Molecular Consortia—Various Structural and Synthetic Concepts for More Effective Therapeutics Synthesis


Abstract
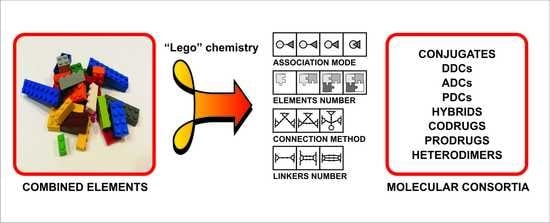
:
1. Introduction
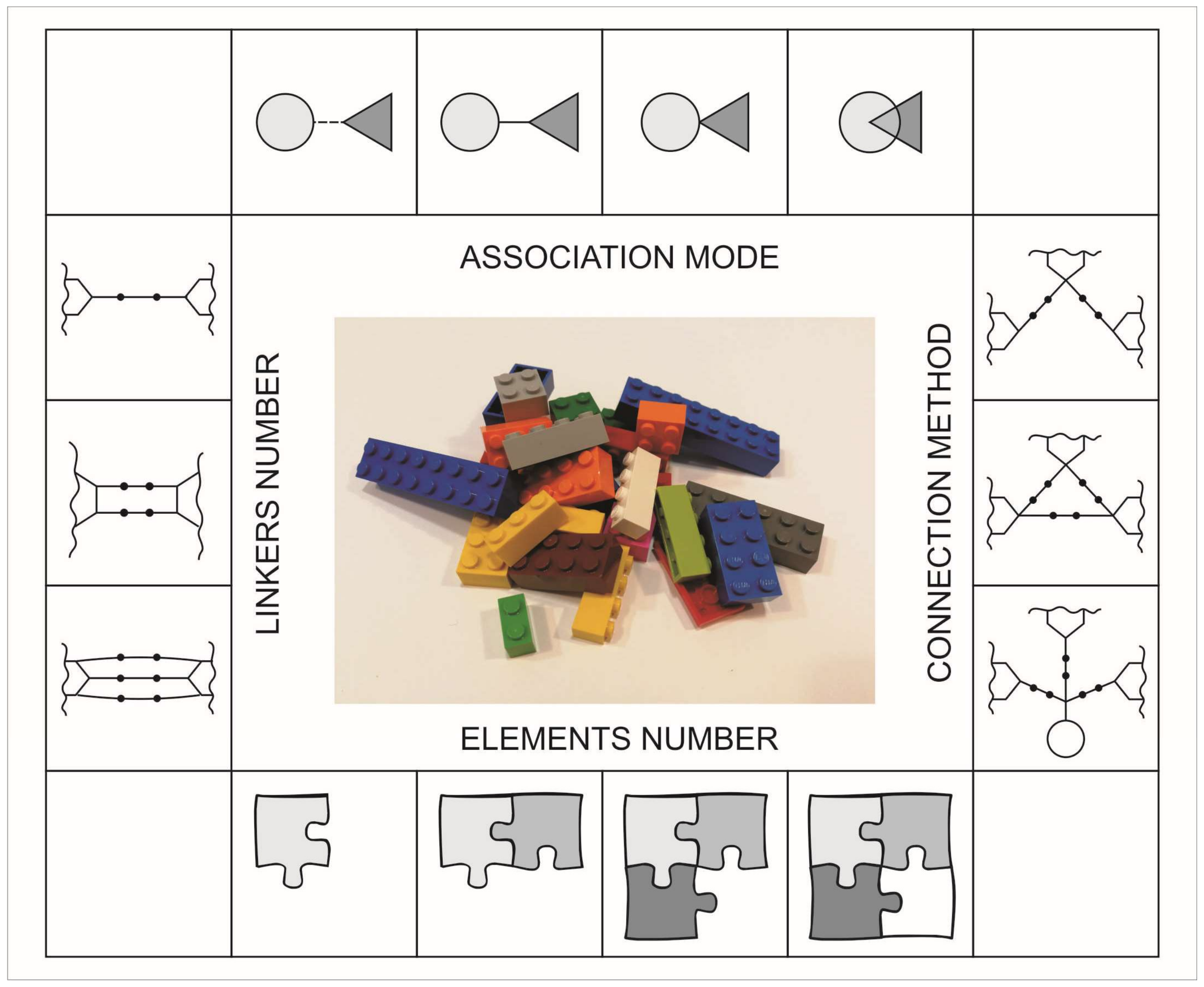
2. Structural Concepts
- Association mode—describes a type of structural connection, interrelation between different selected elements, kind of attachment (direct or indirect) or structural cross-relationship (linked, fused, merged)
- Connection method—selected chemical and pharmaceutical units, depending on the functionality and reactivity, can form linear, pseudolinear or branched, smaller or larger, new structures
- Elements number—the most popular are structures built from two elements (units, puzzles, segments) but there can be many more of them
- Linkers number—the number of intermediaries between selected elements in the structural population.
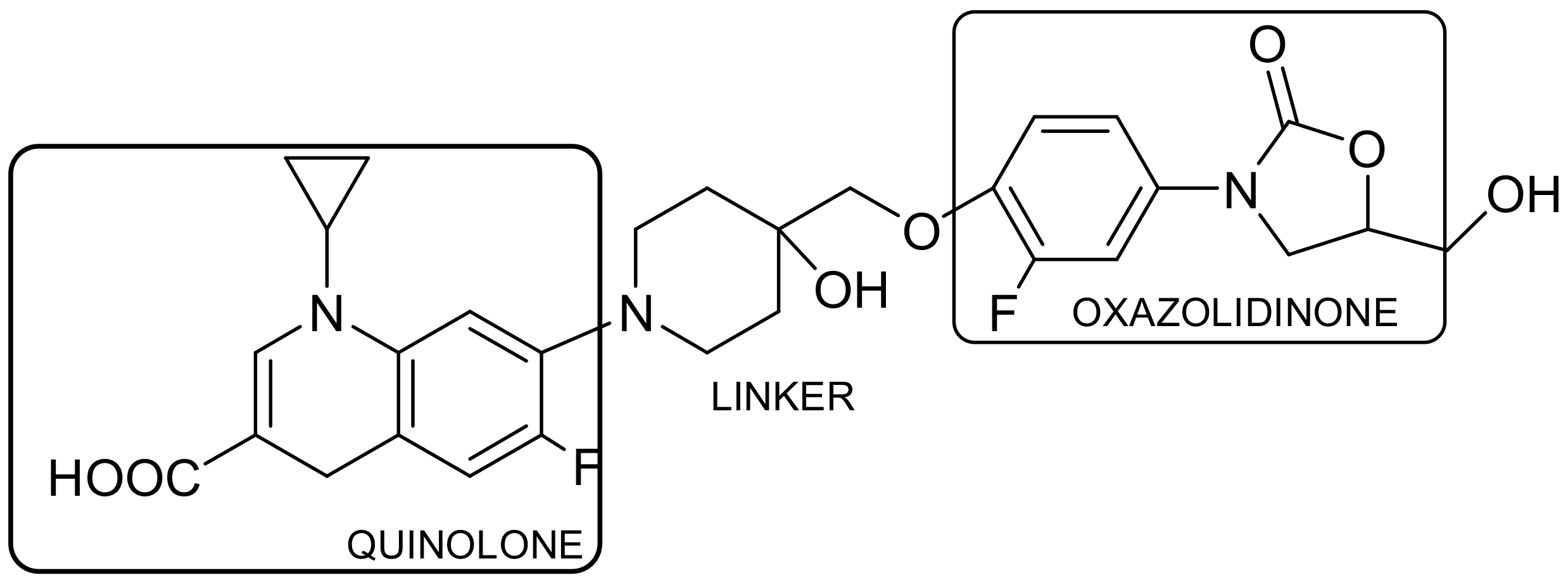
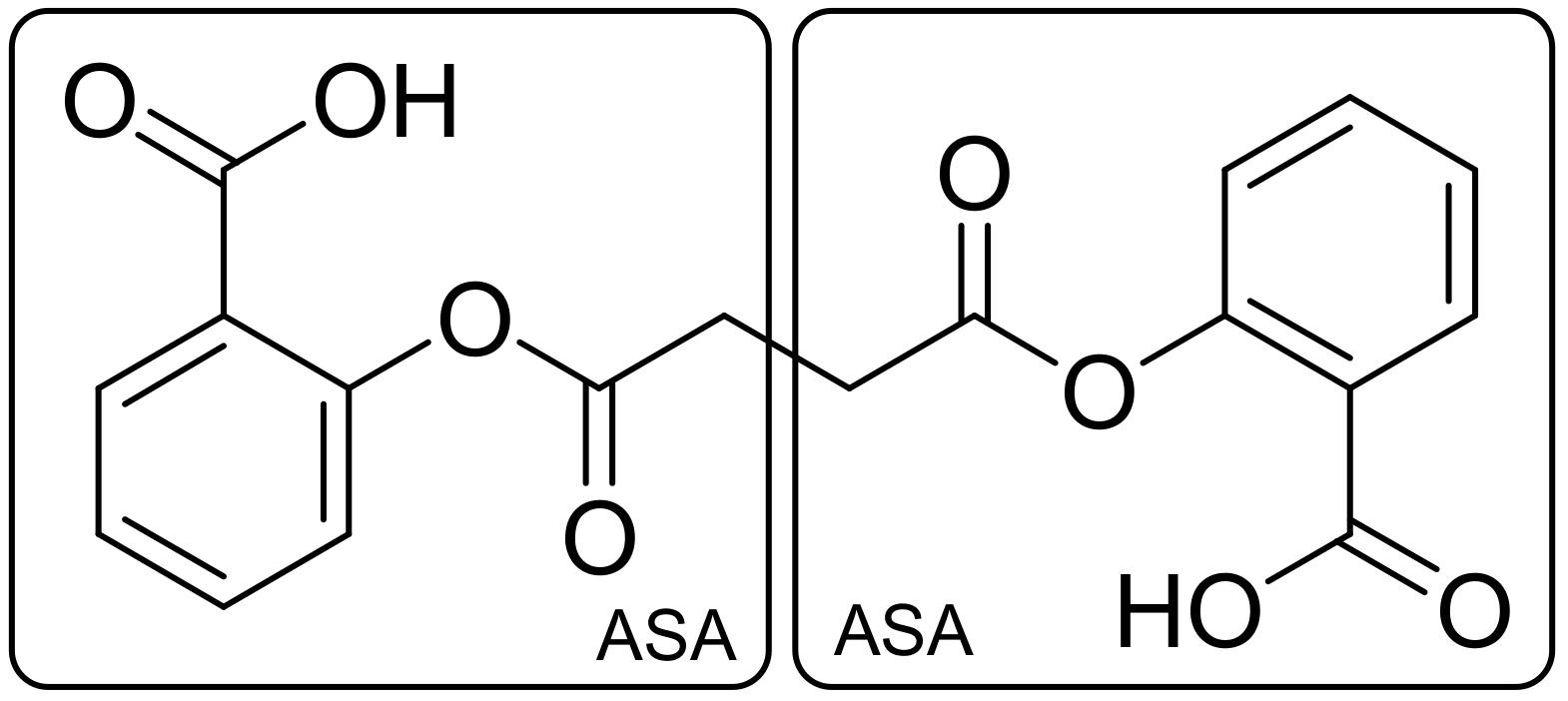
2.1. Drug–Drug Conjugates (DDCs)/Codrugs
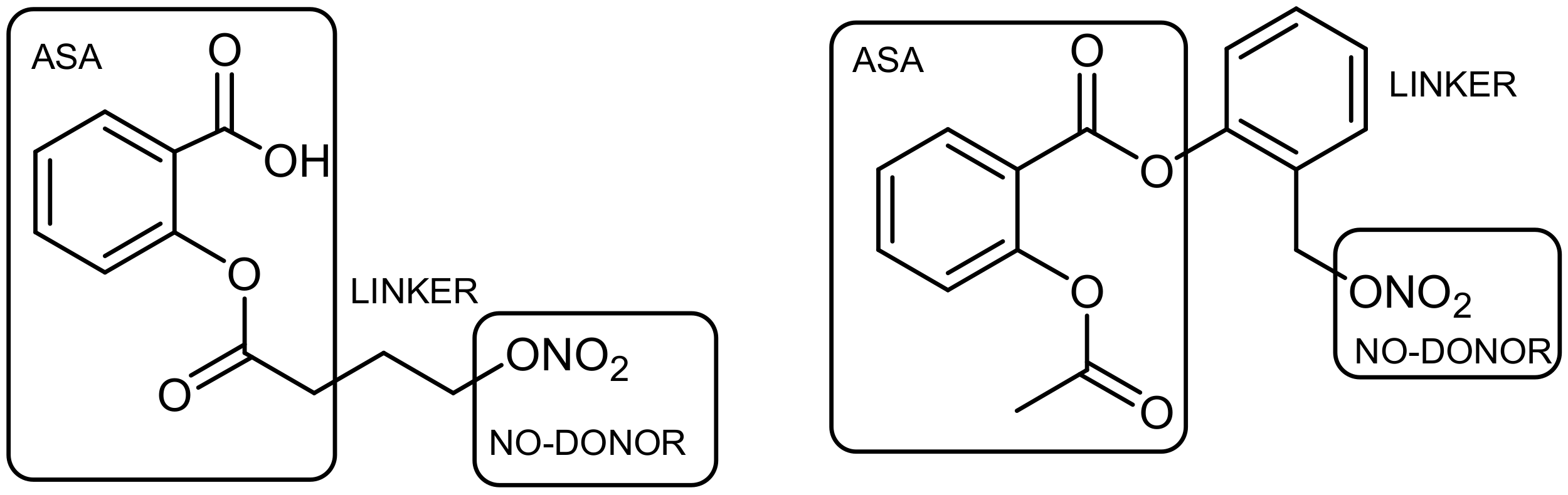
2.1.1. Linker Concept
- cleavable
- non-cleavable.
- difunctional
- trifunctional
- crosslinkers.
- hydrophilic
- hydrophobic.
- homofunctional
- heterofunctional (contain diverse functional groups).
- linear e.g., methylenic or polymeric chain
- non-linear e.g., aromatic, heteroaromatic or non-aromatic cycle
- all the linkers can be available as mono-protected (Boc, Fmoc, and Cbz).
2.1.2. No-Linker (Fused) Concept
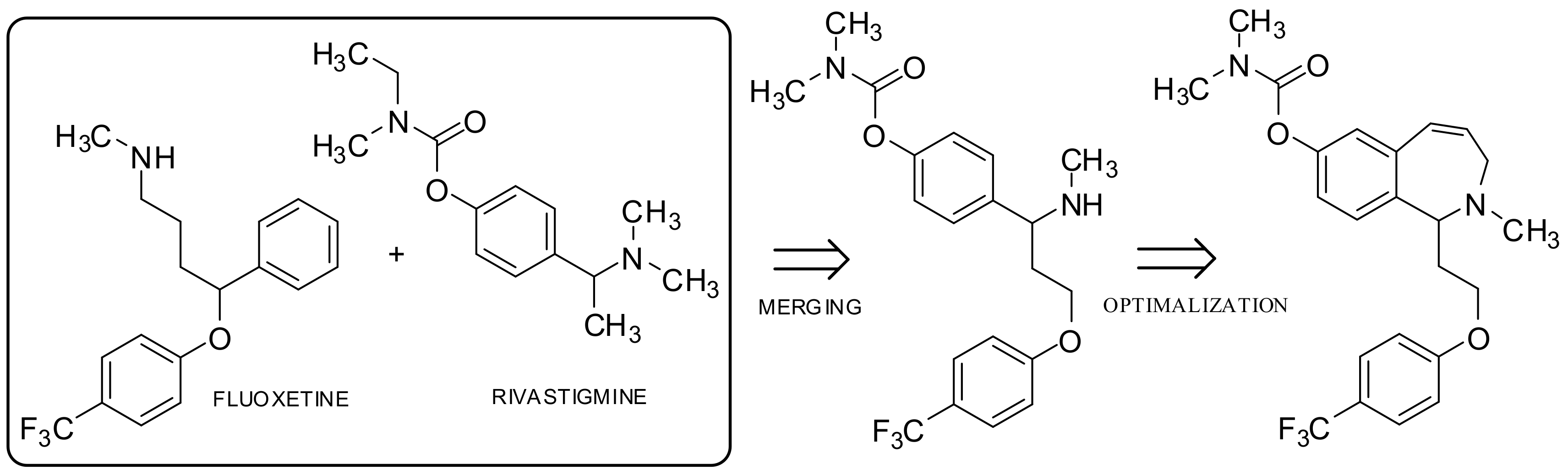
2.1.3. Overlap (Merged) Concept
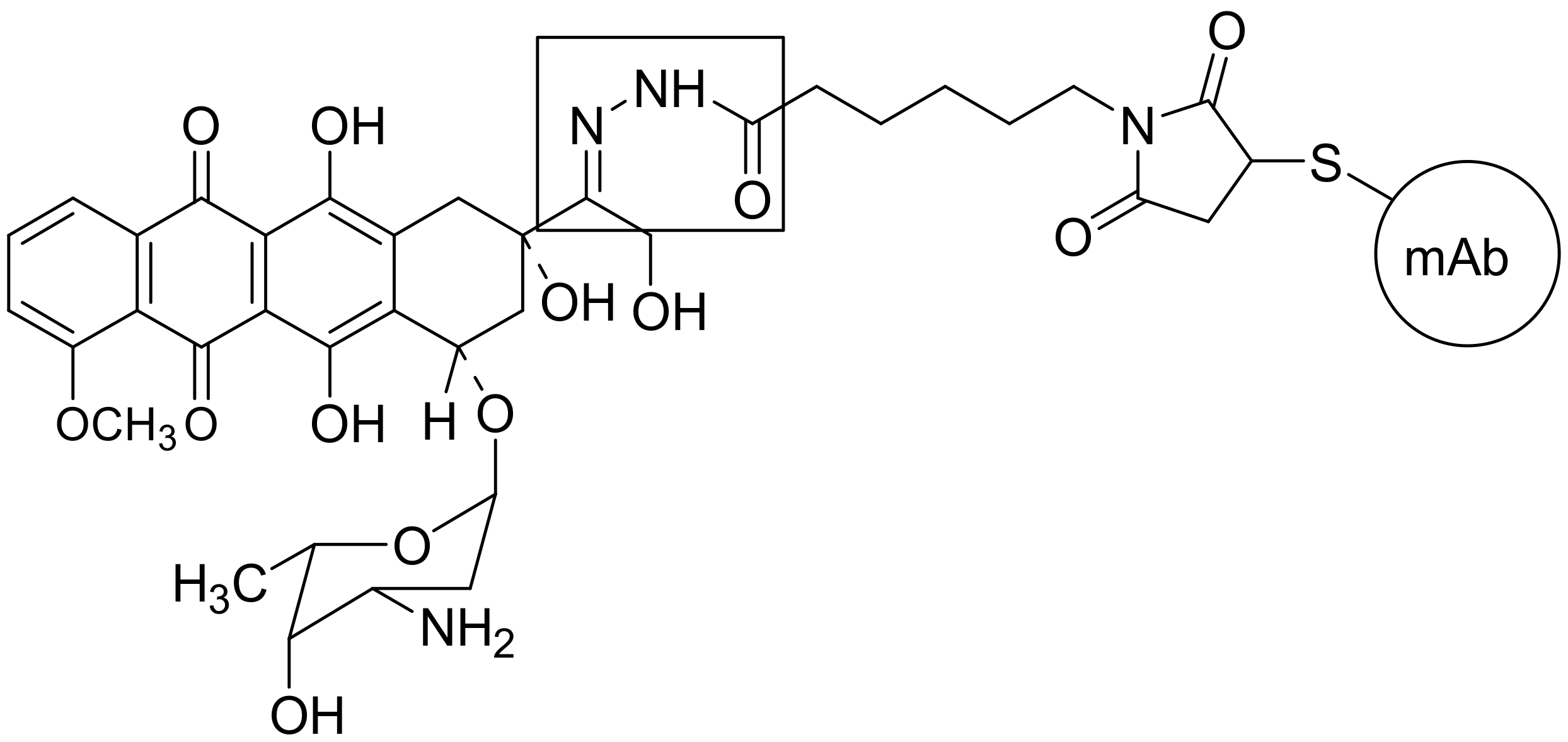
2.2. Antibody-Drug Conjugates (ADCs)
- reducible linkers (glutathione-sensitive) e.g., disulfide linkers.
- peptide-based linkers (protease-sensitive)
- l-glucuronide linkers (glucoronidase-sensitive).
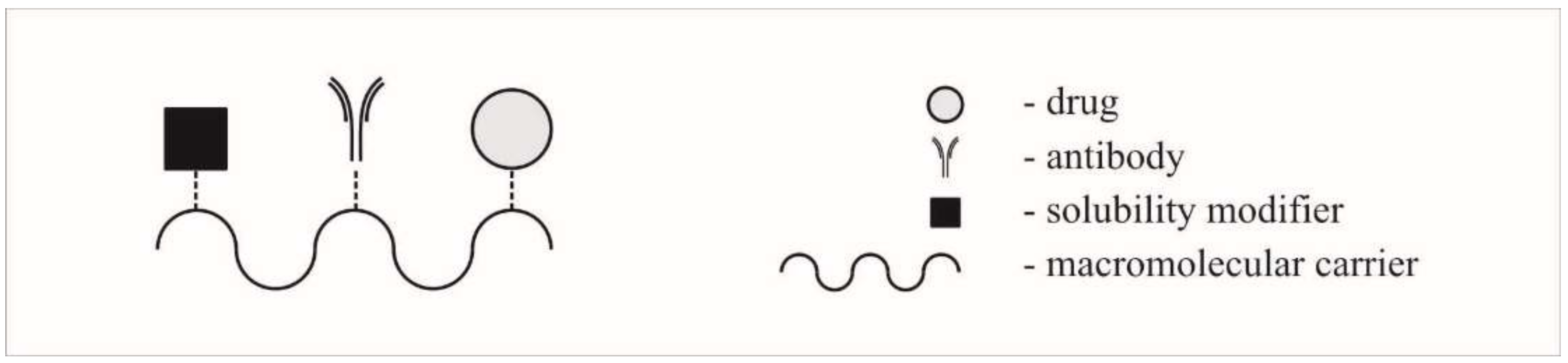
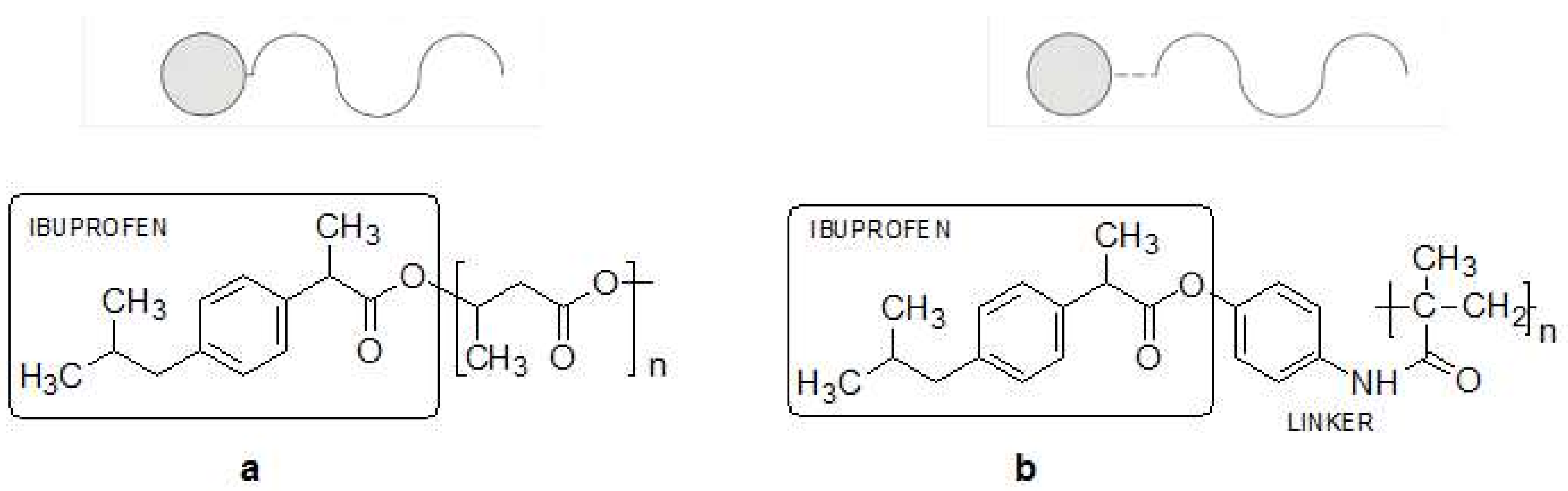
2.3. Polymer-Drug Conjugates (PDCs) (Prodrugs)
- No-linker mode polymer-drug conjugates obtained by cross-reactivity of functional fragments of pharmacophore present in the components, which leads to the formation of a new covalent bond
- Linker mode polymer-drug conjugates obtained by combining the drug and the polymer carrier by appropriately designed linker.
3. Synthetic Hopes in Combined Drug Chemistry
3.1. “Lego” Chemistry Concept
3.2. Green Synthesis Concept
- Application of innovative technology to establish industrial procedures
- Development of environmentally improved routes, synthetic methods, and processes to important products
- Design of new, greener, and safer chemicals and materials
- The use of sustainable resources
- The use of alternatives to chemistry-based solutions
- Development of methodologies and tools for measuring environmental impact
- Development of chemical aspects of renewable energy.
3.3. Complementary Synergy Concept
- synergism by combining various active structures—pharmacomodulation of two biologically active molecules by chemical hybridization methods leads to a new combined structure with interesting biological activity. According to recent literature [6,7,55] compounds obtained derived from different bioactive molecules often characterized by a synergy of their individual component activities
- synergism by synthesis process intensification by means of combining two non-conventional factors: microwaves and ultrasound. These two effects of process intensification have been used to great effect in various chemical processes and engineering applications. There is certainly scope for them to be applied in the drug chemistry field.
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Müller-Schiffa, A.; Sticht, H.; Korth, C. Hybrid compounds. From simple combination to nanomachines. Biodrugs 2012, 26, 21–31. [Google Scholar] [CrossRef] [PubMed]
- Tsogoeva, S.B. Recent progress in the development of synthetic hybrids of natural or unnatural bioactive compounds for medicinal chemistry. Mini-Rev. Med. Chem. 2010, 10, 773–793. [Google Scholar] [CrossRef] [PubMed]
- Decker, M. Design of Hybrid Molecules for Drug Development, 1st ed.; Elsevier: New York, NY, USA, 2017; ISBN 9780081010112. [Google Scholar]
- Muregi, F.W.; Ishih, A. Next-generation antimalarial drugs: Hybrid molecules as a new strategy in drug design. Drug Dev. Res. 2010, 71, 20–32. [Google Scholar] [CrossRef] [PubMed]
- Singh, M.; Kaur, M.; Chadha, N.; Silakari, O. Hybrids: A new paradigm to treat Alzheimer’s disease. Mol. Divers. 2016, 20, 271–297. [Google Scholar] [CrossRef] [PubMed]
- Shaveta, M.S.; Singh, P. Hybrid molecules: The privileged scaffolds for various pharmaceuticals. Eur. J. Med. Chem. 2016, 124, 500–536. [Google Scholar] [CrossRef] [PubMed]
- Bansal, Y.; Silakari, O. Multifunctional New York, NY, USA compounds: Smart molecules for multifactorial diseases. Eur. J. Med. Chem. 2014, 76, 31–42. [Google Scholar] [CrossRef] [PubMed]
- Janout, V.; Bienvenu, C.; Schell, W.; Perfect, J.R.; Regen, S.L. Molecular umbrella-amphotericin B conjugates. Bioconjugate Chem. 2014, 25, 1408–1411. [Google Scholar] [CrossRef] [PubMed]
- Głąbski, T.; Mikołajczyk, J.; Rusek, D. Związki hybrydowe jako potencjalne leki wielofunkcyjne (Hybrid compounds as potential multitarget drugs). Farm. Polska 2013, 69, 422–433. [Google Scholar]
- Contreas, J.M.; Sippl, W. Homo and heterodimer ligands: The twin drug approach. In The Practice of Medicinal Chemistry, 3rd ed.; Elsevier: New York, NY, USA, 2008; pp. 380–402, Chapter 18. [Google Scholar]
- Morphy, R.; Rankovic, Z. Multi-target drugs: Strategies and challenges for medicinal chemists. In The Practice of Medicinal Chemistry, 3rd ed.; Elsevier: New York, NY, USA, 2008; pp. 549–571, Chapter 27. [Google Scholar]
- Fujii, H. Twin and triplet drugs in opioid research. Top. Curr. Chem. 2011, 299, 239–275. [Google Scholar] [CrossRef] [PubMed]
- Morphy, R.; Rancovic, Z. Designed multiple ligands. An emerging drug discovery paradigm. J. Med. Chem. 2005, 48, 6523–6543. [Google Scholar] [CrossRef] [PubMed]
- Wichur, T.; Malawska, B. Ligandy wielofunkcyjne—Nowa strategia poszukiwania leku w terapii chorób o złożonej etiologii (Multifunctional ligands—A new approach in the search for drugs against multi-factorial diseases). Postepy Hig. Med. Dosw. 2015, 69, 1423–1434. [Google Scholar]
- Bojanowski, P.; Lipiński, P.F.J.; Czekała, P.; Plewczyński, D. Leki wielocelowe—Nowy paradygmat w projektowaniu leków. Biul. Wydz. Farm. WUM 2015, 1, 1–10. [Google Scholar]
- Nevozhy, D.; Kańska, U.; Budzyńska, R.; Boratyński, J. Współczesny stan badań nad koniugatami i innymi schematami dostarczania leków w leczeniu schorzeń nowotworowych i innych jednostek chorobowych (Current status of research on conjugates and related drug delivery systems in the treatment of cancer and other diseases). Postępy Hig. Med. Dosw. 2007, 61, 350–360. [Google Scholar]
- Moshin, N.A.; Ahmad, M. Hybrid organic molecules as antiinflamatory agents; a review of structural features and biological activity. Turk. J. Chem. 2018, 42, 1–20. [Google Scholar] [CrossRef]
- Brötz-Oesterhelt, H.; Brenner, N.A. How many modes of action should an antibiotic have? Curr. Opin. Pharmacol. 2008, 8, 564–573. [Google Scholar] [CrossRef] [PubMed]
- Lange, J.H.M.; Coolen, H.K.A.; Van der Neut, M.A.W.; Borst, A.J.M.; Stork, B.; Verveer, P.C.; Kruse, C.G. Design, synthesis, biological properties, and molecular modeling investigations of novel tacrine derivatives with combination of acetylcholinesterase inhibition and cannabinoid CB1 receptor anatagonism. J. Med. Chem. 2010, 53, 1338–1346. [Google Scholar] [CrossRef] [PubMed]
- Hu, M.K.; Wu, L.J.; Hsiao, G.; Yen, M.H. Homodimeric tacrine congeners as acetylcholinesterase inhibitors. J. Med. Chem. 2002, 45, 2277–2282. [Google Scholar] [CrossRef] [PubMed]
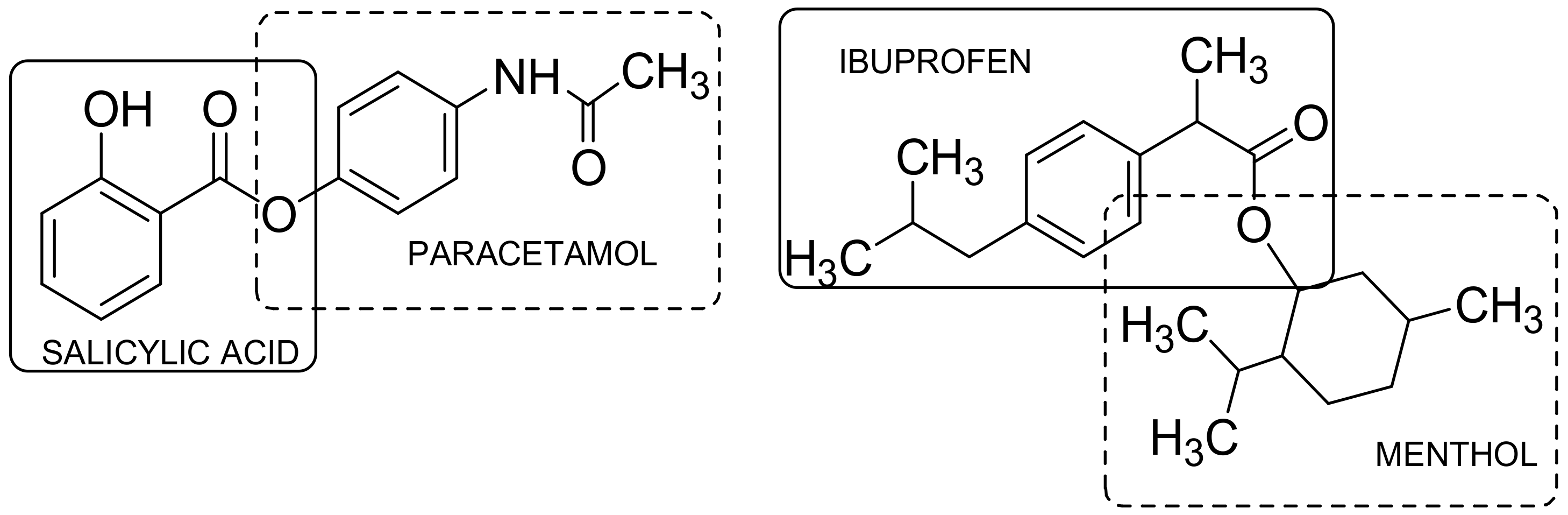
- Redasani, V.K.; Bari, S.B. Synthesis and evaluation of mutual prodrugs of ibuprofen with mentol, thymol and eugenol. Eur. J. Med. Chem. 2012, 56, 134–138. [Google Scholar] [CrossRef] [PubMed]
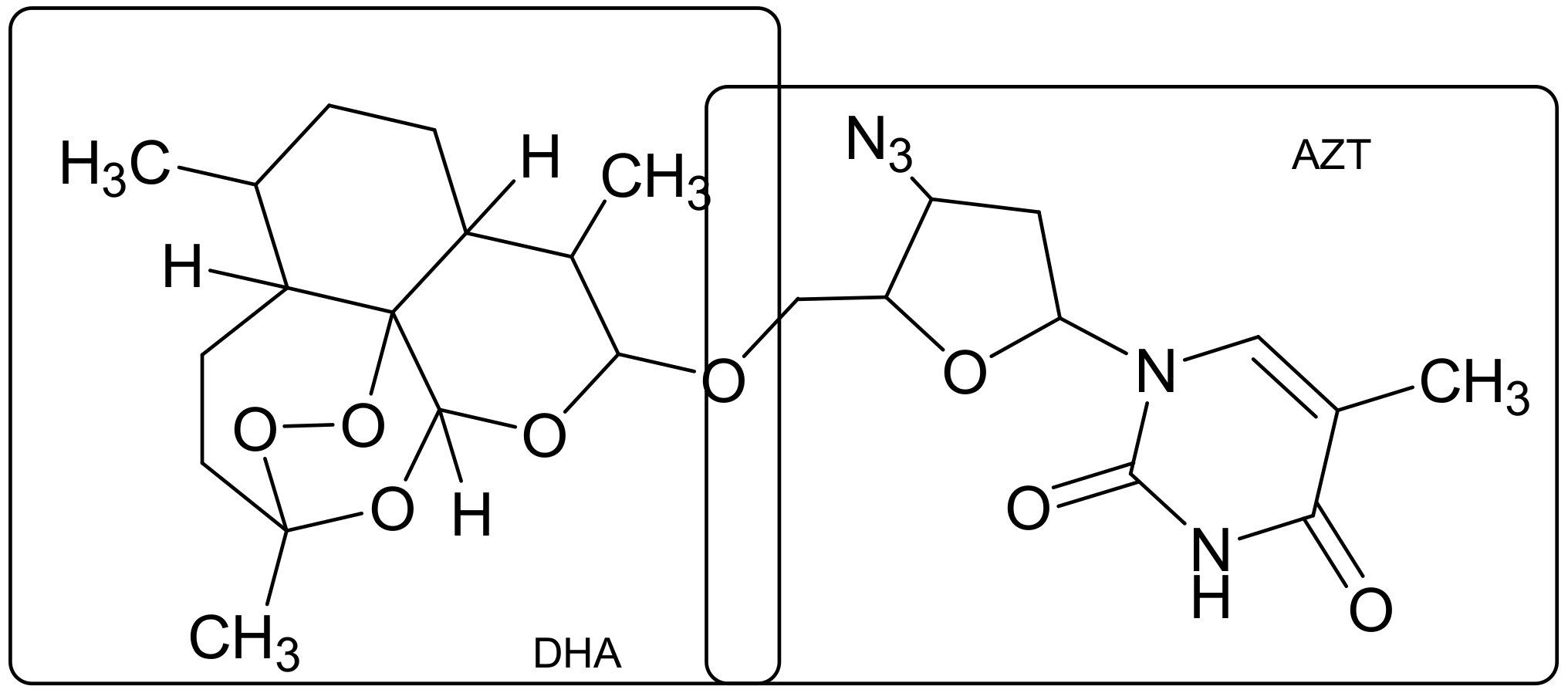
- Aminake, M.N.; Mahajan, A.; Kumar, V.; Hans, R.; Wiesner, L.; Taylor, D.; de Kock, C.; Grobler, A.; Smith, P.J.; Kirschner, M.; et al. Synthesis and evaluation of hybrid drugs for a potential HIV/AIDS-malaria combination therapy. Bioorg. Med. Chem. 2012, 20, 5277–5289. [Google Scholar] [CrossRef] [PubMed]
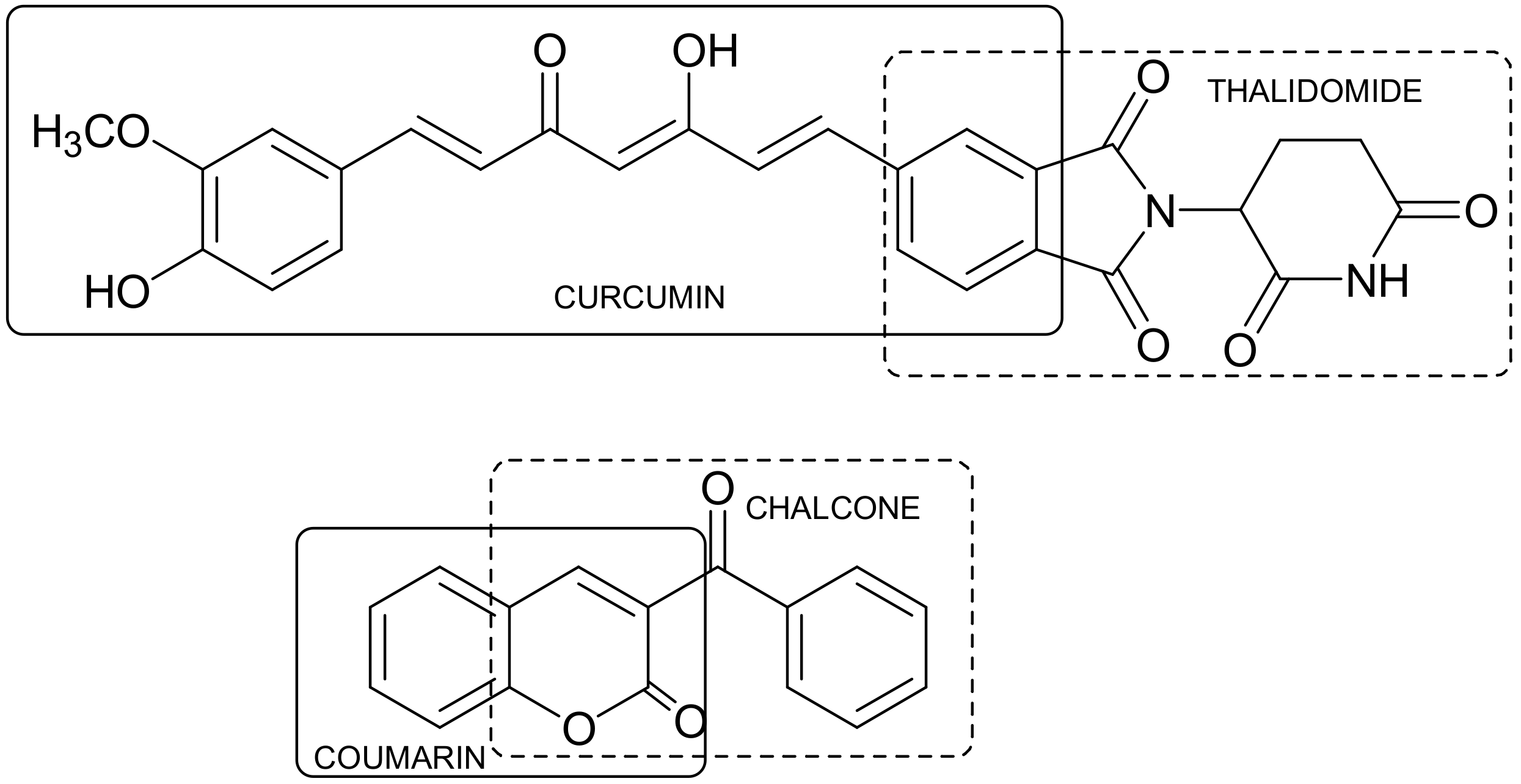
- Liu, K.; Zhang, D.; Chojnacki, J.; Du, Y.; Fu, H.; Grant, S.; Zhang, S. Design and biological characterization of hybrid compounds of curcumin and thalidomide for multiple myeloma. Org. Biomol. Chem. 2013, 11, 4757–4763. [Google Scholar] [CrossRef] [PubMed]
- Teiten, M.H.; Dicato, M.; Diederich, M. Hybrid curcumin compounds: A new strategy for cancer treatment. Molecules 2014, 19, 20839–20863. [Google Scholar] [CrossRef] [PubMed]
- Tailor, N.; Sharma, M. Antioxidant hybrid compounds: A promising therapeutic intervention in oxidative stress induced diseases. Mini-Rev. Med. Chem. 2013, 13, 280–297. [Google Scholar] [CrossRef] [PubMed]
- Pérez-Cruz, F.; Vazquez-Rodrigez, S.; João Matos, M.; Herrera-Morales, A.; Villamena, F.A.; Das, A.; Gopalakrishnan, B.; Olea-Azar, C.; Santana, L.; Uriarte, E. Synthesis and electrochemical and biological studies of novel coumarin-chalcone hybrid compounds. J. Med. Chem. 2013, 56, 6136–6145. [Google Scholar] [CrossRef] [PubMed]
- Sashidhara, K.V.; Kumar, A.; Kumar, M.; Sarkar, J.; Sinha, S. Synthesis and in vitro evaluation of novel coumarin–chalcone hybrids as potential anticancer agents. Bioorg. Med. Chem. Lett. 2010, 20, 7205–7211. [Google Scholar] [CrossRef] [PubMed]
- Koufaki, M.; Detsi, A. Design and synthesis of antioxidant α-lipoic acid hybrids. Methods Mol. Biol. 2010, 594, 297–309. [Google Scholar] [CrossRef] [PubMed]
- Prati, F.; Cavalli, A.; Bolognesi, M.L. Navigating the Chemical Space of Multitarget-Directed Ligands: From Hybrids to Fragments in Alzheimer’s Disease. Molecules 2016, 21, 466. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morphy, R.; Rankovic, Z. Fragments, network biology and designing multiple ligands. Drug Discov. Today 2007, 12, 156–160. [Google Scholar] [CrossRef] [PubMed]
- Toda, N.; Tago, K.; Marumoto, S.; Takami, K.; Ori, M.; Yamada, N.; Koyama, K.; Naruto, S.; Abe, K.; Yamazaki, R.; et al. A conformational restriction approach to the development of dual inhibitors of acetylcholinesterase and serotonin transporter as potential agents for Alzheimer’s disease. Bioorg. Med. Chem. 2003, 11, 4389–4415. [Google Scholar] [CrossRef]
- Scott, A.M.; Wolchok, J.D.; Old, L.J. Antibody therapy of cancer. Nat. Rev. Cancer 2012, 12, 278–287. [Google Scholar] [CrossRef] [PubMed]
- Peters, C.; Brown, S. Antibody-drug conjugates as novel anti-cancer chemotherapeutics. Biosci. Rep. 2015, 35, e00225. [Google Scholar] [CrossRef] [PubMed]
- Flygare, J.A.; Pillow, T.H.; Aristoff, P. Antibody-drug conjugates for the treatment of cancer. Chem. Biol. Drug Des. 2013, 81, 113–121. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.; Jiang, F.; Lu, A.; Zhang, G. Linkers having a crucial role in antybody-drug conjugates. Int. J. Mol. Sci. 2016, 17, 561. [Google Scholar] [CrossRef] [PubMed]
- Pillow, T.H. Novel linkers and connections for antibody-drug conjugates to treat cancer and infectious disease. Pharm. Pat. Anal. 2017, 6, 25–33. [Google Scholar] [CrossRef] [PubMed]
- Jain, N.; Smith, S.W.; Ghone, S.; Tomczuk, B. Current ADC linker chemistry. Pharm. Res. 2015, 32, 3526–3540. [Google Scholar] [CrossRef] [PubMed]
- Bagshawe, K.D.; Sharma, S.K.; Burke, P.J.; Melton, R.G.; Knox, R.J. Developments with targeted enzymes in cancer therapy. Curr. Opin. Immunol. 1999, 11, 579–583. [Google Scholar] [CrossRef]
- Sharma, S.K.; Chester, K.A.; Bagshawe, K.D. Antibody-Directed Enzyme Prodrug Therapy (ADEPT). In Handbook of Therapeutic Antibodies, 2nd ed.; Wiley-VCH Verlag GmbH & Co.: Weinheim, Germany, 2014; Volume 1–4, pp. 475–486. [Google Scholar] [CrossRef]
- Beck, A.; Goetsch, L.; Dumontet, C.; Corvaia, N. Strategies and challenges for the next generation of antibody-drug conjugates. Nat. Rev. Drug Discover. 2017, 16, 315–337. [Google Scholar] [CrossRef] [PubMed]
- Ringsdorf, H. Structure and properties of pharmacologically active polymers. J. Polym. Sci., Polym. Symp. 1975, 51, 135–153. [Google Scholar] [CrossRef]
- Mehra, N.K.; Jain, N.K. Multifunctional hybrid-carbon nanotubes: New horizon in drug delivery and targeting. J. Drug Target. 2016, 2, 297–301. [Google Scholar] [CrossRef] [PubMed]
- Duncan, R.; Vicent, M.J.; Greco, F.; Nicholson, R.I. Polymer-drug conjugates: Towards a novel approach for the treatment of endrocine-related cancer. Endocr. Relat. Cancer 2005, 12, 189–199. [Google Scholar] [CrossRef] [PubMed]
- Haag, R.; Kratz, F. Polymer therapeutics: Concepts and applications. Angew. Chem. Int. Ed. 2006, 45, 1198–1215. [Google Scholar] [CrossRef] [PubMed]
- Harris, J.M.; Martin, N.E.; Modi, M. Pegylation: A novel process for modifying pharmacokinetics. Clin. Pharmacokinet. 2001, 40, 539–551. [Google Scholar] [CrossRef] [PubMed]
- Veronese, F.M.; Pasut, G. PEGylation, successful approach to drug delivery. Drug Discov. Today 2005, 10, 1451–1458. [Google Scholar] [CrossRef]
- Müller, H.J.; Beier, R.; Da Palma, J.; Lanvers, C.; Ahlke, E.; Von Schütz, V.; Gunkel, M.; Horn, A.; Schrappe, M.; Henze, G.; et al. PEG-asparaginase (Oncaspar) 2500 u/m2 BSA in reinduction and relapse treatment in the ALL/NHL-BFM protocols. Cancer Chemother. Pharmacol. 2002, 49, 149–154. [Google Scholar] [CrossRef] [PubMed]
- PEGASYS®. Peginterferon ALFA-2A. Available online: www.pegasys.com (accessed on 23 February 2017).
- Allen, T.M.; Cullis, P.R. Drug delivery systems: Entering the mainstream. Science 2004, 303, 1818–1822. [Google Scholar] [CrossRef] [PubMed]
- Duncan, R. Polymer therapeutics as nanomedicines: New perspectives. Curr. Opin. Biotechnol. 2011, 22, 492–501. [Google Scholar] [CrossRef] [PubMed]
- Zawidlak-Wegrzyńska, B.; Kawalec, M.; Bosek, I.; Łuczyk-Juzwa, M.; Adamus, G.; Rusin, A.; Filipczak, P.; Głowala-Kosińska, M.; Wolańska, K.; Krawczyk, Z.; et al. Synthesis and antiproliferative properties of ibuprofen-oligo(3-hydroxybutyrate) conjugates. Eur. J. Med. Chem. 2010, 45, 1833–1842. [Google Scholar] [CrossRef] [PubMed]
- Liso, P.A.; Rebuelta, M.; San Román, J.; Gallardo, A.; Villar, A.M. Polymeric drugs derived from ibuprofen with improved anti-inflammatory profile. J. Biomed. Mater. Res. 1996, 32, 553–560. [Google Scholar] [CrossRef]
- Wakaskar, R.R. Passive and Active Targeting in Tumor Microenvironment. Int. J. Drug Dev. Res. 2017, 9, 37–41. [Google Scholar]
- Das, N.; Dhanawat, M.; Dash, B.; Nagarwal, R.C.; Shrivastava, S.K. Codrug: An efficient approach for drug optimization. Eur. J. Pharm. Sci. 2010, 41, 571–588. [Google Scholar] [CrossRef] [PubMed]
- Aljuffali, I.A.; Lin, C.F.; Chen, C.H.; Fang, J.Y. The codrug approach for facilitating drug delivery and bioactivity. Expert Opin. Drug Deliv. 2016, 13, 1311–1325. [Google Scholar] [CrossRef] [PubMed]
- Ghawanmeh, A.A.; Chong, K.F.; Sarkar, S.M.; Bakar, M.A.; Othaman, R.; Khalid, R.M. Colchicine prodrugs and codrugs: Chemistry and bioactivities. Eur. J. Med. Chem. 2018, 144, 229–242. [Google Scholar] [CrossRef] [PubMed]
- Maraval, V.; Pyzowski, J.; Caminade, A.-M.; Majoral, J.-P. “Lego” Chemistry for the Straightforward Synthesis of Dendrimers. J. Org. Chem. 2003, 68, 6043–6046. [Google Scholar] [CrossRef] [PubMed]
- Kolb, H.C.; Sharpless, K.B. The growing impact of click chemistry on drug discovery. Drug Discov. Today 2003, 8, 1128–1137. [Google Scholar] [CrossRef]
- Hein, C.D.; Liu, X.-M.; Wang, D. Click Chemistry, a Powerful Tool for Pharmaceutical Sciences. Pharm. Res. 2008, 25, 2216–2230. [Google Scholar] [CrossRef] [PubMed]
- Anastas, P.C.; Warner, J.C. Green Chemistry: Theory and Practice; Oxford University Press: Oxford, UK, 1998. [Google Scholar]
- Cravotto, G.; Cintas, P. The Combined Use of Microwaves and Ultrasound: Improved Tools in Process Chemistry and Organic Synthesis. Chem. Eur. J. 2007, 13, 1902–1909. [Google Scholar] [CrossRef] [PubMed]
- Peng, Y.; Song, G. Simultaneous microwave and ultrasound irradiation: A rapid synthesis of hydrazides. Green Chem. 2001, 3, 302–304. [Google Scholar] [CrossRef]
- Martinez-Guerra, E.; Gude, V.G. Synergistic effect of simultaneous MW and US irradiations on transesterification of waste vegetable oil. Fuel 2014, 137, 100–108. [Google Scholar] [CrossRef]
- Bisi, A.; Gobbi, S.; Belluti, F.; Rampa, A. Design of multifunctional compounds for cardiovascular disease: From natural scaffolds to “classical” multitarget approach. Curr. Med. Chem. 2013, 20, 1759–1782. [Google Scholar] [CrossRef] [PubMed]
- Vlahov, I.R.; Leamon, C.P. Engineering folate—Drug conjugates to target cancer: From chemistry to clinic. Bioconjugate Chem. 2012, 23, 1357–1369. [Google Scholar] [CrossRef] [PubMed]
- Kerru, N.; Singh, P.; Koorbanally, N.; Raj, R.; Kumar, V. Recent advances (2015–2016) in anticancer hybrids. Eur. J. Med. Chem. 2017, 142, 179–212. [Google Scholar] [CrossRef] [PubMed]













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| ASSOCIATION MODE | |||
 | |||
| intermediate | direct | ||
| linker mode | no-linker mode | overlap mode | |
| cleavage linked | stable linked | fused | merged |
 |  |  |  |
| conjugate cleavage | conjugate | fused hybrid/codrugs | merged hybrid (chimera) |
| ASSOCIATION FORMS | |
|---|---|
 |  |
| Association | Duplication/Dimerization |
| drugs: | |
| non-identical twin drugs | identical twin drugs |
| two-pharmacophore drugs | one-pharmacophore twin drugs |
| non-symetrical twin drugs | symmetrical drugs |
| dual acting drugs | |
| hybrid drugs | |
| codrugs | |
| mutual prodrugs | |
| prodrugs | |
| conjugates: | |
| drug-drug conjugates (DDCs) | |
| antybody-drug conjugates (ADCs) | |
| polymer-drug conjugates (PDCs) | |
| other: | |
| heterodimers | homodimers |
| Molecular Consortia Descriptors | Hybrid Drugs | Conjugates | Codrugs | Prodrugs |
|---|---|---|---|---|
| Elements number | two (or more) distinct pharmacophore | two (or more) elements (DDCs, ADCs, PDCs) | two (or more) therapeutic compounds | two (or more) elements, only one is bioactive drug, carrier: inactivepolymer, antibody, gene, virus, enzyme |
| Association mode | direct a | direct a | direct a | direct a |
| indirect b | indirect b | indirect b | indirect b | |
| merged c | ||||
| Transformation (In vivo) | no-enzymatic cleavage | linker dependent selected | enzymatic cleavage | enzymatic cleavage |
| Activity | dual effects, different targets | dual effects, different targets | dual effects from both drugs | single effect (carrier is inactive) |
| Safety | enhancing efficacy, improving safety | improved therapeutic index | improved therapeutic index | additional toxicity depends on carrier |
| Design options | based on non-labile linker | based on labile or non-labile linker | based on specific chemical function | unlimited approach |
| Reaction Characteristics | Ultrasounds (US) | Microwaves (MW) |
|---|---|---|
| Reaction media | aqueous and organic solvents | MW-absorbing liquids; solvent-free protocols |
| Acceleration | variable (from min to h) | high (min, even seconds!) |
| Activation | cavitation (thermal effects) | thermal effects, (specific non-thermal) |
| Scaling up | possible but still a challenge | Possible |
| Chemical effects | selectivity changes mechanistic switching waste reductions | selectivity changes waste reductions |
| Other effects | light emission, cleaning, microstreaming | heating above boiling points, change in solvent properties |
| Method | Time | Yield (%) |
|---|---|---|
| reflux | 9 h | 73 |
| US (50 W) | 1.5 h | 79 |
| MW (200 W) | 18 min | 80 |
| MW + US | 40 s | 84 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pawełczyk, A.; Sowa-Kasprzak, K.; Olender, D.; Zaprutko, L. Molecular Consortia—Various Structural and Synthetic Concepts for More Effective Therapeutics Synthesis. Int. J. Mol. Sci. 2018, 19, 1104. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms19041104
Pawełczyk A, Sowa-Kasprzak K, Olender D, Zaprutko L. Molecular Consortia—Various Structural and Synthetic Concepts for More Effective Therapeutics Synthesis. International Journal of Molecular Sciences. 2018; 19(4):1104. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms19041104
Chicago/Turabian StylePawełczyk, Anna, Katarzyna Sowa-Kasprzak, Dorota Olender, and Lucjusz Zaprutko. 2018. "Molecular Consortia—Various Structural and Synthetic Concepts for More Effective Therapeutics Synthesis" International Journal of Molecular Sciences 19, no. 4: 1104. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms19041104