PGK1 Drives Hepatocellular Carcinoma Metastasis by Enhancing Metabolic Process

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
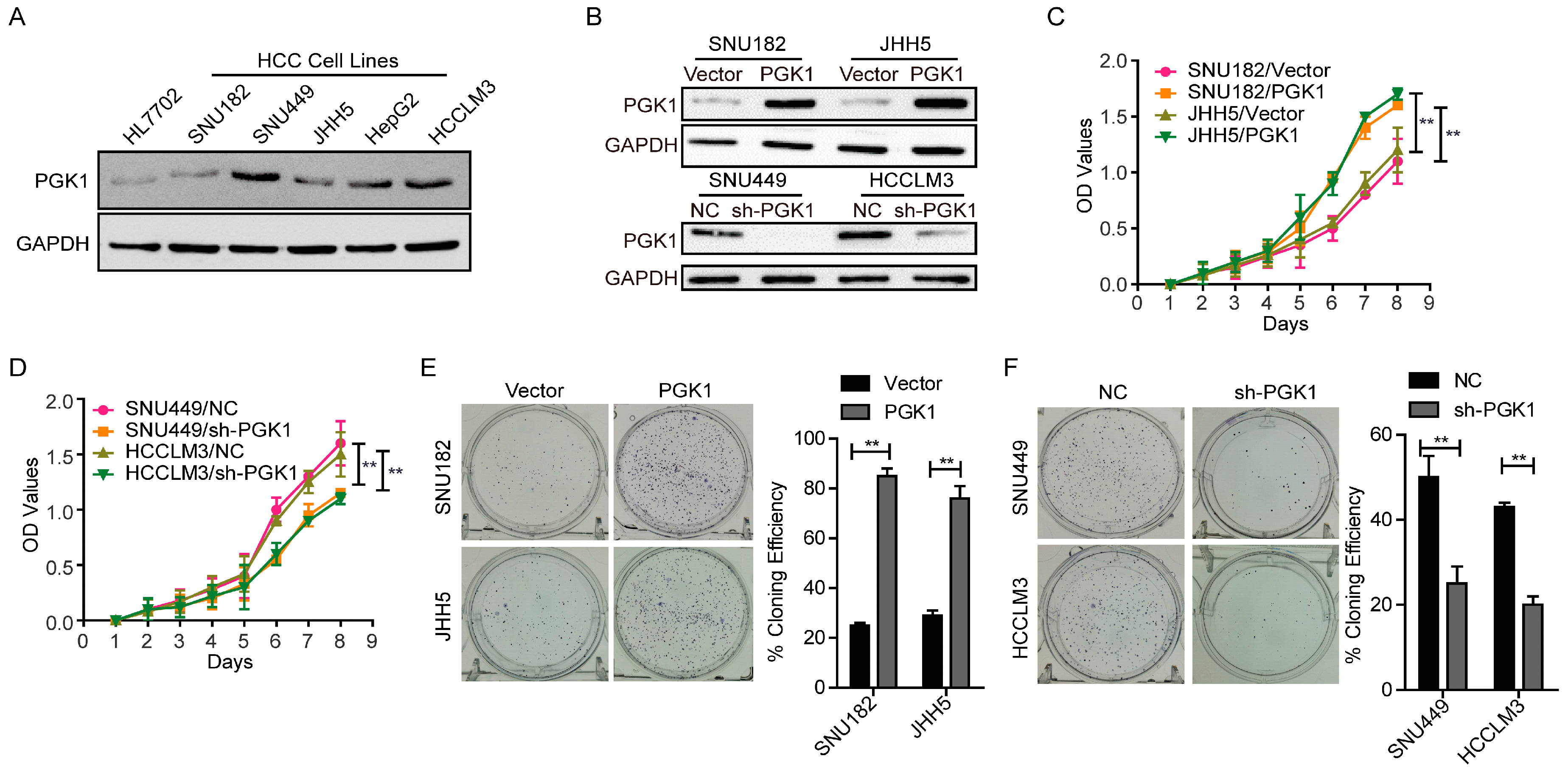
2.1. Phosphoglycerate Kinase 1 (PGK1) Promotes Proliferation in Hepatocellular Carcinoma (HCC) In Vitro
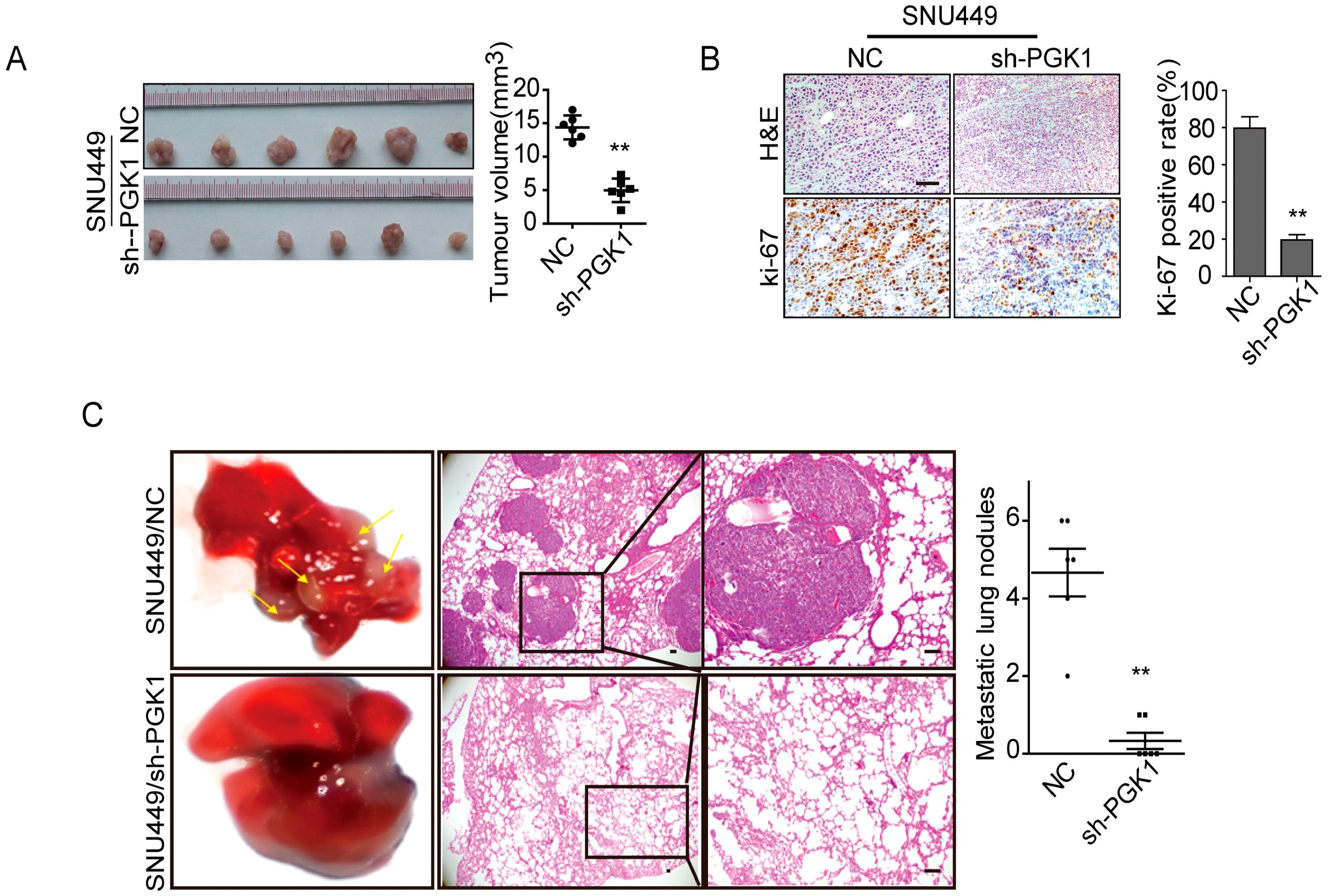
2.2. PGK1 Is Effective in Promoting Tumor Metastasis In Vivo
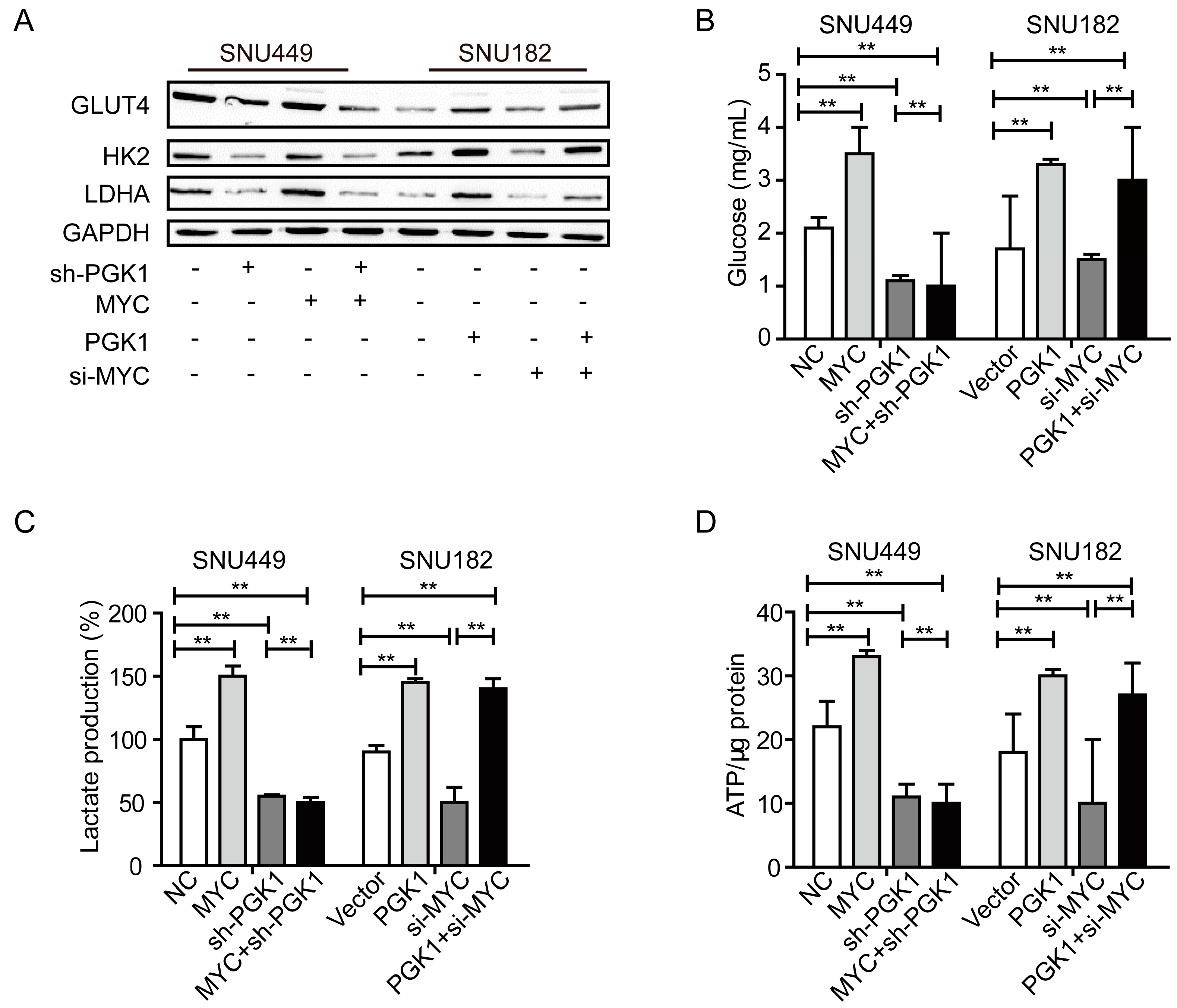
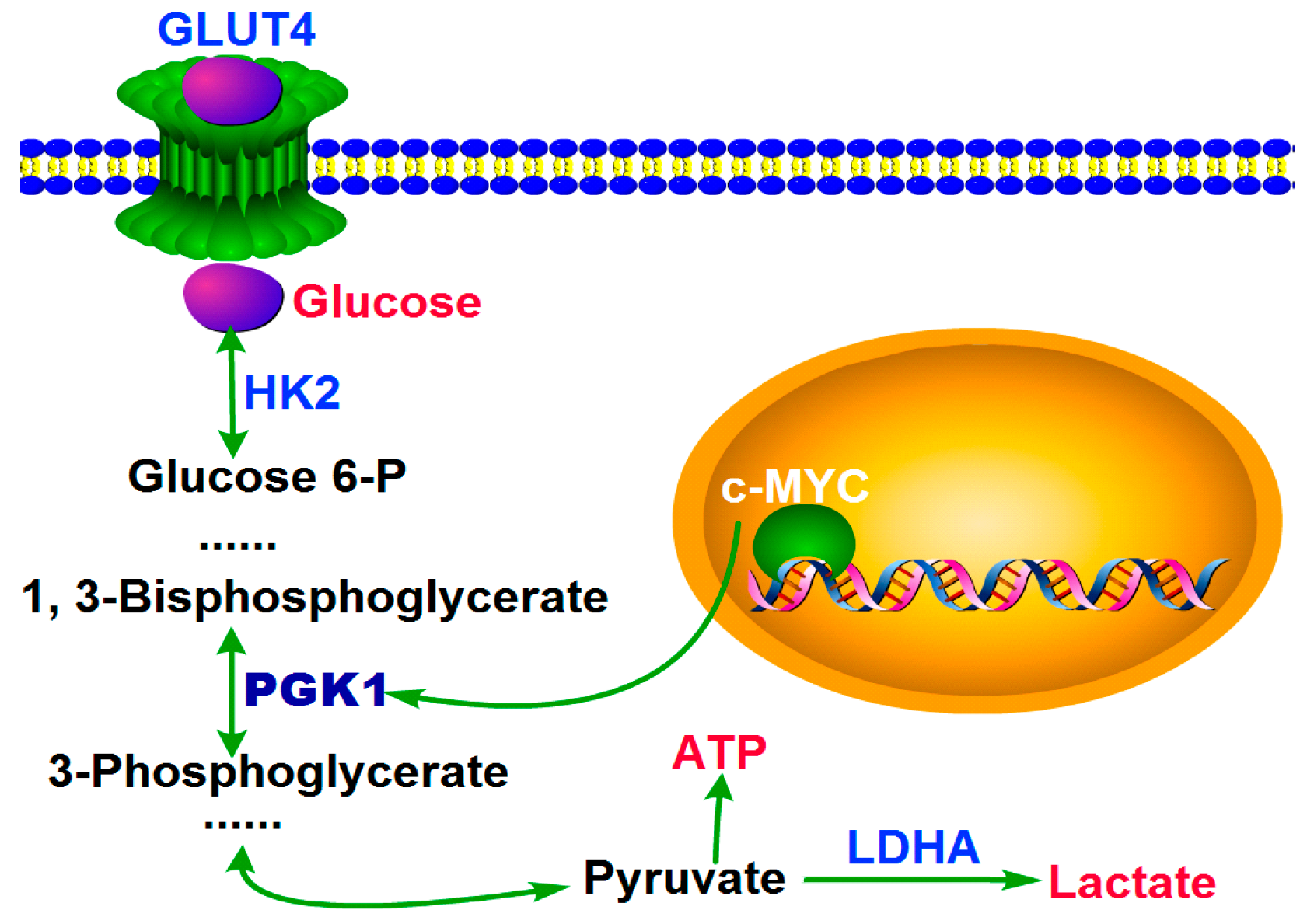
2.3. MYC-Dependent PGK1 Modulates Metabolic Reprogramming of HCC Cells
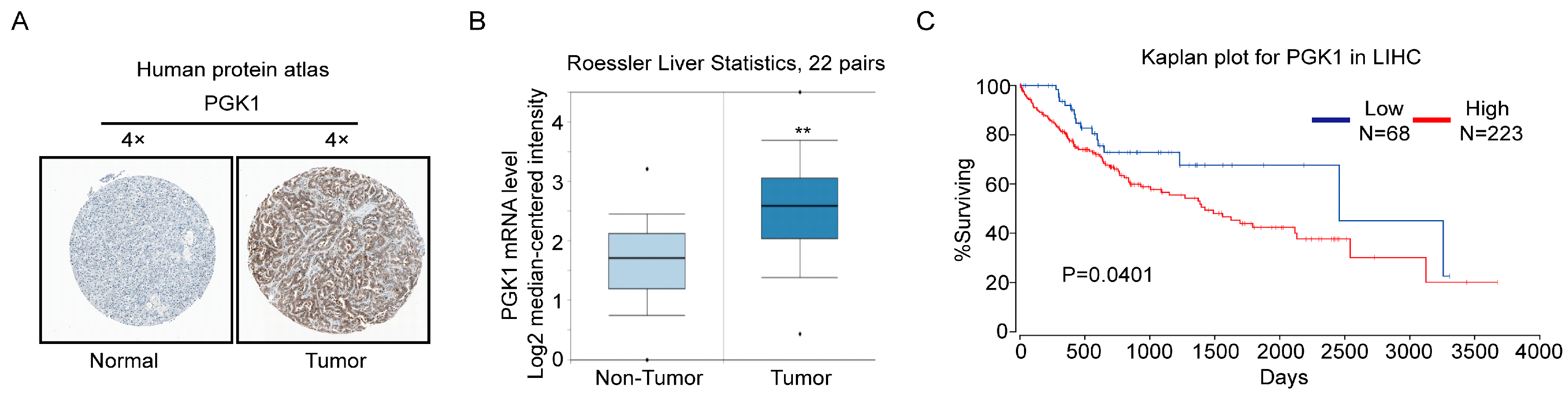
2.4. PGK1 Is Overexpressed in Human Hepatocellular Carcinoma Specimens
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. Cell Transfection with siRNAs and Plasmids
4.3. Western Blotting
4.4. Cell Proliferation Assay
4.5. Plate Colony Formation Assay
4.6. Proliferation and Metastasis in Mouse Model
4.7. Glucose Uptake, Lactate Production, ATP Level Detection
4.8. Analysis of PGK1 Expression in Human HCC
4.9. Statistical Analysis
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Ryerson, A.B.; Eheman, C.R.; Altekruse, S.F.; Ward, J.W.; Jemal, A.; Sherman, R.L.; Henley, S.J.; Holtzman, D.; Lake, A.; Noone, A.M.; et al. Annual report to the Nation on the Status of Cancer, 1975–2012, featuring the increasing incidence of liver cancer. Cancer 2016, 122, 1312–1337. [Google Scholar] [CrossRef] [PubMed]
- Han, T.; Kang, D.; Ji, D.; Wang, X.; Zhan, W.; Fu, M.; Xin, H.B.; Wang, J.B. How does cancer cell metabolism affect tumor migration and invasion? Cell Adhes. Migr. 2013, 7, 395–403. [Google Scholar] [CrossRef] [PubMed]
- Boroughs, L.K.; DeBerardinis, R.J. Metabolic pathways promoting cancer cell survival and growth. Nat. Cell Biol. 2015, 17, 351–359. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Zheng, Y.; Lu, Z. PGK1 is a new member of the protein kinome. Cell Cycle 2016, 15, 1803–1804. [Google Scholar] [CrossRef] [PubMed]
- Qian, X.; Li, X.; Lu, Z. Protein kinase activity of the glycolytic enzyme PGK1 regulates autophagy to promote tumorigenesis. Autophagy 2017, 1–2. [Google Scholar] [CrossRef] [PubMed]
- Sun, S.; Liang, X.; Zhang, X.; Liu, T.; Shi, Q.; Song, Y.; Jiang, Y.; Wu, H.; Jiang, Y.; Lu, X.; et al. Phosphoglycerate kinase-1 is a predictor of poor survival and a novel prognostic biomarker of chemoresistance to paclitaxel treatment in breast cancer. Br. J. Cancer 2015, 112, 1332–1339. [Google Scholar] [CrossRef] [PubMed]
- Ameis, H.M.; Drenckhan, A.; von Loga, K.; Escherich, G.; Wenke, K.; Izbicki, J.R.; Reinshagen, K.; Gros, S.J. PGK1 as predictor of CXCR4 expression, bone marrow metastases and survival in neuroblastoma. PLoS ONE 2013, 8, e83701. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, S.S.; Glatzle, J.; Bajaeifer, K.; Bühler, S.; Lehmann, T.; Königsrainer, I.; Vollmer, J.P.; Sipos, B.; Ahmad, S.S.; Northoff, H.; et al. Phosphoglycerate kinase 1 as a promoter of metastasis in colon cancer. Int. J. Oncol. 2013, 43, 586–590. [Google Scholar] [CrossRef] [PubMed]
- Jang, C.H.; Lee, I.A.; Ha, Y.R.; Lim, J.; Sung, M.K.; Lee, S.J.; Kim, J.S. PGK1 induction by a hydrogen peroxide treatment is suppressed by antioxidants in human colon carcinoma cells. Biosci. Biotechnol. Biochem. 2008, 72, 1799–1808. [Google Scholar] [CrossRef] [PubMed]
- Younghun, J.; Yusuke, S.; Jianhua, W.; Jingcheng, W.; Zhuo, W.; Pedersen, E.A.; Lee, C.H.; Hall, C.L.; Hogg, P.J.; Krebsbach, P.H.; et al. Taichman expression of PGK1 by prostate cancer cells induces bone formation. Mol. Cancer Res. 2009, 7, 1595–1604. [Google Scholar] [CrossRef]
- Zieker, D.; Königsrainer, I.; Traub, F.; Nieselt, K.; Knapp, B.; Schillinger, C.; Stirnkorb, C.; Fend, F.; Northoff, H.; Kupka, S.; et al. PGK1 a potential marker for peritoneal dissemination in gastric cancer. Cell. Physiol. Biochem. 2008, 21, 429–436. [Google Scholar] [CrossRef] [PubMed]
- Zieker, D.; Königsrainer, I.; Tritschler, I.; Löffler, M.; Beckert, S.; Traub, F.; Nieselt, K.; Bühler, S.; Weller, M.; Gaedcke, J.; et al. Phosphoglycerate kinase 1 a promoting enzyme for peritoneal dissemination in gastric cancer. Int. J. Cancer 2010, 126, 1513–1520. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ai, J.; Huang, H.; Lv, X.; Tang, Z.; Chen, M.; Chen, T.; Duan, W.; Sun, H.; Li, Q.; Tan, R.; et al. FLNA and PGK1 are two potential markers for progression in hepatocellular carcinoma. Cell. Physiol. Biochem. 2011, 27, 207–216. [Google Scholar] [CrossRef] [PubMed]
- Hundhausen, C.; Boesch-Saadatmandi, C.; Matzner, N.; Lang, F.; Blank, R.; Wolffram, S.; Blaschek, W.; Rimbach, G. Ochratoxin a lowers mRNA levels of genes encoding for key proteins of liver cell metabolism. Cancer Genom. Proteom. 2008, 5, 319–332. [Google Scholar]
- Okino, S.T.; Chichester, C.H.; Whitlock, J.P., Jr. Hypoxia-inducible mammalian gene expression analyzed in vivo at a TATA-driven promoter and at an initiator-driven promoter. J. Biol. Chem. 1998, 273, 23837–23843. [Google Scholar] [CrossRef] [PubMed]
- Hu, H.; Zhu, W.; Qin, J.; Chen, M.; Gong, L.; Li, L.; Liu, X.; Tao, Y.; Yin, H.; Zhou, H.; et al. Acetylation of PGK1 promotes liver cancer cell proliferation and tumorigenesis. Hepatology 2017, 65, 515–528. [Google Scholar] [CrossRef] [PubMed]
- Valentini, G.; Maggi, M.; Pey, A.L. Protein stability, folding and misfolding in human PGK1 deficiency. Biomolecules 2013, 3, 1030–1052. [Google Scholar] [CrossRef] [PubMed]
- Schneider, C.C.; Archid, R.; Fischer, N.; Bühler, S.; Venturelli, S.; Berger, A.; Burkard, M.; Kirschniak, A.; Bachmann, R.; Königsrainer, A.; et al. Metabolic alteration—Overcoming therapy resistance in gastric cancer via PGK-1 inhibition in a combined therapy with standard chemotherapeutics. Int. J. Surg. 2015, 22, 92–98. [Google Scholar] [CrossRef] [PubMed]
- Shichijo, S.; Azuma, K.; Komatsu, N.; Ito, M.; Maeda, Y.; Ishihara, Y.; Itoh, K. Two proliferation-related proteins, TYMS and PGK1, could be new cytotoxic T lymphocyte-directed tumor-associated antigens of HLA-A2+ colon cancer. Clin. Cancer Res. 2004, 10, 5828–5836. [Google Scholar] [CrossRef] [PubMed]
- Wahlstrom, T.; Henriksson, M.A. Impact of MYC in regulation of tumor cell metabolism. Biochim. Biophys. Acta 2015, 1849, 563–569. [Google Scholar] [CrossRef] [PubMed]
- Tang, S.W.; Chang, W.H.; Su, Y.C.; Chen, Y.C.; Lai, Y.H.; Wu, P.T.; Hsu, C.I.; Lin, W.C.; Lai, M.K.; Lin, J.Y. MYC pathway is activated in clear cell renal cell carcinoma and essential for proliferation of clear cell renal cell carcinoma cells. Cancer Lett. 2009, 273, 35–43. [Google Scholar] [CrossRef] [PubMed]
- Xu, D.; Aka, J.A.; Wang, R.; Lin, S.X. 17beta-hydroxysteroid dehydrogenase type 5 is negatively correlated to apoptosis inhibitor GRP78 and tumor-secreted protein PGK1, and modulates breast cancer cell viability and proliferation. J. Steroid Biochem. Mol. Biol. 2017, 171, 270–280. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Shim, H.; Dolde, C.; Lewis, B.C.; Wu, C.S.; Dang, G.; Jungmann, R.A.; Dalla-Favera, R.; Dang, C.V. c-MYC transactivation of LDH-A: Implications for tumor metabolism and growth. Proc. Natl. Acad. Sci. USA 1997, 94, 6658–6663. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Wang, Y.; Zeller, K.I.; Potter, J.J.; Wonsey, D.R.; O’Donnell, K.A.; Kim, J.W.; Yustein, J.T.; Lee, L.A.; Dang, C.V. MYC stimulates nuclearly encoded mitochondrial genes and mitochondrial biogenesis. Mol. Cell. Biol. 2005, 25, 6225–6234. [Google Scholar] [CrossRef] [PubMed]
- Wise, D.R.; DeBerardinis, R.J.; Mancuso, A.; Sayed, N.; Zhang, X.Y.; Pfeiffer, H.K.; Nissim, I.; Daikhin, E.; Yudkoff, M.; McMahon, S.B.; et al. MYC regulates a transcriptional program that stimulates mitochondrial glutaminolysis and leads to glutamine addiction. Proc. Natl. Acad. Sci. USA 2008, 105, 18782–18787. [Google Scholar] [CrossRef] [PubMed]
- Gao, P.; Tchernyshyov, I.; Chang, T.C.; Lee, Y.S.; Kita, K.; Ochi, T.; Zeller, K.I.; de Marzo, A.M.; van Eyk, J.E.; Mendell, J.T.; et al. c-MYC suppression of miR-23a/b enhances mitochondrial glutaminase expression and glutamine metabolism. Nature 2009, 458, 762–765. [Google Scholar] [CrossRef] [PubMed]
- DeBerardinis, R.J.; Cheng, T. Q’s next: The diverse functions of glutamine in metabolism, cell biology and cancer. Oncogene 2010, 29, 313–324. [Google Scholar] [CrossRef] [PubMed]
- Yun, J.; Rago, C.; Cheong, I.; Pagliarini, R.; Angenendt, P.; Rajagopalan, H.; Schmidt, K.; Willson, J.K.; Markowitz, S.; Zhou, S.; et al. Glucose deprivation contributes to the development of KRAS pathway mutations in tumor cells. Science 2009, 325, 1555–1559. [Google Scholar] [CrossRef] [PubMed]
- Le, A.; Lane, A.N.; Hamaker, M.; Bose, S.; Gouw, A.; Barbi, J.; Tsukamoto, T.; Rojas, C.J.; Slusher, B.S.; Zhang, H.; et al. Glucose-independent glutamine metabolism via TCA cycling for proliferation and survival in B cells. Cell Metab. 2012, 15, 110–121. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.; Sudderth, J.; Dang, T.; Bachoo, R.M.; McDonald, J.G.; DeBerardinis, R.J. Glioblastoma cells require glutamate dehydrogenase to survive impairments of glucose metabolism or Akt signaling. Cancer Res. 2009, 69, 7986–7993. [Google Scholar] [CrossRef] [PubMed]
- Cheng, T.; Sudderth, J.; Yang, C.; Mullen, A.R.; Jin, E.S.; Matés, J.M.; DeBerardinis, R.J. Pyruvate carboxylase is required for glutamine-independent growth of tumor cells. Proc. Natl. Acad. Sci. USA 2011, 108, 8674–8679. [Google Scholar] [CrossRef] [PubMed]
- Birsoy, K.; Possemato, R.; Lorbeer, F.K.; Bayraktar, E.C.; Thiru, P.; Yucel, B.; Wang, T.; Chen, W.W.; Clish, C.B.; Sabatini, D.M. Metabolic determinants of cancer cell sensitivity to glucose limitation and biguanides. Nature 2014, 508, 108–112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caro, P.; Kishan, A.U.; Norberg, E.; Stanley, I.A.; Chapuy, B.; Ficarro, S.B.; Polak, K.; Tondera, D.; Gounarides, J.; Yin, H.; et al. Metabolic signatures uncover distinct targets in molecular subsets of diffuse large B cell lymphoma. Cancer Cell 2012, 22, 547–560. [Google Scholar] [CrossRef] [PubMed]
- Gatenby, R.A.; Gawlinski, E.T.; Gmitro, A.F.; Kaylor, B.; Gillies, R.J. Acid-mediated tumor invasion: A multidisciplinary study. Cancer Res. 2006, 66, 5216–5223. [Google Scholar] [CrossRef] [PubMed]
- Anaya, J. OncoLnc: Linking TCGA survival data to mRNAs, miRNAs, and lncRNAs. PeerJ 2016, 32, 1–8. [Google Scholar] [CrossRef]





© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xie, H.; Tong, G.; Zhang, Y.; Liang, S.; Tang, K.; Yang, Q. PGK1 Drives Hepatocellular Carcinoma Metastasis by Enhancing Metabolic Process. Int. J. Mol. Sci. 2017, 18, 1630. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms18081630
Xie H, Tong G, Zhang Y, Liang S, Tang K, Yang Q. PGK1 Drives Hepatocellular Carcinoma Metastasis by Enhancing Metabolic Process. International Journal of Molecular Sciences. 2017; 18(8):1630. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms18081630
Chicago/Turabian StyleXie, Huijun, Guihui Tong, Yupei Zhang, Shu Liang, Kairui Tang, and Qinhe Yang. 2017. "PGK1 Drives Hepatocellular Carcinoma Metastasis by Enhancing Metabolic Process" International Journal of Molecular Sciences 18, no. 8: 1630. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms18081630