Google Scholar] [CrossRef] [PubMed]
- Soda, M.; Choi, Y.L.; Enomoto, M.; Takada, S.; Yamashita, Y.; Ishikawa, S.; Fujiwara, S.; Watanabe, H.; Kurashina, K.; Hatanaka, H.; et al. Identification of the transforming EML4–ALK fusion gene in non-small-cell lung cancer. Nature 2007, 448, 561–566. [Google Scholar] [CrossRef] [PubMed]
- Juchum, M.; Gunther, M.; Laufer, S.A. Fighting cancer drug resistance: Opportunities and challenges for mutation-specific EGFR inhibitors. Drug Resist. Update 2015, 20, 12–28. [Google Scholar] [CrossRef] [PubMed]
- Sullivan, I.; Planchard, D. ALK inhibitors in non-small cell lung cancer: The latest evidence and developments. Ther. Adv. Med. Oncol. 2016, 8, 32–47. [Google Scholar] [CrossRef] [PubMed]
- Cortes, J.E.; Kantarjian, H.; Shah, N.P.; Bixby, D.; Mauro, M.J.; Flinn, I.; O'Hare, T.; Hu, S.; Narasimhan, N.I.; Rivera, V.M.; et al. Ponatinib in refractory Philadelphia chromosome-positive leukemias. N. Engl. J. Med. 2012, 367, 2075–2088. [Google Scholar] [CrossRef] [PubMed]
- Janne, P.A.; Yang, J.C.; Kim, D.W.; Planchard, D.; Ohe, Y.; Ramalingam, S.S.; Ahn, M.J.; Kim, S.W.; Su, W.C.; Horn, L.; et al. AZD9291 in EGFR inhibitor-resistant non-small-cell lung cancer. N. Engl. J. Med. 2015, 372, 1689–1699. [Google Scholar] [CrossRef] [PubMed]
- Sequist, L.V.; Soria, J.C.; Goldman, J.W.; Wakelee, H.A.; Gadgeel, S.M.; Varga, A.; Papadimitrakopoulou, V.; Solomon, B.J.; Oxnard, G.R.; Dziadziuszko, R.; et al. Rociletinib in EGFR-mutated non-small-cell lung cancer. N. Engl. J. Med. 2015, 372, 1700–1709. [Google Scholar] [CrossRef] [PubMed]
- Shaw, A.T.; Kim, D.W.; Mehra, R.; Tan, D.S.; Felip, E.; Chow, L.Q.; Camidge, D.R.; Vansteenkiste, J.; Sharma, S.; De Pas, T.; et al. Ceritinib in ALK-rearranged non-small-cell lung cancer. N. Engl. J. Med. 2014, 370, 1189–1197. [Google Scholar] [CrossRef] [PubMed]
- Gadgeel, S.M.; Gandhi, L.; Riely, G.J.; Chiappori, A.A.; West, H.L.; Azada, M.C.; Morcos, P.N.; Lee, R.M.; Garcia, L.; Yu, L.; et al. Safety and activity of alectinib against systemic disease and brain metastases in patients with crizotinib-resistant ALK-rearranged non-small-cell lung cancer (AF-002JG): Results from the dose-finding portion of a phase 1/2 study. Lancet Oncol. 2014, 15, 1119–1128. [Google Scholar] [CrossRef]
- Gettinger, S.N.; Bazhenova, L.A.; Langer, C.J.; Salgia, R.; Gold, K.A.; Rosell, R.; Shaw, A.T.; Weiss, G.J.; Tugnait, M.; Narasimhan, N.I.; et al. Activity and safety of brigatinib in ALK-rearranged non-small-cell lung cancer and other malignancies: A single-arm, open-label, phase 1/2 trial. Lancet Oncol. 2016, 17, 1683–1696. [Google Scholar] [CrossRef]
- Lindahl, E. Molecular dynamics simulations. Methods Mol. Biol. 2015, 1215, 3–26. [Google Scholar] [PubMed]
- Adcock, S.A.; McCammon, J.A. Molecular dynamics: Survey of methods for simulating the activity of proteins. Chem. Rev. 2006, 106, 1589–1615. [Google Scholar] [CrossRef] [PubMed]
- Sneha, P.; Doss, C.G. Molecular Dynamics: New Frontier in Personalized Medicine. Adv. Protein Chem. Struct. Biol. 2016, 102, 181–224. [Google Scholar] [PubMed]
- Zhong, H.; Huang, W.; He, G.; Peng, C.; Wu, F.; Ouyang, L. Molecular dynamics simulation of tryptophan hydroxylase-1: Binding modes and free energy analysis to phenylalanine derivative inhibitors. Int. J. Mol. Sci. 2013, 14, 9947–9962. [Google Scholar] [CrossRef] [PubMed]
- Shaw, A.T.; Friboulet, L.; Leshchiner, I.; Gainor, J.F.; Bergqvist, S.; Brooun, A.; Burke, B.J.; Deng, Y.L.; Liu, W.; Dardaei, L.; et al. Resensitization to Crizotinib by the Lorlatinib ALK Resistance Mutation L1198F. N. Engl. J. Med. 2016, 374, 54–61. [Google Scholar] [CrossRef] [PubMed]
- Bossi, R.T.; Saccardo, M.B.; Ardini, E.; Menichincheri, M.; Rusconi, L.; Magnaghi, P.; Orsini, P.; Avanzi, N.; Borgia, A.L.; Nesi, M.; et al. Crystal structures of anaplastic lymphoma kinase in complex with ATP competitive inhibitors. Biochemistry 2010, 49, 6813–6825. [Google Scholar] [CrossRef] [PubMed]
- Heuckmann, J.M.; Holzel, M.; Sos, M.L.; Heynck, S.; Balke-Want, H.; Koker, M.; Peifer, M.; Weiss, J.; Lovly, C.M.; Grutter, C.; et al. ALK mutations conferring differential resistance to structurally diverse ALK inhibitors. Clin. Cancer Res. 2011, 17, 7394–7401. [Google Scholar] [CrossRef] [PubMed]
- Tartari, C.J.; Gunby, R.H.; Coluccia, A.M.; Sottocornola, R.; Cimbro, B.; Scapozza, L.; Donella-Deana, A.; Pinna, L.A.; Gambacorti-Passerini, C. Characterization of some molecular mechanisms governing autoactivation of the catalytic domain of the anaplastic lymphoma kinase. J. Biol. Chem. 2008, 283, 3743–3750. [Google Scholar] [CrossRef] [PubMed]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef] [PubMed]
- Janoueix-Lerosey, I.; Lequin, D.; Brugieres, L.; Ribeiro, A.; de Pontual, L.; Combaret, V.; Raynal, V.; Puisieux, A.; Schleiermacher, G.; Pierron, G.; et al. Somatic and germline activating mutations of the ALK kinase receptor in neuroblastoma. Nature 2008, 455, 967–970. [Google Scholar] [CrossRef] [PubMed]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [PubMed]
- Pronk, S.; Pall, S.; Schulz, R.; Larsson, P.; Bjelkmar, P.; Apostolov, R.; Shirts, M.R.; Smith, J.C.; Kasson, P.M.; van der Spoel, D.; et al. GROMACS 4.5: A high-throughput and highly parallel open source molecular simulation toolkit. Bioinformatics 2013, 29, 845–854. [Google Scholar] [CrossRef] [PubMed]
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.D.; Impey, R.W.; Klein, M.L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983, 79, 926–935. [Google Scholar] [CrossRef]
- Wang, J.; Wolf, R.M.; Caldwell, J.W.; Kollman, P.A.; Case, D.A. Development and testing of a general amber force field. J. Comput. Chem. 2004, 25, 1157–1174. [Google Scholar] [CrossRef] [PubMed]
- Darden, T.; York, D.; Pedersen, L. Particle mesh Ewald: An Nlog(N) method for Ewald sums in large systems. J. Chem. Phys. 1993, 98, 10089–10092. [Google Scholar] [CrossRef]
- Hess, B. P-LINCS: A Parallel Linear Constraint Solver for Molecular Simulation. J. Chem. Theory Comput. 2008, 4, 116–122. [Google Scholar] [CrossRef] [PubMed]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Li, J.; Zhou, N.; Cai, P.; Bao, J. In Silico Screening Identifies a Novel Potential PARP1 Inhibitor Targeting Synthetic Lethality in Cancer Treatment. Int. J. Mol. Sci. 2016, 17, 258. [Google Scholar] [CrossRef] [PubMed]
- Kumari, R.; Kumar, R.; Lynn, A. g_mmpbsa—A GROMACS Tool for High-Throughput MM-PBSA Calculations. J. Chem. Inf. Model. 2014, 54, 1951–1962. [Google Scholar] [CrossRef] [PubMed]
- Homeyer, N.; Gohlke, H. Free Energy Calculations by the Molecular Mechanics Poisson-Boltzmann Surface Area Method. Mol. Inf. 2014, 31, 114–122. [Google Scholar] [CrossRef] [PubMed]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Distance to the Inhibitor (nm) | ||||||||
|---|---|---|---|---|---|---|---|---|
| ALK–Crizotinib | ALK–Lorlatinib | |||||||
| 0 ns | 5–10 ns | 20–25 ns | 25–30 ns | 0 ns | 5–10 ns | 20–25 ns | 25–30 ns | |
| Glu1197 | 3.02 | 2.53 | 2.68 | 2.66 | 2.81 | 2.94 | 3.02 | 3.02 |
| Met1199 | 2.96 | 2.81 | 2.84 | 2.74 | 2.96 | 2.60 | 2.70 | 2.84 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, J.; Sun, R.; Wu, Y.; Song, M.; Li, J.; Yang, Q.; Chen, X.; Bao, J.; Zhao, Q. L1198F Mutation Resensitizes Crizotinib to ALK by Altering the Conformation of Inhibitor and ATP Binding Sites. Int. J. Mol. Sci. 2017, 18, 482. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms18030482
Li J, Sun R, Wu Y, Song M, Li J, Yang Q, Chen X, Bao J, Zhao Q. L1198F Mutation Resensitizes Crizotinib to ALK by Altering the Conformation of Inhibitor and ATP Binding Sites. International Journal of Molecular Sciences. 2017; 18(3):482. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms18030482
Chicago/Turabian StyleLi, Jian, Rong Sun, Yuehong Wu, Mingzhu Song, Jia Li, Qianye Yang, Xiaoyi Chen, Jinku Bao, and Qi Zhao. 2017. "L1198F Mutation Resensitizes Crizotinib to ALK by Altering the Conformation of Inhibitor and ATP Binding Sites" International Journal of Molecular Sciences 18, no. 3: 482. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms18030482