The Janus Face of VEGF in Stroke


1
Department of Pharmaceutical Biosciences, School of Pharmacy, University of Oslo, 0371 Oslo, Norway
2
Institute for Behavioral Sciences, Faculty of Health Sciences, OsloMet—Oslo Metropolitan University, 0166 Oslo, Norway
*
Author to whom correspondence should be addressed.
Int. J. Mol. Sci. 2018, 19(5), 1362; https://0-doi-org.brum.beds.ac.uk/10.3390/ijms19051362
Submission received: 28 March 2018
/
Revised: 27 April 2018
/
Accepted: 1 May 2018
/
Published: 4 May 2018
(This article belongs to the Special Issue Vascular Endothelial Growth Factor)
{kind=link}
{kind=link}
{kind=link}
Abstract
:The family of vascular endothelial growth factors (VEGFs) are known for their regulation of vascularization. In the brain, VEGFs are important regulators of angiogenesis, neuroprotection and neurogenesis. Dysregulation of VEGFs is involved in a large number of neurodegenerative diseases and acute neurological insults, including stroke. Stroke is the main cause of acquired disabilities, and normally results from an occlusion of a cerebral artery or a hemorrhage, both leading to focal ischemia. Neurons in the ischemic core rapidly undergo necrosis. Cells in the penumbra are exposed to ischemia, but may be rescued if adequate perfusion is restored in time. The neuroprotective and angiogenic effects of VEGFs would theoretically make VEGFs ideal candidates for drug therapy in stroke. However, contradictory to what one might expect, endogenously upregulated levels of VEGF as well as the administration of exogenous VEGF is detrimental in acute stroke. This is probably due to VEGF-mediated blood–brain-barrier breakdown and vascular leakage, leading to edema and increased intracranial pressure as well as neuroinflammation. The key to understanding this Janus face of VEGF function in stroke may lie in the timing; the harmful effect of VEGFs on vessel integrity is transient, as both VEGF preconditioning and increased VEGF after the acute phase has a neuroprotective effect. The present review discusses the multifaceted action of VEGFs in stroke prevention and therapy.
1. Introduction
Millions of people suffer a stroke every year, and stroke is the main cause of disabilities among adults. The two major causes of stroke are an occlusion of a precerebral or cerebral artery or a hemorrhage. Both cause ischemia, which rapidly leads to necrosis in the stroke core. The fate of the area surrounding the necrotic core, the penumbra, depends largely on the time before reperfusion is restored. Neural death is proportional to the degree of loss in perfusion, and early reperfusion is essential to prevent extensive neural damage [1,2,3,4]. Brain injury after stroke occurs as a result of a complex series of pathophysiological events, including excitotoxicity, oxidative stress, vasopermeability of the blood–brain barrier (BBB), and inflammation, leading to cell death. Growth factors are important regulators of protection and recovery after ischemia, and the combined action of growth factors regulates angiogenesis, neuroprotection, neurogenesis as well as the migration of neuronal stem cells into the ischemic area, and their proliferation into functional neurons. One important family of growth factors is the family of vascular endothelial growth factors (VEGFs). Due to their upregulation in the ischemic brain and their strong angiogenic and neuroprotective properties [5,6,7,8,9], the administration of VEGFs per se, or substances that regulate VEGFs or VEGF receptor actions, are considered interesting potential treatment strategies in stroke.
The mammalian VEGF family consists of five members, VEGF-A, -B, -C, -D, and placental growth factor (PIGF). Of these, VEGF-A has gained most attention. VEGF-A is pro-angiogenic and neuroprotective and induces neurogenesis [10]. Therefore, systemic treatment with VEGF-A in stroke has gained considerable interest. However, such VEGF-A treatment has shown somewhat disappointing and inconsistent effects in pre-clinical trials. This is probably because VEGF-A has a dual role in the regulation of the vascular endothelium [11]: firstly, it stimulates the proliferation and migration of endothelial cells, leading to the formation of new vessels. This results in a more efficient network of collaterals, which may bypass the occluded vessel and thereby rescue the penumbra. However, VEGF-A also increases vascular permeability [12]. The latter effect may cause brain edema and increase intracranial pressure, which is detrimental in the acute phase of stroke [13]. Increased vascular permeability also allows the entry of molecules and immune cells that are normally prohibited by the blood–brain barrier (BBB), causing neuroinflammation.
2. The VEGF Receptor Family
The canonical VEGF signal is initiated when VEGFs bind to VEGF receptors, namely VEGFR-1 (fms-like tyrosine kinase-1; flt-1), VEGFR-2 (fetal liver kinase-1 (flk-1)/kinase domain receptor (KDR)) or VEGFR-3 (flt-4). These receptors belong to the family of receptor tyrosine kinases. The overall structure of these receptors, and the intracellular signaling cascades they initiate, are well preserved through evolution [14], indicating that they regulate important functions in development as well as in adult health and disease. Non-canonical VEGF signaling also exists, for example when VEGFs activate other receptors than the VEGFRs. One such receptor family is the family of neuropilin (NRP) receptors, which consists of NRP-1 and NRP-2. The NRPs are not “true” receptors, as they lack intrinsic enzymatic activity and therefore cannot be activated by the binding of agonists. These are termed co-receptors for the VEGFs. Members of the VEGF family bind to the VEGFRs or co-receptors with different affinities: VEGF-A primarily binds to VEGFR-1, VEGFR-2, NRP-1 and NRP-2, while VEGF-B binds to VEGFR-1 and NRP-1. VEGF-C and VEGF-D predominantly binds to VEGFR-3, VEGFR-2 and PIGF binds VEGFR-1, NRP-2 and NRP-1 (for review, see [15]).
The VEGF receptors and co-receptors have different properties and activate different intracellular signaling pathways. VEGF receptor signaling has been reviewed elsewhere in detail [16]. Although VEGF-A also activates other receptors, the main focus of this review is on the effects that are initiated by the binding of VEGF-A to VEGFR-2, as they are the most important in stroke. VEGFR-1 may act as a decoy receptor [17,18], the main effect of which is to prevent the binding of VEGF-A to VEGFR-2. Angiogenesis therefore requires a balanced action of VEGFR-1 and VEGFR-2. Depending on the residue of the intracellular phosphorylation, the activation of VEGFR-2 triggers different intracellular signaling cascades. Although these pathways regulate partly overlapping functions in the cell, each pathway can explain at least one of the main functions of VEGF in stroke. The phosphatidylinositol-3-kinases-protein/kinase B (PI3K/Akt) pathway is a main survival pathway, the MEK–MAPK pathway regulates proliferation and migration, and the Src-suppressed C kinase substrate (SSeCKS) pathway mediates vasopermeability, all of which are important effects of VEGF-A in stroke.
3. VEGF-A in the Prevention and Therapy of Stroke
3.1. VEGF-A Expression
In the healthy adult mammalian brain, VEGF-A is expressed at low levels and is observed sporadically in parenchymal cells in most brain regions [19]. VEGFR-2 is expressed on neurons and vascular endothelial cells [20,21,22], while VEGFR-1 is found in the vasculature, in the choroid plexus and on glial cells.
In response to stroke, VEGF-A as well as its receptors VEGFR-1 and VEGFR-2 are up-regulated [23,24]. The increase occurs predominantly in the stroke/penumbra area. However, an increase in the cortical areas that are functionally and/or behaviorally related to the area of the infarction has also been reported [25,26]. In stroke, VEGF-A is increased in astrocytes, neurons and endothelial cells, and in the penumbra, compared to both the infarcted area and the contralateral hemisphere [22,24,27,28,29]. As for VEGF-A itself, the expression of VEGFR-2 in the vasculature increases in the penumbra compared to the contralateral hemisphere [24]. VEGFR-1 increase has been reported in the penumbra, but also in the pia mater, and on the vessel invading the core region of the infarcted area [23]. In the penumbra, the upregulation of VEGFR-1 was almost exclusively in reactive astrocytes [30,31] and in sprouting vessels [23]. The increase in VEGF-A and VEGF-receptors is reported to start as early as 2–4 h after the onset of stroke and to last for at least 28 days [28]. The increase starts later in astrocytes than in neurons [32]. According to Zan and coworkers [33], the increase in VEGF-A in response to ischemia is biphasic. The authors reported a first peak in VEGF-A at 6 h after reperfusion that normalized within 12 h, only to peak again at seven days after reperfusion. VEGF-A levels were reported to return to baseline after two weeks.
What regulates VEGF-A levels in the brain in response to stroke? Hypoxia-inducible factor 1 (HIF-1) is a pivotal transcription factor in the hypoxic brain. It consists of an inducible subunit HIF-1α and a constitutive subunit, HIF-1β. Under normoxic conditions, HIF-1α is rapidly ubiqutinylated and degraded. However, hypoxia stabilizes HIF-1α, leading to an upregulation of HIF-1α in the hypoxic/ischemic brain [34,35]. HIF-1α is a prominent activator of VEGF-A gene expression [23,36]. The direct connection between HIF-1α and its target-gene VEGF-A in hypoxia/ischemia is well documented [37,38,39]. An indirect mechanism may also be involved, where HIF-1α induces erythropoietin (EPO) [40], which in turn increases the secretion of VEGF [41]. Inflammatory cytokines may also regulate VEGF-A in stroke [42,43]. In addition, we had recently shown that lactate increases VEGF-A expression via the lactate receptor HCAR1, which causes HCAR1-dependent angiogenesis in the brain [44].
3.2. VEGF-A in the Treatment of Stroke
The current state of knowledge regarding the role of VEGF-A in stroke is almost exclusively based on animal models. VEGF-A has multiple protective effects, including the promotion of angiogenesis, neurogenesis, and neuroprotection, leading to improved functional recovery [45]. VEGF-A is significantly upregulated in the brain of the naked mole rat (Heterocephalus glaber), where it contributes to their exceptional intrinsic hypoxia tolerance [46,47]. VEGF-A is therefore a very interesting candidate for therapeutic treatment in ischemic stroke [48]. The involvement of VEGF-A in protective and harmful mechanisms in stroke is discussed below.
3.2.1. Effects of VEGF-A in Angiogenesis
Increased angiogenesis is highly important for the neuroprotective effects of VEGFs in stroke, and the upregulation of VEGF-A and VEGFR-2 in the penumbra is directly correlated to neuro-vascularization [27,28,49]. In the normal brain, the administration of VEGF-A causes an upregulation of VEGFR-1 and VEGFR-2 and a significant increase in cerebral vascularization [22]. Furthermore, the transplantation of stem cells that overexpress VEGF-A has been shown to cause angiogenesis of the host nervous tissue [50]. VEGF-A regulates angiogenesis in the brain by the combined action of VEGFR-1 and VEGFR-2, where the activation of the latter increases angiogenesis, and the activation of the former decreases it (for details see below). Together these receptors ensure that sprouting angiogenesis in the brain is a carefully regulated process. When VEGF-A binds to VEGFR-2, phosphoinositide 3-kinase (PI3K) is activated; this kinase is a central component in the angiogenic process. The molecular link between VEGFR-2 and PI3K is not well described, but Axl, a member of the TAM family of receptor tyrosine kinases, seems to be involved [51]. PI3K activates Kinase B (Akt) [52], which promotes migration of the endothelial cells of the BBB [53,54,55,56]. Activation of the Akt pathway by VEGF-A after stroke has been extensively demonstrated [57,58,59,60]. In a recent study [61], CRISPR/Cas9-mediated depletion of VEGFR-2 was shown to completely block VEGF-induced phosphorylation of Akt in human retinal microvascular endothelial cells. Consequently, the proliferation, migration and tube formation of these cells in vitro was inhibited. This demonstrates the dependency of angiogenesis on the VEGFR-2-PI3K-Akt pathway.
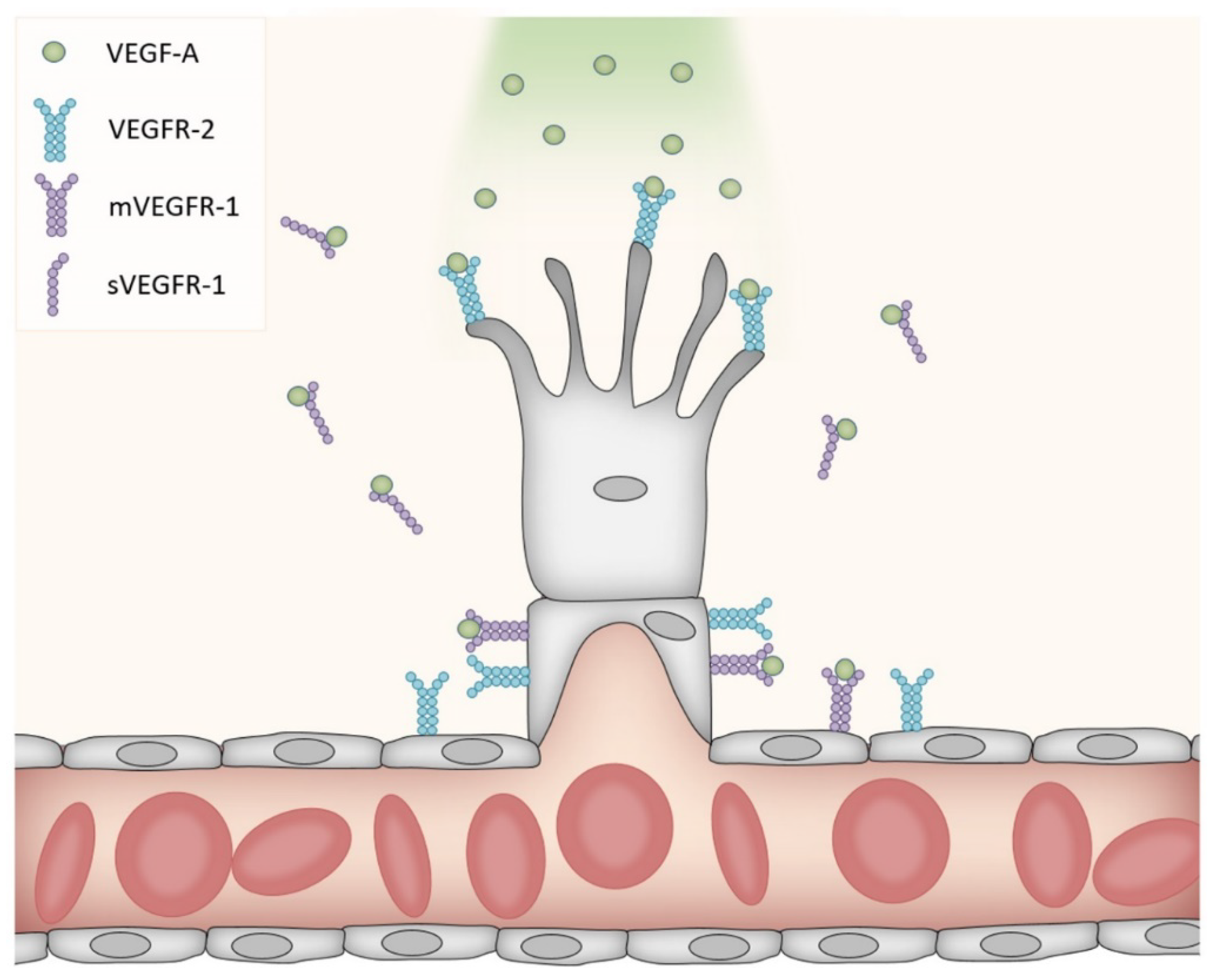
Further downstream mechanisms of phosphorylated Akt (pAkt) include the activation of nitric oxide synthase (NOS). This enzyme catalyzes the conversion of the amino acid l-arginine to nitric oxide (NO). Four isoforms of NOS are described: endothelial NOS (eNOS), inducible NOS (iNOS), neuronal NOS (nNOS) and mitochondrial NOS (mtNOS) [62,63]. pAkt-induced phosphorylation of eNOS at Ser1177, with the subsequent increase in NOS activity, may regulate cerebrovascular functions through several mechanism (for a review of eNOS in cerebrovascular diseases, see [64]). While the role of VEGFR-2 in angiogenesis is well described, the detailed mechanism involved in VEGFR-1 signaling is less known. A reduction of VEGFR-2-mediated pathways seems to be an important effect; alternative splicing of VEGFR-1 results in a membrane-bound form and a soluble form. The latter is secreted from endothelial cells and may modulate the amount of VEGF-A available for binding to VEGFR-2 [65,66]. In addition, VEGFR-1 in the membrane of endothelial cells antagonizes the angiogenic function of VEGFR-2 on the same cells, and VEGFR-1 activation thereby limits vascular growth [67,68]. VEGFR-1 on endothelial cells binds VEGF-A with high affinity, but displays low kinase activity. In fact, deleting the kinase domain without affecting the ligand binding region produced no detectable abnormalities in the density of blood vessels [69]. However, the genetic deletion of VEGFR-1 led to vessel overgrowth and the formation of dysfunctional vessels [68,70]. VEGFR-1 knock-out mice died early in the embryonal period [69], highlighting the importance of VEGFR-1 in addition to VEGFR-2 for proper vascularization. While the secreted VEGFR-1 isoform, but not the membrane-bound isoform, regulates branching [71], both isoforms regulate the mitotic properties of endothelial cells. The current belief is therefore that the secreted VEGFR-1 inactivates VEGF-A on both sides of the sprout, thereby providing the path of higher VEGF-A concentration that guides the sprouting vessel in the proper direction (Figure 1).
Interestingly, VEGF-mediated angiogenesis seems not to be restricted to the ischemia area, as an increase in VEGF-A and a corresponding vascularization have been observed even in the contralesional hemisphere. In fact, Wang et al. [72] found that VEGF-A-induced angiogenesis may lead to a hemodynamic steal phenomena where blood flow is reduced in the ischemic areas but increased in areas outside the lesion [72]. They suggest that VEGF-A protects neurons from ischemic cell death by a direct action on the neurons rather than only by promoting angiogenesis.
In addition to the endothelial cells, other cell types also express VEGF-A receptors, and may contribute to angiogenesis. Pericytes, the contractile cells that wrap around the abluminal surface of the endothelial cells of the vessels, also express VEGFRs. These cells are suggested to play a role in the formation of stable vascular networks. The inhibition of pericyte-specific VEGFR-1 signaling results in the loss of branches and the enlargement of vessels, suggesting that pericytes promote endothelial sprouting [73]. This has only been reported in the retina; the extent to which the pericytes regulate angiogenesis in the brain in response to stroke remains to be investigated.
3.2.2. Effects of VEGF-A on Vasodilation
Outside the central nervous system (CNS), VEGF-A has been shown to have a vasodilative effect, increasing blood flow when expressed under ischemic conditions. For instance, in an ischemic limb model in rabbits, it was shown that the co-application of VEGF-A with serotonin in the iliac artery increased blood flow by more than 100% [74]. In isolated coronary arteries, VEGF-A leads to a slow rise of cytosolic calcium in endothelial cells and an endothelium-dependent relaxation of the arteries [75]. As described above, VEGF may activate the VEGFR-2–PI3K–Akt–eNOS pathway to induce angiogenesis. However, the same pathway is also involved in more acute effects. For instance, eNOS is responsible for vasodilation after hypoxia/ischemia, leading to increased cerebral blood flow [76]. This effect is believed to be mediated through cyclic guanosine monophosphate (cGMP), which escapes from the endothelial cells and causes the relaxation of smooth muscle cells in the vicinity [77]. Furthermore, a systematic review of the effects of NO donors in animals models of stroke [78] concluded that NO donors improved cerebral blood flow and decreased the infarction volume. Further demonstrating the relationship between eNOS and stroke progression, eNOS knock-out mice displayed decreased cerebral blood flow and developed larger cerebral infarctions than wild-type mice [79]. Furthermore, during the first 30 min after a middle cerebral artery occlusion (MCAO) in rats, the administration of the NO precursor l-arginine, or NO donors (sodium nitroprusside (SNP) and 3-morpholino sydnonimine) improved cerebral blood flow and prevented tissue necrosis [79,80,81]. Even though high levels of VEGFR-2 and eNOS were reported 1–3 days after MCAO [82], an acute increase in blood flow in the brain in response to VEGF-A was not detected [83,84]. One explanation may be that a possible vasodilative effect of eNOS is counteracted by capillary pericyte constriction in ischemia [85].
3.2.3. Acute Effects of VEGF-A on Vasopermeability
Increased vascular permeability is an early event in stroke. Leaky blood vessels are known to induce edema, which in turn hampers perfusion and therefore result in more pronounced neuronal death [86]. This effect is largely mediated through the action of VEGF-A–VEGFR-2 and the Src pathway, although activation of the PI3K–Akt–eNOS pathway also plays a role in the increased permeability of the BBB seen in acute stroke [87]. The non-receptor tyrosine kinase Src is transiently upregulated in the ischemic brain as early as 3 h after reperfusion [33]. The family of Src kinases consists of proto-oncogenic, non-receptor tyrosine kinases. Src-activation is regulated by a number of different signals, including VEGF-A receptor actions. An increase in Src phosphorylation during the acute phase of ischemia is associated with VEGF-induced vascular permeability [33,88,89]. Src activation then returns to basal levels within the first day, before a second increase occurs 3–7 days after reperfusion [33]. The association of the Src pathway with VEGF-A seems to be bidirectional: under ischemic conditions; Src can regulate the expression of VEGF-A [90], as inhibition of Src decreases VEGF-A levels [33] and consequently VEGF-A-induced vascular permeability [91]. The result is reduced brain edema and reduced lesion volume [92,93]. On the other hand, Src knock-out mice as well as wild-type mice treated with an Src inhibitor are resistant to VEGF-induced vasopermeability and edema [88,91,94]. The latter findings indicate that Src acts downstream of VEGF-A as opposed to the other way around. Activation of the VEGF-A–Src pathway may underlie the unfavorable effects of VEGF-A in the early treatment of stroke. One important VEGF-A–VEGRF-2–Src-mediated mechanism that underlies vasopermeability is the regulation of the adhesion junctions and tight junctions between endothelial cells (Figure 2). For instance, VEGF-A triggers endocytosis of a key cell-adhesion molecule, VE-cadherin, via a VEGFR-2–Src pathway that involves the subsequent phosphorylation of the small GTP-binding protein Rac and the GTPase-activated kinase PAK (p21 activated kinase). Activated PAK phosphorylates the internal tail of VE-cadherin, leading to its internalization and subsequently the disruption of the intercellular junctions of the BBB [95,96,97,98]. In addition to endothelial cell-derived VEGF-A, astrocyte-derived VEGF-A may also contribute to BBB leakage in early stroke, as it has been demonstrated in cultures that ischemic neurons activate astrocytes to increase their VEGF-A production, which in turn induces endothelial barrier disruption [99].
In the context of VEGF-A-mediated BBB disruption early in stroke, inflammation may be an important factor. The neuroinflammatory response after stroke contributes to neural damage, but also plays an important role in neurogenesis, as reviewed in [100]. As mentioned above, VEGF-A seems to be upregulated in response to inflammatory cytokines in the CNS [42,43]. Growing evidence suggests that the ERK pathway contributes to neuroinflammation and neuronal death in ischemic stroke [101], possibly via the regulation of pro-inflammatory cytokines [102]. More research is needed to unravel the direct involvement of VEGF-A in neuroinflammation after stroke.
3.2.4. Effects of VEGF-A on Neuroprotection
Despite the name, VEGF-A does not only act on the vascular endothelium. Instead, VEGF-A acts on several other cell types, including neurons [103]. This has been demonstrated in numerous studies using a diverse array of neuronal preparations [30,104,105,106,107,108]. VEGF-A promotes neuronal survival in cell culture models of stroke, including the oxygen-and-glucose deprivation model [105] and the excitotoxicity model [109,110]. Most of these direct neuronal effects of VEGF-A have been ascribed to the activation of the PI3K-Akt pathway described above, and the mitogen-activated protein kinase (MAPK) cascade. The latter involves the MAP kinase kinase (MEK) and its effector MAP kinase/extracellular signal-regulated kinase (ERK). Both pathways are activated by several signals, including in response to VEGF-A-induced activation of VEGFR-2 [58]. The effect of the MEK–ERK pathway on neuroprotection remains controversial, as the stimulation of cell growth and proliferation [111] as well as neuronal death [112,113,114,115] by this pathway have been described. In vivo, neuroprotective effects of VEGF-A have also been demonstrated in MCAO models of stroke. Local application of VEGF-A to the surface of the reperfused brain reduced the infarction volume in rats [116]. Further demonstrating a protective effect of VEGF-A, the intraventricular infusion of an anti-VEGF-A antibody led to an increased lesion volume [117]. From the in vivo studies, it is not possible to distinguish the direct protective actions of VEGF-A acting on neuronal VEGFRs from the indirect effects mediated through the endothelial VEGFRs.
3.2.5. Neurogenesis
Neurogenesis in the adult brain occurs in two niches: the subventricular zone (SVZ) of the lateral ventricles and the subgranular zone (SGZ) of the dentate gyrus. Although a recent study [118] challenged the concept of adult neurogenesis in the SGZ of humans, most studies show that both niches are sources of neurogenesis throughout adulthood [119,120,121,122,123,124]. Cerebral ischemia stimulates neurogenesis in both of these niches [125,126]. An increased VEGF-A level is probably an important elicitor, as enhanced VEGF-A alone induces neurogenesis in both of these regions [127,128,129]. In transgenic mice that overexpress VEGF-A, not only neurogenesis, but also the migration of newly formed neurons to the peri-infarcted cortex, is increased [128]. This suggests that VEGF-A-induced neurogenesis can replace some of the neurons that die during a stroke. Many reports of neurogenesis describe increased levels of the neural proliferation marker BrdU and the immature neuronal marker doublecortin in the dentate gyrus of the hippocampus as a result of increased VEGF [130,131,132]. Hippocampal neural stem and progenitor cells (NSPS) even may produce VEGF-A in order to maintain the NSPC pool in the subgranular zone [133]. The proliferative actions of VEGF-A seem to require the activation of both ERK and Akt signaling cascades [132].
VEGFR-2 is the main VEGF-A receptor involved in neurogenesis [105,130,134,135]. After cerebral ischemia, neuroblasts expressing VEGFR-2 migrate along vessels in the ischemic area. Furthermore, the blockage of VEGFR-2 reduced neurogenesis in an animal model of stroke [134]. VEGF-A stimulated the expansion of neural stem cells, whereas the blockage of VEGFR-2 activity reduced neural stem cell expansion [135]. Increased numbers of migrating and developing neurons in the penumbra correlated with VEGF-A and VEGFR-2 [49]. VEGF-A is colocalised with the DNA repair factor ERCC6 in neurons but not in astrocytes after MCAO [136], suggesting a direct role in neuronal repair. The inhibition of astrocytes with fluorocitrate reduces VEGF-A-mediated increases in neuronal proliferation markers in newborn neurons after MCAO, suggesting that the VEGF-mediated increase of newly generated neurons is caused by the transdifferentiation of astrocytes into neurons [137].
3.3. VEGF-A in Stroke Prevention—Exercise and Preconditioning
3.3.1. Timing and Dosage
As described, VEGF-A induces both detrimental (BBB disruption) and beneficial (angiogenesis, neuroprotection and neurogenesis) processes in the ischemic brain. Therefore, whether VEGF-A is neuroprotective or neurotoxic depends on which of these processes dominate. The timing, the dosage and even on the route of administration of VEGF-A after stroke have an influence on the outcome (Figure 3).
Excessive levels of VEGF-A early after stroke increase BBB leakage in the ischemic brain, causing edema and subsequently elevated intracranial pressure that obstruct blood supply (as presented above). In addition, leaky vessels in the penumbra disturb the homeostasis of the nervous tissue, as the molecules and immune cells that are normally prohibited from entering the brain may now pass the BBB. Together, these mechanisms may aggravate neural damage [11,57]. To avoid detrimental effects, intravenous VEGF-A should not be administered between 1–3 and 24 h after stroke onset [11,57]. VEGF-A application later than one day after stroke onset seems to always lead to neuroprotection, increased vascular volume, decreased lesion volume, enhanced neural cell proliferation; even behavioral recovery from stroke is improved [8,127,138].
The route of VEGF-A administration seems to make a difference, as topical (directly on the cortical stroke area) or intracerebroventricular application prevents neural damage as well as BBB leakage, even when applied early after stroke [57,116]. Systemic administration of VEGF-A, as described above, often causes more of the unwanted effects. Furthermore, it has been shown that low doses (less than 2.4 ng/day) infused into the internal carotid artery do not affect endothelial proliferation or changes in vascularization [135,139] and that high doses (~10 ng/day) may lead to neural damage, despite eliciting increased vascularization [139].
3.3.2. Hypoxic/Ischemic Preconditioning
The central principle in preconditioning is that mild forms of stress induce tolerance to an otherwise lethal injury (reviewed in [140]). Hypoxic/ischemic preconditioning means that a brief episode or a mild form of hypoxia/ischemia prior to a stroke will reduce the damage produced by the stroke, and was first described by Kitagawa and co-workers [141]. In laboratory animals, this type of preconditioning is well known to increase the resistance of the brain to hypoxic/ischemic insult [142,143,144,145,146,147]. However, the translation of the preconditioning research to the human setting is challenging. First of all, the average stroke patient is elderly, often suffers from other diseases and uses medications, all of which are factors that may influence the efficacy of the preconditioning. Secondly, the fact that a stroke may occur without warning makes it difficult to administer the preconditioning at a suitable time prior to the stroke. For these reasons, preconditioning in the prevention of stroke has a limited place in clinical practice and the optimal preconditioning strategy remains to be established.
The underlying mechanisms of hypoxia/ischemia-induced preconditioning involve an increase in HIF-1α [148] and its target genes EPO and VEGF, leading to vascularization [36,149]. Hypoxic preconditioning elicits HIF-1α-dependent upregulation of genes, including VEGF-A, not only during the preconditioning period, but also at an elevated rate during a subsequent ischemia, suggesting that the treatment modifies the brain’s genomic response to ischemia [150]. Ischemic preconditioning in vivo has been shown to protect the hippocampus from ischemic/reperfusion damage by increasing both the expression of VEGF-A and VEGFR-2 [151]. Hypoxic preconditioning in vitro leads to increased levels of VEGF-A, VEGFR-2, pAkt, and pERK in neurons, and the inhibition of VEGFR-2 negates the activation of Akt [152]. Elevated levels of VEGF-A are associated with an increase in collateral formation [153,154], reducing the extent of perfusion-loss in stroke. In line with this, preconditioning by VEGF-A injections increases cerebral perfusion, reduces stroke-induced neural damage [155], and increases neurogenesis even for months after the treatment [156]. Furthermore, the preconditioning event does not need to be present in the organ that is protected, as remote ischemic preconditioning (rIPC) also protects organs from ischemic damage by raising systemic VEGF-A levels [157]. Laboratory experiments have shown that rIPC reduces brain infarction [148,158,159,160]. Two pilot clinical trials have confirmed that rIPC is feasible in people at risk of stroke, and significantly reduces stroke prevalence [161,162].
The fact that a mild or short ischemic/hypoxic event can initiate protective mechanisms that prepare the brain for a larger event of the same kind is comprehensible. However, it appears that most events that produce mild stress in the brain induce ischemic tolerance, regardless of the type of stress [163,164,165,166]. Volatile anaesthetics, for example, induce ischemic tolerance without causing hypoxia/ischemia (reviewed in [167]). The mechanism behind the protective effects of volatile anaesthetics is largely unknown, but HIF-1α seems to be a key mediator also in this context. The involvement of VEGF-A during preconditioning with volatile anaesthetics is also unknown. One study suggested that an increase of VEGF in the acute phase of ischemia after such preconditioning may underlie part of the protective effect [168].
In summary, the HIF-1α–VEGF-A–VEGFR-2–Akt pathway is part of the protective mechanism in hypoxic preconditioning, but one has to keep in mind that a number of additional factors including a number of additional HIF target genes and heat-shock proteins are also involved [169].
3.3.3. Exercise
Exercise is one of the best preventive strategies in stroke, as it induces some of the same mechanisms as seen in hypoxic/ischemic preconditioning. Therefore, exercise may be seen as a means of preconditioning in itself. Pre-ischemic exercise leads to increased VEGF-mediated angiogenesis and reduced brain damage after ischemic stroke [170,171,172,173,174]. The underlying mechanisms are not completely understood, but an increase in eNOS seems to be important [175,176,177,178]. Exercise and oxygen-glucose-deprivation (OGD) induce VEGF-A/VEGFR-2-mediated cAMP response element-binding protein (CREB) phosphorylation as a shared pathway in the protection of both endothelial cells and neurons [179]. In animal studies, treadmill exercise has been reported to be more efficient than exercise in running wheels at inducing protection against stroke [180], suggesting that higher intensities are needed. Lactate, a partial exercise mimetic [181] may be involved. We have recently shown that the lactate receptor HCAR-1 in the brain [182] is responsible for the increased VEGF-A levels and angiogenesis induced by exercise or lactate injections [44].
4. VEGF-A in Human Cerebral Stroke
Despite a number of publications discussing the clinical use of VEGF in cerebral stroke, the data supporting a protective role of VEGF-A is almost exclusively based on animal models. Clinical studies are sparse and mainly focus on intrinsic VEGF levels as biomarkers for progression after cerebral stroke. Serum VEGF-A levels in humans increase after stroke [183,184], however, how VEGF-A levels correlate to the severity of the stroke remains to be elucidated. One study found that increased VEGF-A levels might be used as a predictor for improved stoke recovery [185]. Another study, however, found that VEGF-A levels correlated positively with stroke severity in cardioembolic infarction, while a negative correlation to neurological severity was found in atherothrombotic infarction [184]. The current data therefore makes it difficult to determine in which settings VEGF-A manipulation would be beneficial. Additional clinical studies have been performed (e.g., ClinicalTrials.gov identifier: NCT02157896 and NCT00134433), but the results have not been published yet. The challenge in using VEGF-A manipulation in clinical trials probably lies in the multifaceted action of VEGF-A described above. Before VEGF-A can be safely manipulated in a clinical setting, the time window in which the beneficial effects of increased VEGF-A outweigh the detrimental effects needs to be identified. The same is true for the optimal dosage and the route of administration.
5. Conclusions
Stroke results from the occlusion of a precerebral or cerebral artery or an intracerebral hemorrhage, both of which lead to focal hypoxia/ischemia and necrosis of the ischemic strike core. Cells in the penumbra may be rescued if adequate perfusion is restored in time. Growth factors affect the recovery after stroke. VEGF-A is a key regulator of angiogenesis, neuroprotection, and neurogenesis. In animal models of stroke, treatment with VEGF-A per se, or with medications that augment VEGF-A effects, reduces the lesion volume. However, VEGF-A treatment has shown somewhat inconsistent results. The timing of the VEGF-A increase as well as the route of administration are important factors to consider when judging the effectiveness of VEGF-A treatment in stroke. During the acute phase, increased VEGF-A induces BBB breakdown and vascular leakage, which lead to disturbed homeostasis, the invasion of peripheral immune cells, and edema. These harmful effects of VEGF-A on vessel integrity are transient, as both VEGF-A preconditioning and increased VEGF-A after the acute phase has a neuroprotective effect (Figure 3). VEGF-A therefore has a Janus face in the treatment of stroke. Further investigations are needed to increase the safety of VEGF-A treatment and to find strategies to enhance the angiogenic, neuroprotective and neurogenic properties, while avoiding the detrimental effects.
Acknowledgments
The present work was supported by the Research Council of Norway, grant # 262647.
Conflicts of Interest
The authors declare no conflict of interest.
Abbreviations
| VEGF | Vascular Endothelial Growth Factor |
| ERK | Extracellular Signal-Regulated Kinase |
| PI3K | Phosphoinositide 3-Kinase |
| HIF | Hypoxia Inducible Factor |
References
- Baron, J.-C.; Yamauchi, H.; Fujioka, M.; Endres, M. Selective neuronal loss in ischemic stroke and cerebrovascular disease. J. Cereb. Blood Flow Metab. 2014, 34, 2–18. [Google Scholar] [CrossRef] [PubMed]
- Krupinski, J.; Kaluza, J.; Kumar, P.; Wang, M.; Kumar, S. Prognostic value of blood vessel density in ischaemic stroke. Lancet 1993, 342, 742. [Google Scholar] [CrossRef]
- Marchal, G.; Serrati, C.; Rioux, P.; Petit-Taboue, M.C.; Viader, F.; de la Sayette, V.; Le Doze, F.; Lochon, P.; Derlon, J.M.; Orgogozo, J.M.; et al. Pet imaging of cerebral perfusion and oxygen consumption in acute ischaemic stroke: Relation to outcome. Lancet 1993, 341, 925–927. [Google Scholar] [CrossRef]
- Krupinski, J.; Kaluza, J.; Kumar, P.; Kumar, S.; Wang, J.M. Role of angiogenesis in patients with cerebral ischemic stroke. Stroke 1994, 25, 1794–1798. [Google Scholar] [CrossRef] [PubMed]
- Namiecinska, M.; Marciniak, K.; Nowak, J.Z. VEGF as an angiogenic, neurotrophic, and neuroprotective factor. Postepy Hig. Med. Dosw. 2005, 59, 573–583. [Google Scholar]
- Gora-Kupilas, K.; Josko, J. The neuroprotective function of vascular endothelial growth factor (VEGF). Folia Neuropathol. 2005, 43, 31–39. [Google Scholar] [PubMed]
- Cao, L.; Jiao, X.; Zuzga, D.S.; Liu, Y.; Fong, D.M.; Young, D.; During, M.J. VEGF links hippocampal activity with neurogenesis, learning and memory. Nat. Genet. 2004, 36, 827–835. [Google Scholar] [CrossRef] [PubMed]
- Dzietko, M.; Derugin, N.; Wendland, M.F.; Vexler, Z.S.; Ferriero, D.M. Delayed VEGF treatment enhances angiogenesis and recovery after neonatal focal rodent stroke. Transl. Stroke Res. 2013, 4, 189–200. [Google Scholar] [CrossRef] [PubMed]
- Nishijima, K.; Ng, Y.-S.; Zhong, L.; Bradley, J.; Schubert, W.; Jo, N.; Akita, J.; Samuelsson, S.J.; Robinson, G.S.; Adamis, A.P.; et al. Vascular endothelial growth factor-a is a survival factor for retinal neurons and a critical neuroprotectant during the adaptive response to ischemic injury. Am. J. Pathol. 2007, 171, 53–67. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Zechariah, A.; Qu, Y.; Hermann, D.M. Effects of vascular endothelial growth factor in ischemic stroke. J. Neurosci. Res. 2012, 90, 1873–1882. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.G.; Zhang, L.; Jiang, Q.; Zhang, R.; Davies, K.; Powers, C.; Bruggen, N.V.; Chopp, M. VEGF enhances angiogenesis and promotes blood-brain barrier leakage in the ischemic brain. J. Clin. Investig. 2000, 106, 829–838. [Google Scholar] [CrossRef] [PubMed]
- Weis, S.M.; Cheresh, D.A. Pathophysiological consequences of VEGF-induced vascular permeability. Nature 2005, 437, 497–504. [Google Scholar] [CrossRef] [PubMed]
- Van Bruggen, N.; Thibodeaux, H.; Palmer, J.T.; Lee, W.P.; Fu, L.; Cairns, B.; Tumas, D.; Gerlai, R.; Williams, S.-P.; Campagne, M.V.L.; et al. Vegf antagonism reduces edema formation and tissue damage after ischemia/reperfusion injury in the mouse brain. J. Clin. Investig. 1999, 104, 1613–1620. [Google Scholar] [CrossRef] [PubMed]
- Grassot, J.; Gouy, M.; Perriere, G.; Mouchiroud, G. Origin and molecular evolution of receptor tyrosine kinases with immunoglobulin-like domains. Mol. Biol. Evol. 2006, 23, 1232–1241. [Google Scholar] [CrossRef] [PubMed]
- Ruiz de Almodovar, C.; Lambrechts, D.; Mazzone, M.; Carmeliet, P. Role and therapeutic potential of VEGF in the nervous system. Physiol. Rev. 2009, 89, 607–648. [Google Scholar] [CrossRef] [PubMed]
- Simons, M.; Gordon, E.; Claesson-Welsh, L. Mechanisms and regulation of endothelial VEGF receptor signalling. Nat. Rev. Mol. Cell Biol. 2016, 17, 611–625. [Google Scholar] [CrossRef] [PubMed]
- Park, J.E.; Chen, H.H.; Winer, J.; Houck, K.A.; Ferrara, N. Placenta growth factor. Potentiation of vascular endothelial growth factor bioactivity, in vitro and in vivo, and high affinity binding to flt-1 but not to flk-1/KDR. J. Biol. Chem. 1994, 269, 25646–25654. [Google Scholar] [PubMed]
- Meyer, R.D.; Mohammadi, M.; Rahimi, N. A single amino acid substitution in the activation loop defines the decoy characteristic of VEGFR-1/flt-1. J. Biol. Chem. 2006, 281, 867–875. [Google Scholar] [CrossRef] [PubMed]
- Maharaj, A.S.; Saint-Geniez, M.; Maldonado, A.E.; D’Amore, P.A. Vascular endothelial growth factor localization in the adult. Am. J. Pathol. 2006, 168, 639–648. [Google Scholar] [CrossRef] [PubMed]
- Sun, F.J.; Wei, Y.J.; Li, S.; Guo, W.; Chen, X.; Liu, S.Y.; He, J.J.; Yin, Q.; Yang, H.; Zhang, C.Q. Elevated expression of VEGF-c and its receptors, VEGFR-2 and VEGFR-3, in patients with mesial temporal lobe epilepsy. J. Mol. Neurosci. 2016, 59, 241–250. [Google Scholar] [CrossRef] [PubMed]
- Ferrara, N.; Gerber, H.P.; LeCouter, J. The biology of VEGF and its receptors. Nat. Med. 2003, 9, 669–676. [Google Scholar] [CrossRef] [PubMed]
- Krum, J.M.; Mani, N.; Rosenstein, J.M. Angiogenic and astroglial responses to vascular endothelial growth factor administration in adult rat brain. Neuroscience 2002, 110, 589–604. [Google Scholar] [CrossRef]
- Marti, H.J.; Bernaudin, M.; Bellail, A.; Schoch, H.; Euler, M.; Petit, E.; Risau, W. Hypoxia-induced vascular endothelial growth factor expression precedes neovascularization after cerebral ischemia. Am. J. Pathol. 2000, 156, 965–976. [Google Scholar] [CrossRef]
- Issa, R.; Krupinski, J.; Bujny, T.; Kumar, S.; Kaluza, J.; Kumar, P. Vascular endothelial growth factor and its receptor, KDR, in human brain tissue after ischemic stroke. Lab. Investig. 1999, 79, 417–425. [Google Scholar] [PubMed]
- Stowe, A.M.; Plautz, E.J.; Eisner-Janowicz, I.; Frost, S.B.; Barbay, S.; Zoubina, E.V.; Dancause, N.; Taylor, M.D.; Nudo, R.J. Vegf protein associates to neurons in remote regions following cortical infarct. J. Cereb. Blood Flow Metab. 2007, 27, 76–85. [Google Scholar] [CrossRef] [PubMed]
- Stowe, A.M.; Plautz, E.J.; Nguyen, P.; Frost, S.B.; Eisner-Janowicz, I.; Barbay, S.; Dancause, N.; Sensarma, A.; Taylor, M.D.; Zoubina, E.V.; et al. Neuronal HIF-1α protein and VEGFR-2 immunoreactivity in functionally related motor areas following a focal m1 infarct. J. Cereb. Blood Flow Metab. 2008, 28, 612–620. [Google Scholar] [CrossRef] [PubMed]
- Guan, W.; Somanath, P.R.; Kozak, A.; Goc, A.; El-Remessy, A.B.; Ergul, A.; Johnson, M.H.; Alhusban, A.; Soliman, S.; Fagan, S.C. Vascular protection by angiotensin receptor antagonism involves differential VEGF expression in both hemispheres after experimental stroke. PLoS ONE 2011, 6, e24551. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.G.; Zhang, L.; Tsang, W.; Soltanian-Zadeh, H.; Morris, D.; Zhang, R.; Goussev, A.; Powers, C.; Yeich, T.; Chopp, M. Correlation of VEGF and angiopoietin expression with disruption of blood–brain barrier and angiogenesis after focal cerebral ischemia. J. Cereb. Blood Flow Metab. 2002, 22, 379–392. [Google Scholar] [CrossRef] [PubMed]
- Margaritescu, O.; Pirici, D.; Margaritescu, C. Vegf expression in human brain tissue after acute ischemic stroke. Rom. J. Morphol. Embryol. 2011, 52, 1283–1292. [Google Scholar] [PubMed]
- Khaibullina, A.A.; Rosenstein, J.M.; Krum, J.M. Vascular endothelial growth factor promotes neurite maturation in primary CNS neuronal cultures. Brain Res. Dev. Brain Res. 2004, 148, 59–68. [Google Scholar] [CrossRef] [PubMed]
- Krum, J.M.; Khaibullina, A. Inhibition of endogenous VEGF impedes revascularization and astroglial proliferation: Roles for VEGF in brain repair. Exp. Neurol. 2003, 181, 241–257. [Google Scholar] [CrossRef]
- Lee, M.Y.; Ju, W.K.; Cha, J.H.; Son, B.C.; Chun, M.H.; Kang, J.K.; Park, C.K. Expression of vascular endothelial growth factor MRNA following transient forebrain ischemia in rats. Neurosci. Lett. 1999, 265, 107–110. [Google Scholar] [CrossRef]
- Zan, L.; Zhang, X.; Xi, Y.; Wu, H.; Song, Y.; Teng, G.; Li, H.; Qi, J.; Wang, J. Src regulates angiogenic factors and vascular permeability after focal cerebral ischemia-reperfusion. Neuroscience 2014, 262, 118–128. [Google Scholar] [CrossRef] [PubMed]
- Josko, J.; Mazurek, M. Transcription factors having impact on vascular endothelial growth factor (VEGF) gene expression in angiogenesis. Med. Sci. Monit. 2004, 10, Ra89–Ra98. [Google Scholar] [PubMed]
- Dery, M.A.; Michaud, M.D.; Richard, D.E. Hypoxia-inducible factor 1: Regulation by hypoxic and non-hypoxic activators. Int. J. Biochem. Cell Biol. 2005, 37, 535–540. [Google Scholar] [CrossRef] [PubMed]
- Bernaudin, M.; Nedelec, A.-S.; Divoux, D.; MacKenzie, E.T.; Petit, E.; Schumann-Bard, P. Normobaric hypoxia induces tolerance to focal permanent cerebral ischemia in association with an increased expression of hypoxia-inducible factor-1 and its target genes, erythropoietin and VEGF, in the adult mouse brain. J. Cereb. Blood Flow Metab. 2002, 22, 393–403. [Google Scholar] [CrossRef] [PubMed]
- Oosthuyse, B.; Moons, L.; Storkebaum, E.; Beck, H.; Nuyens, D.; Brusselmans, K.; Van Dorpe, J.; Hellings, P.; Gorselink, M.; Heymans, S.; et al. Deletion of the hypoxia-response element in the vascular endothelial growth factor promoter causes motor neuron degeneration. Nat. Genet. 2001, 28, 131–138. [Google Scholar] [CrossRef] [PubMed]
- Hu, C.-J.; Wang, L.-Y.; Chodosh, L.A.; Keith, B.; Simon, M.C. Differential roles of hypoxia-inducible factor 1α (HIF-1α) and HIF-2α in hypoxic gene regulation. Mol. Cell. Biol. 2003, 23, 9361–9374. [Google Scholar] [CrossRef] [PubMed]
- Manalo, D.J.; Rowan, A.; Lavoie, T.; Natarajan, L.; Kelly, B.D.; Ye, S.Q.; Garcia, J.G.; Semenza, G.L. Transcriptional regulation of vascular endothelial cell responses to hypoxia by HIF-1. Blood 2005, 105, 659–669. [Google Scholar] [CrossRef] [PubMed]
- Fandrey, J. Oxygen-dependent and tissue-specific regulation of erythropoietin gene expression. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2004, 286, R977–R988. [Google Scholar] [CrossRef] [PubMed]
- Xiong, Y.; Zhang, Y.; Mahmood, A.; Meng, Y.; Qu, C.; Chopp, M. Erythropoietin mediates neurobehavioral recovery and neurovascular remodeling following traumatic brain injury in rats by increasing expression of vascular endothelial growth factor. Transl. Stroke Res. 2011, 2, 619–632. [Google Scholar] [CrossRef] [PubMed]
- You, T.; Bi, Y.; Li, J.; Zhang, M.; Chen, X.; Zhang, K.; Li, J. IL-17 induces reactive astrocytes and up-regulation of vascular endothelial growth factor (VEGF) through JAK/STAT signaling. Sci. Rep. 2017, 7, 41779. [Google Scholar] [CrossRef] [PubMed]
- Nagineni, C.N.; Kommineni, V.K.; William, A.; Detrick, B.; Hooks, J.J. Regulation of VEGF expression in human retinal cells by cytokines: Implications for the role of inflammation in age-related macular degeneration. J. Cell. Physiol. 2012, 227, 116–126. [Google Scholar] [CrossRef] [PubMed]
- Morland, C.; Andersson, K.A.; Haugen, O.P.; Hadzic, A.; Kleppa, L.; Gille, A.; Rinholm, J.E.; Palibrk, V.; Diget, E.H.; Kennedy, L.H.; et al. Exercise induces cerebral VEGF and angiogenesis via the lactate receptor hcar1. Nat. Commun. 2017, 8, 15557. [Google Scholar] [CrossRef] [PubMed]
- Thau-Zuchman, O.; Shohami, E.; Alexandrovich, A.G.; Leker, R.R. Vascular endothelial growth factor increases neurogenesis after traumatic brain injury. J. Cereb. Blood Flow Metab. 2010, 30, 1008–1016. [Google Scholar] [CrossRef] [PubMed]
- Larson, J.; Drew, K.L.; Folkow, L.P.; Milton, S.L.; Park, T.J. No oxygen? No problem! Intrinsic brain tolerance to hypoxia in vertebrates. J. Exp. Biol. 2014, 217, 1024–1039. [Google Scholar] [CrossRef] [PubMed]
- Xiao, B.; Wang, S.; Yang, G.; Sun, X.; Zhao, S.; Lin, L.; Cheng, J.; Yang, W.; Cong, W.; Sun, W.; et al. HIF-1α contributes to hypoxia adaptation of the naked mole rat. Oncotarget 2017, 8, 109941–109951. [Google Scholar] [CrossRef] [PubMed]
- Manoonkitiwongsa, P.S. Critical questions for preclinical trials on safety and efficacy of vascular endothelial growth factor-based therapeutic angiogenesis for ischemic stroke. CNS Neurol. Disord. Drug Targets 2011, 10, 215–234. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Zhang, C.; Jiang, H.; Li, Y.; Zhang, L.; Robin, A.; Katakowski, M.; Lu, M.; Chopp, M. Atorvastatin induction of VEGF and BDNF promotes brain plasticity after stroke in mice. J. Cereb. Blood Flow Metab. 2005, 25, 281–290. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.J.; Kim, K.S.; Park, I.H.; Kim, S.U. Human neural stem cells over-expressing VEGF provide neuroprotection, angiogenesis and functional recovery in mouse stroke model. PLoS ONE 2007, 2, e156. [Google Scholar] [CrossRef] [PubMed]
- Ruan, G.X.; Kazlauskas, A. Axl is essential for VEGF-A-dependent activation of PI3K/Akt. EMBO J. 2012, 31, 1692–1703. [Google Scholar] [CrossRef] [PubMed]
- Koch, S.; Claesson-Welsh, L. Signal transduction by vascular endothelial growth factor receptors. Cold Spring Harb. Perspect. Med. 2012, 2, a006502. [Google Scholar] [CrossRef] [PubMed]
- Kureishi, Y.; Luo, Z.; Shiojima, I.; Bialik, A.; Fulton, D.; Lefer, D.J.; Sessa, W.C.; Walsh, K. The HMG-COA reductase inhibitor simvastatin activates the protein kinase AKT and promotes angiogenesis in normocholesterolemic animals. Nat. Med. 2000, 6, 1004–1010. [Google Scholar] [CrossRef] [PubMed]
- Morales-Ruiz, M.; Fulton, D.; Sowa, G.; Languino, L.R.; Fujio, Y.; Walsh, K.; Sessa, W.C. Vascular endothelial growth factor-stimulated actin reorganization and migration of endothelial cells is regulated via the serine/threonine kinase AKT. Circ. Res. 2000, 86, 892–896. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Jiang, H.; Lu, D.; Qu, C.; Xiong, Y.; Zhou, D.; Chopp, M.; Mahmood, A. Induction of angiogenesis and modulation of vascular endothelial growth factor receptor-2 by simvastatin after traumatic brain injury. Neurosurgery 2011, 68, 1363–1371. [Google Scholar] [CrossRef] [PubMed]
- Radisavljevic, Z.; Avraham, H.; Avraham, S. Vascular endothelial growth factor up-regulates ICAM-1 expression via the phosphatidylinositol 3 OH-kinase/AKT/Nitric oxide pathway and modulates migration of brain microvascular endothelial cells. J. Biol. Chem. 2000, 275, 20770–20774. [Google Scholar] [CrossRef] [PubMed]
- Kaya, D.; Gursoy-Ozdemir, Y.; Yemisci, M.; Tuncer, N.; Aktan, S.; Dalkara, T. Vegf protects brain against focal ischemia without increasing blood–brain permeability when administered intracerebroventricularly. J. Cereb. Blood Flow Metab. 2005, 25, 1111–1118. [Google Scholar] [CrossRef] [PubMed]
- Kilic, E.; Kilic, U.; Wang, Y.; Bassetti, C.L.; Marti, H.H.; Hermann, D.M. The phosphatidylinositol-3 kinase/akt pathway mediates VEGF’s neuroprotective activity and induces blood brain barrier permeability after focal cerebral ischemia. FASEB J. 2006, 20, 1185–1187. [Google Scholar] [CrossRef] [PubMed]
- Dewil, M.; Lambrechts, D.; Sciot, R.; Shaw, P.J.; Ince, P.G.; Robberecht, W.; Van den Bosch, L. Vascular endothelial growth factor counteracts the loss of phospho-akt preceding motor neurone degeneration in amyotrophic lateral sclerosis. Neuropathol. Appl. Neurobiol. 2007, 33, 499–509. [Google Scholar] [CrossRef] [PubMed]
- Tolosa, L.; Mir, M.; Olmos, G.; Llado, J. Vascular endothelial growth factor protects motoneurons from serum deprivation-induced cell death through phosphatidylinositol 3-kinase-mediated p38 mitogen-activated protein kinase inhibition. Neuroscience 2009, 158, 1348–1355. [Google Scholar] [CrossRef] [PubMed]
- Wu, W.; Duan, Y.; Ma, G.; Zhou, G.; Park-Windhol, C.; D’Amore, P.A.; Lei, H. Aav-crispr/cas9-mediated depletion of VEGFR2 blocks angiogenesis in vitro. Investig. Ophthalmol. Vis. Sci. 2017, 58, 6082–6090. [Google Scholar] [CrossRef] [PubMed]
- Guix, F.X.; Uribesalgo, I.; Coma, M.; Munoz, F.J. The physiology and pathophysiology of nitric oxide in the brain. Prog. Neurobiol. 2005, 76, 126–152. [Google Scholar] [CrossRef] [PubMed]
- Andrew, P.J.; Mayer, B. Enzymatic function of nitric oxide synthases. Cardiovasc. Res. 1999, 43, 521–531. [Google Scholar] [CrossRef]
- Zhu, J.; Song, W.; Li, L.; Fan, X. Endothelial nitric oxide synthase: A potential therapeutic target for cerebrovascular diseases. Mol. Brain 2016, 9, 30. [Google Scholar] [CrossRef] [PubMed]
- Kendall, R.L.; Thomas, K.A. Inhibition of vascular endothelial cell growth factor activity by an endogenously encoded soluble receptor. Proc. Natl. Acad. Sci. USA 1993, 90, 10705–10709. [Google Scholar] [CrossRef] [PubMed]
- Roberts, D.M.; Kearney, J.B.; Johnson, J.H.; Rosenberg, M.P.; Kumar, R.; Bautch, V.L. The vascular endothelial growth factor (VEGF) receptor flt-1 (VEGFR-1) modulates flk-1 (VEGFR-2) signaling during blood vessel formation. Am. J. Pathol. 2004, 164, 1531–1535. [Google Scholar] [CrossRef]
- Chappell, J.C.; Taylor, S.M.; Ferrara, N.; Bautch, V.L. Local guidance of emerging vessel sprouts requires soluble flt-1. Dev. Cell 2009, 17, 377–386. [Google Scholar] [CrossRef] [PubMed]
- Kearney, J.B.; Ambler, C.A.; Monaco, K.A.; Johnson, N.; Rapoport, R.G.; Bautch, V.L. Vascular endothelial growth factor receptor flt-1 negatively regulates developmental blood vessel formation by modulating endothelial cell division. Blood 2002, 99, 2397–2407. [Google Scholar] [CrossRef] [PubMed]
- Hiratsuka, S.; Minowa, O.; Kuno, J.; Noda, T.; Shibuya, M. Flt-1 lacking the tyrosine kinase domain is sufficient for normal development and angiogenesis in mice. Proc. Natl. Acad. Sci. USA 1998, 95, 9349–9354. [Google Scholar] [CrossRef] [PubMed]
- Fong, G.H.; Rossant, J.; Gertsenstein, M.; Breitman, M.L. Role of the flt-1 receptor tyrosine kinase in regulating the assembly of vascular endothelium. Nature 1995, 376, 66–70. [Google Scholar] [CrossRef] [PubMed]
- Kappas, N.C.; Zeng, G.; Chappell, J.C.; Kearney, J.B.; Hazarika, S.; Kallianos, K.G.; Patterson, C.; Annex, B.H.; Bautch, V.L. The VEGF receptor flt-1 spatially modulates flk-1 signaling and blood vessel branching. J. Cell Biol. 2008, 181, 847–858. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Kilic, E.; Kilic, U.; Weber, B.; Bassetti, C.L.; Marti, H.H.; Hermann, D.M. VEGF overexpression induces post-ischaemic neuroprotection, but facilitates haemodynamic steal phenomena. Brain 2005, 128, 52–63. [Google Scholar] [CrossRef] [PubMed]
- Eilken, H.M.; Dieguez-Hurtado, R.; Schmidt, I.; Nakayama, M.; Jeong, H.W.; Arf, H.; Adams, S.; Ferrara, N.; Adams, R.H. Pericytes regulate VEGF-induced endothelial sprouting through VEGFR1. Nat. Commun. 2017, 8, 1574. [Google Scholar] [CrossRef] [PubMed]
- Bauters, C.; Asahara, T.; Zheng, L.P.; Takeshita, S.; Bunting, S.; Ferrara, N.; Symes, J.F.; Isner, J.M. Recovery of disturbed endothelium-dependent flow in the collateral-perfused rabbit ischemic hindlimb after administration of vascular endothelial growth factor. Circulation 1995, 91, 2802–2809. [Google Scholar] [CrossRef] [PubMed]
- Ku, D.D.; Zaleski, J.K.; Liu, S.; Brock, T.A. Vascular endothelial growth factor induces EDRF-dependent relaxation in coronary arteries. Am. J. Physiol. 1993, 265, H586–H592. [Google Scholar] [CrossRef] [PubMed]
- Bolanos, J.P.; Almeida, A. Roles of nitric oxide in brain hypoxia-ischemia. Biochim. Biophys. Acta 1999, 1411, 415–436. [Google Scholar] [CrossRef]
- Rees, D.D.; Palmer, R.M.; Moncada, S. Role of endothelium-derived nitric oxide in the regulation of blood pressure. Proc. Natl. Acad. Sci. USA 1989, 86, 3375–3378. [Google Scholar] [CrossRef] [PubMed]
- Willmot, M.; Gray, L.; Gibson, C.; Murphy, S.; Bath, P.M. A systematic review of nitric oxide donors and l-arginine in experimental stroke; effects on infarct size and cerebral blood flow. Nitric Oxide 2005, 12, 141–149. [Google Scholar] [CrossRef] [PubMed]
- Huang, Z.; Huang, P.L.; Ma, J.; Meng, W.; Ayata, C.; Fishman, M.C.; Moskowitz, M.A. Enlarged infarcts in endothelial nitric oxide synthase knockout mice are attenuated by nitro-l-arginine. J. Cereb. Blood Flow Metab. 1996, 16, 981–987. [Google Scholar] [CrossRef] [PubMed]
- Morikawa, E.; Moskowitz, M.A.; Huang, Z.; Yoshida, T.; Irikura, K.; Dalkara, T. L-arginine infusion promotes nitric oxide-dependent vasodilation, increases regional cerebral blood flow, and reduces infarction volume in the rat. Stroke 1994, 25, 429–435. [Google Scholar] [CrossRef] [PubMed]
- Salom, J.B.; Orti, M.; Centeno, J.M.; Torregrosa, G.; Alborch, E. Reduction of infarct size by the no donors sodium nitroprusside and spermine/no after transient focal cerebral ischemia in rats. Brain Res. 2000, 865, 149–156. [Google Scholar] [CrossRef]
- Moisan, A.; Favre, I.M.; Rome, C.; Grillon, E.; Naegele, B.; Barbieux, M.; De Fraipont, F.; Richard, M.J.; Barbier, E.L.; Rémy, C.; et al. Microvascular plasticity after experimental stroke: A molecular and MRI study. Cerebrovasc. Dis. 2014, 38, 344–353. [Google Scholar] [CrossRef] [PubMed]
- Harrigan, M.R.; Ennis, S.R.; Sullivan, S.E.; Keep, R.F. Effects of intraventricular infusion of vascular endothelial growth factor on cerebral blood flow, edema, and infarct volume. Acta Neurochir. (Wien.) 2003, 145, 49–53. [Google Scholar] [CrossRef] [PubMed]
- Takeuchi, H.; Tomita, H.; Taki, Y.; Kikuchi, Y.; Ono, C.; Yu, Z.; Sekiguchi, A.; Nouchi, R.; Kotozaki, Y.; Nakagawa, S.; et al. The VEGF gene polymorphism impacts brain volume and arterial blood volume. Hum. Brain Mapp. 2017. [Google Scholar] [CrossRef] [PubMed]
- Hall, C.N.; Reynell, C.; Gesslein, B.; Hamilton, N.B.; Mishra, A.; Sutherland, B.A.; O’Farrell, F.M.; Buchan, A.M.; Lauritzen, M.; Attwell, D. Capillary pericytes regulate cerebral blood flow in health and disease. Nature 2014, 508, 55–60. [Google Scholar] [CrossRef] [PubMed]
- Ames, A., 3rd; Wright, R.L.; Kowada, M.; Thurston, J.M.; Majno, G. Cerebral ischemia. II. The no-reflow phenomenon. Am. J. Pathol. 1968, 52, 437–453. [Google Scholar] [PubMed]
- Pedram, A.; Razandi, M.; Levin, E.R. Deciphering vascular endothelial cell growth factor/vascular permeability factor signaling to vascular permeability. Inhibition by atrial natriuretic peptide. J. Biol. Chem. 2002, 277, 44385–44398. [Google Scholar] [CrossRef] [PubMed]
- Paul, R.; Zhang, Z.G.; Eliceiri, B.P.; Jiang, Q.; Boccia, A.D.; Zhang, R.L.; Chopp, M.; Cheresh, D.A. Src deficiency or blockade of Src activity in mice provides cerebral protection following stroke. Nat. Med. 2001, 7, 222–227. [Google Scholar] [CrossRef] [PubMed]
- He, Y.X.; Liu, J.; Guo, B.; Wang, Y.X.; Pan, X.; Li, D.; Tang, T.; Chen, Y.; Peng, S.; Bian, Z.; et al. Src inhibitor reduces permeability without disturbing vascularization and prevents bone destruction in steroid-associated osteonecrotic lesions in rabbits. Sci. Rep. 2015, 5, 8856. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.W.; Kim, W.J.; Choi, Y.K.; Song, H.S.; Son, M.J.; Gelman, I.H.; Kim, Y.J.; Kim, K.W. Ssecks regulates angiogenesis and tight junction formation in blood-brain barrier. Nat. Med. 2003, 9, 900–906. [Google Scholar] [CrossRef] [PubMed]
- Bella, A.J.; Lin, G.; Tantiwongse, K.; Garcia, M.; Lin, C.S.; Brant, W.; Lue, T.F. Brain-derived neurotrophic factor (BDNF) acts primarily via the JAK/STAT pathway to promote neurite growth in the major pelvic ganglion of the rat: Part i. J. Sex. Med. 2006, 3, 815–820. [Google Scholar] [CrossRef] [PubMed]
- Akiyama, C.; Yuguchi, T.; Nishio, M.; Tomishima, T.; Fujinaka, T.; Taniguchi, M.; Nakajima, Y.; Kohmura, E.; Yoshimine, T. Src family kinase inhibitor pp1 reduces secondary damage after spinal cord compression in rats. J. Neurotrauma 2004, 21, 923–931. [Google Scholar] [CrossRef] [PubMed]
- Lennmyr, F.; Ericsson, A.; Gerwins, P.; Akterin, S.; Ahlstrom, H.; Terent, A. Src family kinase-inhibitor pp2 reduces focal ischemic brain injury. Acta Neurol. Scand. 2004, 110, 175–179. [Google Scholar] [CrossRef] [PubMed]
- Eliceiri, B.P.; Paul, R.; Schwartzberg, P.L.; Hood, J.D.; Leng, J.; Cheresh, D.A. Selective requirement for Src kinases during VEGF-induced angiogenesis and vascular permeability. Mol. Cell 1999, 4, 915–924. [Google Scholar] [CrossRef]
- Gavard, J.; Gutkind, J.S. Vegf controls endothelial-cell permeability by promoting the beta-arrestin-dependent endocytosis of ve-cadherin. Nat. Cell Biol. 2006, 8, 1223–1234. [Google Scholar] [CrossRef] [PubMed]
- Weis, S.; Shintani, S.; Weber, A.; Kirchmair, R.; Wood, M.; Cravens, A.; McSharry, H.; Iwakura, A.; Yoon, Y.-S.; Himes, N.; et al. Src blockade stabilizes a FLK/cadherin complex, reducing edema and tissue injury following myocardial infarction. J. Clin. Investig. 2004, 113, 885–894. [Google Scholar] [CrossRef] [PubMed]
- Wallez, Y.; Vilgrain, I.; Huber, P. Angiogenesis: The ve-cadherin switch. Trends Cardiovasc. Med. 2006, 16, 55–59. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mukherjee, S.; Tessema, M.; Wandinger-Ness, A. Vesicular trafficking of tyrosine kinase receptors and associated proteins in the regulation of signaling and vascular function. Circ. Res. 2006, 98, 743–756. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.N.; Pan, R.; Qin, X.J.; Yang, W.L.; Qi, Z.; Liu, W.; Liu, K.J. Ischemic neurons activate astrocytes to disrupt endothelial barrier via increasing VEGF expression. J. Neurochem. 2014, 129, 120–129. [Google Scholar] [CrossRef] [PubMed]
- Tobin, M.K.; Bonds, J.A.; Minshall, R.D.; Pelligrino, D.A.; Testai, F.D.; Lazarov, O. Neurogenesis and inflammation after ischemic stroke: What is known and where we go from here. J. Cereb. Blood Flow Metab. 2014, 34, 1573–1584. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.; Nan, G. The extracellular signal-regulated kinase 1/2 pathway in neurological diseases: A potential therapeutic target (review). Int. J. Mol. Med. 2017, 39, 1338–1346. [Google Scholar] [CrossRef] [PubMed]
- Maddahi, A.; Povlsen, G.K.; Edvinsson, L. Regulation of enhanced cerebrovascular expression of proinflammatory mediators in experimental subarachnoid hemorrhage via the mitogen-activated protein kinase kinase/extracellular signal-regulated kinase pathway. J. Neuroinflamm. 2012, 9, 274. [Google Scholar] [CrossRef] [PubMed]
- Greenberg, D.A.; Jin, K. From angiogenesis to neuropathology. Nature 2005, 438, 954–959. [Google Scholar] [CrossRef] [PubMed]
- Silverman, W.F.; Krum, J.M.; Mani, N.; Rosenstein, J.M. Vascular, glial and neuronal effects of vascular endothelial growth factor in mesencephalic explant cultures. Neuroscience 1999, 90, 1529–1541. [Google Scholar] [CrossRef]
- Jin, K.L.; Mao, X.O.; Greenberg, D.A. Vascular endothelial growth factor: Direct neuroprotective effect in in vitro ischemia. Proc. Natl. Acad. Sci. USA 2000, 97, 10242–10247. [Google Scholar] [CrossRef] [PubMed]
- Ogunshola, O.O.; Antic, A.; Donoghue, M.J.; Fan, S.Y.; Kim, H.; Stewart, W.B.; Madri, J.A.; Ment, L.R. Paracrine and autocrine functions of neuronal vascular endothelial growth factor (VEGF) in the central nervous system. J. Biol. Chem. 2002, 277, 11410–11415. [Google Scholar] [CrossRef] [PubMed]
- Rosenstein, J.M.; Mani, N.; Khaibullina, A.; Krum, J.M. Neurotrophic effects of vascular endothelial growth factor on organotypic cortical explants and primary cortical neurons. J. Neurosci. 2003, 23, 11036–11044. [Google Scholar] [CrossRef] [PubMed]
- Jin, K.; Mao, X.O.; Greenberg, D.A. Vascular endothelial growth factor stimulates neurite outgrowth from cerebral cortical neurons via rho kinase signaling. J. Neurobiol. 2006, 66, 236–242. [Google Scholar] [CrossRef] [PubMed]
- Matsuzaki, H.; Tamatani, M.; Yamaguchi, A.; Namikawa, K.; Kiyama, H.; Vitek, M.P.; Mitsuda, N.; Tohyama, M. Vascular endothelial growth factor rescues hippocampal neurons from glutamate-induced toxicity: Signal transduction cascades. FASEB J. 2001, 15, 1218–1220. [Google Scholar] [CrossRef] [PubMed]
- Svensson, B.; Peters, M.; Konig, H.G.; Poppe, M.; Levkau, B.; Rothermundt, M.; Arolt, V.; Kogel, D.; Prehn, J.H. Vascular endothelial growth factor protects cultured rat hippocampal neurons against hypoxic injury via an antiexcitotoxic, caspase-independent mechanism. J. Cereb. Blood Flow Metab. 2002, 22, 1170–1175. [Google Scholar] [CrossRef] [PubMed]
- Irving, E.A.; Barone, F.C.; Reith, A.D.; Hadingham, S.J.; Parsons, A.A. Differential activation of MAPK/ERK and p38/SAPK in neurones and glia following focal cerebral ischaemia in the rat. Brain Res. Mol. Brain Res. 2000, 77, 65–75. [Google Scholar] [CrossRef]
- Runden, E.; Seglen, P.O.; Haug, F.M.; Ottersen, O.P.; Wieloch, T.; Shamloo, M.; Laake, J.H. Regional selective neuronal degeneration after protein phosphatase inhibition in hippocampal slice cultures: Evidence for a map kinase-dependent mechanism. J. Neurosci. 1998, 18, 7296–7305. [Google Scholar] [CrossRef] [PubMed]
- Murray, B.; Alessandrini, A.; Cole, A.J.; Yee, A.G.; Furshpan, E.J. Inhibition of the p44/42 map kinase pathway protects hippocampal neurons in a cell-culture model of seizure activity. Proc. Natl. Acad. Sci. USA 1998, 95, 11975–11980. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Jin, K. Current perspectives on the link between neuroinflammation and neurogenesis. Metab. Brain Dis. 2015, 30, 355–365. [Google Scholar] [CrossRef] [PubMed]
- Alessandrini, A.; Namura, S.; Moskowitz, M.A.; Bonventre, J.V. Mek1 protein kinase inhibition protects against damage resulting from focal cerebral ischemia. Proc. Natl. Acad. Sci. USA 1999, 96, 12866–12869. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, T.; Abe, K.; Itoyama, Y. Reduction of ischemic damage by application of vascular endothelial growth factor in rat brain after transient ischemia. J. Cereb. Blood Flow Metab. 1998, 18, 887–895. [Google Scholar] [CrossRef] [PubMed]
- Bao, W.L.; Lu, S.D.; Wang, H.; Sun, F.Y. Intraventricular vascular endothelial growth factor antibody increases infarct volume following transient cerebral ischemia. Zhongguo Yao Li Xue Bao 1999, 20, 313–318. [Google Scholar] [PubMed]
- Sorrells, S.F.; Paredes, M.F.; Cebrian-Silla, A.; Sandoval, K.; Qi, D.; Kelley, K.W.; James, D.; Mayer, S.; Chang, J.; Auguste, K.I.; et al. Human hippocampal neurogenesis drops sharply in children to undetectable levels in adults. Nature 2018, 555, 377–381. [Google Scholar] [CrossRef] [PubMed]
- Eriksson, P.S.; Perfilieva, E.; Bjork-Eriksson, T.; Alborn, A.M.; Nordborg, C.; Peterson, D.A.; Gage, F.H. Neurogenesis in the adult human hippocampus. Nat. Med. 1998, 4, 1313–1317. [Google Scholar] [CrossRef] [PubMed]
- Ming, G.-L.; Song, H. Adult neurogenesis in the mammalian brain: Significant answers and significant questions. Neuron 2011, 70, 687–702. [Google Scholar] [CrossRef] [PubMed]
- Ernst, A.; Frisén, J. Adult neurogenesis in humans- common and unique traits in mammals. PLoS Biol. 2015, 13, e1002045. [Google Scholar] [CrossRef] [PubMed]
- Gage, F.H. Neurogenesis in the adult brain. J. Neurosci. 2002, 22, 612–613. [Google Scholar] [CrossRef] [PubMed]
- Gage, F.H.; Kempermann, G.; Palmer, T.D.; Peterson, D.A.; Ray, J. Multipotent progenitor cells in the adult dentate gyrus. J. Neurobiol. 1998, 36, 249–266. [Google Scholar] [CrossRef]
- Nilsson, M.; Perfilieva, E.; Johansson, U.; Orwar, O.; Eriksson, P.S. Enriched environment increases neurogenesis in the adult rat dentate gyrus and improves spatial memory. J. Neurobiol. 1999, 39, 569–578. [Google Scholar] [CrossRef]
- Jin, K.; Wang, X.; Xie, L.; Mao, X.O.; Zhu, W.; Wang, Y.; Shen, J.; Mao, Y.; Banwait, S.; Greenberg, D.A. Evidence for stroke-induced neurogenesis in the human brain. Proc. Natl. Acad. Sci. USA 2006, 103, 13198–13202. [Google Scholar] [CrossRef] [PubMed]
- Jin, K.; Minami, M.; Lan, J.Q.; Mao, X.O.; Batteur, S.; Simon, R.P.; Greenberg, D.A. Neurogenesis in dentate subgranular zone and rostral subventricular zone after focal cerebral ischemia in the rat. Proc. Natl. Acad. Sci. USA 2001, 98, 4710–4715. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Jin, K.; Xie, L.; Childs, J.; Mao, X.O.; Logvinova, A.; Greenberg, D.A. Vegf-induced neuroprotection, neurogenesis, and angiogenesis after focal cerebral ischemia. J. Clin. Investig. 2003, 111, 1843–1851. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Jin, K.; Mao, X.O.; Xie, L.; Banwait, S.; Marti, H.H.; Greenberg, D.A. Vegf-overexpressing transgenic mice show enhanced post-ischemic neurogenesis and neuromigration. J. Neurosci. Res. 2007, 85, 740–747. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.Q.; Cui, H.R.; Yang, S.Z.; Sun, H.P.; Qiu, M.H.; Feng, X.Y.; Sun, F.Y. Vegf enhance cortical newborn neurons and their neurite development in adult rat brain after cerebral ischemia. Neurochem. Int. 2009, 55, 629–636. [Google Scholar] [CrossRef] [PubMed]
- Jin, K.; Zhu, Y.; Sun, Y.; Mao, X.O.; Xie, L.; Greenberg, D.A. Vascular endothelial growth factor (VEGF) stimulates neurogenesis in vitro and in vivo. Proc. Natl. Acad. Sci. USA 2002, 99, 11946–11950. [Google Scholar] [CrossRef] [PubMed]
- Segi-Nishida, E.; Warner-Schmidt, J.L.; Duman, R.S. Electroconvulsive seizure and VEGF increase the proliferation of neural stem-like cells in rat hippocampus. Proc. Natl. Acad. Sci. USA 2008, 105, 11352–11357. [Google Scholar] [CrossRef] [PubMed]
- Fournier, N.M.; Lee, B.; Banasr, M.; Elsayed, M.; Duman, R.S. Vascular endothelial growth factor regulates adult hippocampal cell proliferation through MEK/ERK- and pi3k/AKT-dependent signaling. Neuropharmacology 2012, 63, 642–652. [Google Scholar] [CrossRef] [PubMed]
- Kirby, E.D.; Kuwahara, A.A.; Messer, R.L.; Wyss-Coray, T. Adult hippocampal neural stem and progenitor cells regulate the neurogenic niche by secreting VEGF. Proc. Natl. Acad. Sci. USA 2015, 112, 4128–4133. [Google Scholar] [CrossRef] [PubMed]
- Li, W.-L.; Fraser, J.L.; Yu, S.P.; Zhu, J.; Jiang, Y.-J.; Wei, L. The role of VEGF/VEGFR2 signaling in peripheral stimulation-induced cerebral neurovascular regeneration after ischemic stroke in mice. Exp. Brain Res. 2011, 214, 503. [Google Scholar] [CrossRef] [PubMed]
- Schanzer, A.; Wachs, F.P.; Wilhelm, D.; Acker, T.; Cooper-Kuhn, C.; Beck, H.; Winkler, J.; Aigner, L.; Plate, K.H.; Kuhn, H.G. Direct stimulation of adult neural stem cells in vitro and neurogenesis in vivo by vascular endothelial growth factor. Brain Pathol. 2004, 14, 237–248. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.J.; Bao, W.L.; Qiu, M.H.; Zhang, L.M.; Lu, S.D.; Huang, Y.L.; Sun, F.Y. Role of vascular endothelial growth factor in neuronal DNA damage and repair in rat brain following a transient cerebral ischemia. J. Neurosci. Res. 2002, 70, 140–149. [Google Scholar] [CrossRef] [PubMed]
- Shen, S.-W.; Duan, C.-L.; Chen, X.-H.; Wang, Y.-Q.; Sun, X.; Zhang, Q.-W.; Cui, H.-R.; Sun, F.-Y. Neurogenic effect of VEGF is related to increase of astrocytes transdifferentiation into new mature neurons in rat brains after stroke. Neuropharmacology 2016, 108, 451–461. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.-P.; Liu, H.-J.; Liu, X.-F. VEGF promotes angiogenesis and functional recovery in stroke rats. J. Investig. Surg. 2010, 23, 149–155. [Google Scholar] [CrossRef] [PubMed]
- Manoonkitiwongsa, P.S.; Schultz, R.L.; McCreery, D.B.; Whitter, E.F.; Lyden, P.D. Neuroprotection of ischemic brain by vascular endothelial growth factor is critically dependent on proper dosage and may be compromised by angiogenesis. J. Cereb. Blood Flow Metab. 2004, 24, 693–702. [Google Scholar] [CrossRef] [PubMed]
- Koch, S.; Sacco, R.L.; Perez-Pinzon, M.A. Preconditioning the brain: Moving on to the next frontier of neurotherapeutics. Stroke 2012, 43, 1455–1457. [Google Scholar] [CrossRef] [PubMed]
- Kitagawa, K.; Matsumoto, M.; Tagaya, M.; Hata, R.; Ueda, H.; Niinobe, M.; Handa, N.; Fukunaga, R.; Kimura, K.; Mikoshiba, K.; et al. ‘Ischemic tolerance’ phenomenon found in the brain. Brain Res. 1990, 528, 21–24. [Google Scholar] [CrossRef] [Green Version]
- Dirnagl, U.; Becker, K.; Meisel, A. Preconditioning and tolerance against cerebral ischaemia: From experimental strategies to clinical use. Lancet Neurol. 2009, 8, 398–412. [Google Scholar] [CrossRef]
- Koch, S. Preconditioning the human brain: Practical considerations for proving cerebral protection. Transl. Stroke Res. 2010, 1, 161–169. [Google Scholar] [CrossRef] [PubMed]
- Pan, J.; Li, X.; Peng, Y. Remote ischemic conditioning for acute ischemic stroke: Dawn in the darkness. Rev. Neurosci. 2016, 27, 501–510. [Google Scholar] [CrossRef] [PubMed]
- Kirino, T.; Tsujita, Y.; Tamura, A. Induced tolerance to ischemia in gerbil hippocampal neurons. J. Cereb. Blood Flow Metab. 1991, 11, 299–307. [Google Scholar] [CrossRef] [PubMed]
- Stagliano, N.E.; Perez-Pinzon, M.A.; Moskowitz, M.A.; Huang, P.L. Focal ischemic preconditioning induces rapid tolerance to middle cerebral artery occlusion in mice. J. Cereb. Blood Flow Metab. 1999, 19, 757–761. [Google Scholar] [CrossRef] [PubMed]
- Perez-Pinzon, M.A.; Xu, G.P.; Dietrich, W.D.; Rosenthal, M.; Sick, T.J. Rapid preconditioning protects rats against ischemic neuronal damage after 3 but not 7 days of reperfusion following global cerebral ischemia. J. Cereb. Blood Flow Metab. 1997, 17, 175–182. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Liu, C.; Du, X.; Liu, M.; Ji, X.; Du, H.; Zhao, H. Hypoxia inducible factor 1α plays a key role in remote ischemic preconditioning against stroke by modulating inflammatory responses in rats. J. Am. Heart Assoc. 2018, 7. [Google Scholar] [CrossRef] [PubMed]
- Kawata, H.; Yoshida, K.; Kawamoto, A.; Kurioka, H.; Takase, E.; Sasaki, Y.; Hatanaka, K.; Kobayashi, M.; Ueyama, T.; Hashimoto, T.; et al. Ischemic preconditioning upregulates vascular endothelial growth factor mrna expression and neovascularization via nuclear translocation of protein kinase c epsilon in the rat ischemic myocardium. Circ. Res. 2001, 88, 696–704. [Google Scholar] [CrossRef] [PubMed]
- Tang, Y.; Pacary, E.; Freret, T.; Divoux, D.; Petit, E.; Schumann-Bard, P.; Bernaudin, M. Effect of hypoxic preconditioning on brain genomic response before and following ischemia in the adult mouse: Identification of potential neuroprotective candidates for stroke. Neurobiol. Dis. 2006, 21, 18–28. [Google Scholar] [CrossRef] [PubMed]
- Park, Y.S.; Cho, J.H.; Kim, I.H.; Cho, G.S.; Cho, J.H.; Park, J.H.; Ahn, J.H.; Chen, B.H.; Shin, B.N.; Shin, M.C.; et al. Effects of ischemic preconditioning on VEGF and PFLK-1 immunoreactivities in the gerbil ischemic hippocampus after transient cerebral ischemia. J. Neurol. Sci. 2014, 347, 179–187. [Google Scholar] [CrossRef] [PubMed]
- Wick, A.; Wick, W.; Waltenberger, J.; Weller, M.; Dichgans, J.; Schulz, J.B. Neuroprotection by hypoxic preconditioning requires sequential activation of vascular endothelial growth factor receptor and Akt. J. Neurosci. 2002, 22, 6401–6407. [Google Scholar] [CrossRef] [PubMed]
- Clayton, J.A.; Chalothorn, D.; Faber, J.E. Vascular endothelial growth factor-A specifies formation of native collaterals and regulates collateral growth in ischemia. Circ. Res. 2008, 103, 1027–1036. [Google Scholar] [CrossRef] [PubMed]
- Lucitti, J.L.; Mackey, J.K.; Morrison, J.C.; Haigh, J.J.; Adams, R.H.; Faber, J.E. Formation of the collateral circulation is regulated by vascular endothelial growth factor-A and a disintegrin and metalloprotease family members 10 and 17. Circ. Res. 2012, 111, 1539–1550. [Google Scholar] [CrossRef] [PubMed]
- Zechariah, A.; ElAli, A.; Doeppner, T.R.; Jin, F.; Hasan, M.R.; Helfrich, I.; Mies, G.; Hermann, D.M. Vascular endothelial growth factor promotes pericyte coverage of brain capillaries, improves cerebral blood flow during subsequent focal cerebral ischemia, and preserves the metabolic penumbra. Stroke 2013, 44, 1690–1697. [Google Scholar] [CrossRef] [PubMed]
- Licht, T.; Rothe, G.; Kreisel, T.; Wolf, B.; Benny, O.; Rooney, A.G.; Ffrench-Constant, C.; Enikolopov, G.; Keshet, E. VEGF preconditioning leads to stem cell remodeling and attenuates age-related decay of adult hippocampal neurogenesis. Proc. Natl. Acad. Sci. USA 2016, 113, E7828–E7836. [Google Scholar] [CrossRef] [PubMed]
- Limani, P.; Linecker, M.; Oberkofler, C.E.; Barmettler, G.; Kaech, A.; Graf, R.; Humar, B.; Clavien, P.A. Remote ischemic preconditioning: A novel strategy in rescuing older livers from ischemia-reperfusion injury in a rodent model. Ann. Surg. 2016, 264, 797–803. [Google Scholar] [CrossRef] [PubMed]
- Hess, D.C.; Blauenfeldt, R.A.; Andersen, G.; Hougaard, K.D.; Hoda, M.N.; Ding, Y.; Ji, X. Remote ischaemic conditioning-a new paradigm of self-protection in the brain. Nat. Rev. Neurol. 2015, 11, 698–710. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.J.; Chen, C.; Li, X.R.; Ran, Y.Y.; Xu, T.; Zhang, Y.; Geng, X.K.; Zhang, Y.; Du, H.S.; Leak, R.K.; et al. Remote ischemic preconditioning-mediated neuroprotection against stroke is associated with significant alterations in peripheral immune responses. CNS Neurosci. Ther. 2016, 22, 43–52. [Google Scholar] [CrossRef] [PubMed]
- Wei, D.; Ren, C.; Chen, X.; Zhao, H. The chronic protective effects of limb remote preconditioning and the underlying mechanisms involved in inflammatory factors in rat stroke. PLoS ONE 2012, 7, e30892. [Google Scholar] [CrossRef] [PubMed]
- Meng, R.; Asmaro, K.; Meng, L.; Liu, Y.; Ma, C.; Xi, C.; Li, G.; Ren, C.; Luo, Y.; Ling, F.; et al. Upper limb ischemic preconditioning prevents recurrent stroke in intracranial arterial stenosis. Neurology 2012, 79, 1853–1861. [Google Scholar] [CrossRef] [PubMed]
- Mi, T.; Yu, F.; Ji, X.; Sun, Y.; Qu, D. The interventional effect of remote ischemic preconditioning on cerebral small vessel disease: A pilot randomized clinical trial. Eur. Neurol. 2016, 76, 28–34. [Google Scholar] [CrossRef] [PubMed]
- Yanamoto, H.; Hashimoto, N.; Nagata, I.; Kikuchi, H. Infarct tolerance against temporary focal ischemia following spreading depression in rat brain. Brain Res. 1998, 784, 239–249. [Google Scholar] [CrossRef]
- Tasaki, K.; Ruetzler, C.A.; Ohtsuki, T.; Martin, D.; Nawashiro, H.; Hallenbeck, J.M. Lipopolysaccharide pre-treatment induces resistance against subsequent focal cerebral ischemic damage in spontaneously hypertensive rats. Brain Res. 1997, 748, 267–270. [Google Scholar] [CrossRef]
- Nawashiro, H.; Tasaki, K.; Ruetzler, C.A.; Hallenbeck, J.M. TNF-α pretreatment induces protective effects against focal cerebral ischemia in mice. J. Cereb. Blood Flow Metab. 1997, 17, 483–490. [Google Scholar] [CrossRef] [PubMed]
- Ohtsuki, T.; Ruetzler, C.A.; Tasaki, K.; Hallenbeck, J.M. Interleukin-1 mediates induction of tolerance to global ischemia in gerbil hippocampal ca1 neurons. J. Cereb. Blood Flow Metab. 1996, 16, 1137–1142. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Lotz, C.; Roewer, N.; Broscheit, J.-A. Comparison of volatile anesthetic-induced preconditioning in cardiac and cerebral system: Molecular mechanisms and clinical aspects. Eur. J. Med. Res. 2018, 23, 10. [Google Scholar] [CrossRef] [PubMed]
- Zitta, K.; Meybohm, P.; Bein, B.; Ohnesorge, H.; Steinfath, M.; Scholz, J.; Albrecht, M. Cytoprotective effects of the volatile anesthetic sevoflurane are highly dependent on timing and duration of sevoflurane conditioning: Findings from a human, in vitro hypoxia model. Eur. J. Pharmacol. 2010, 645, 39–46. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.Q.; Sheng, R.; Qin, Z.H. The neuroprotective mechanism of brain ischemic preconditioning. Acta Pharmacol. Sin. 2009, 30, 1071–1080. [Google Scholar] [CrossRef] [PubMed]
- Endres, M.; Gertz, K.; Lindauer, U.; Katchanov, J.; Schultze, J.; Schrock, H.; Nickenig, G.; Kuschinsky, W.; Dirnagl, U.; Laufs, U. Mechanisms of stroke protection by physical activity. Ann. Neurol. 2003, 54, 582–590. [Google Scholar] [CrossRef] [PubMed]
- Ding, Y.H.; Luan, X.D.; Li, J.; Rafols, J.A.; Guthinkonda, M.; Diaz, F.G.; Ding, Y. Exercise-induced overexpression of angiogenic factors and reduction of ischemia/reperfusion injury in stroke. Curr. Neurovasc. Res. 2004, 1, 411–420. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Hu, X.; Luo, J.; Li, L.; Chen, X.; Huang, R.; Pei, Z. Physical exercise improves functional recovery through mitigation of autophagy, attenuation of apoptosis and enhancement of neurogenesis after MCAO in rats. BMC Neurosci. 2013, 14, 46. [Google Scholar] [CrossRef] [PubMed]
- Ding, Y.H.; Li, J.; Zhou, Y.; Rafols, J.A.; Clark, J.C.; Ding, Y. Cerebral angiogenesis and expression of angiogenic factors in aging rats after exercise. Curr. Neurovasc. Res. 2006, 3, 15–23. [Google Scholar] [CrossRef] [PubMed]
- Tahamtan, M.; Allahtavakoli, M.; Abbasnejad, M.; Roohbakhsh, A.; Taghipour, Z.; Taghavi, M.; Khodadadi, H.; Shamsizadeh, A. Exercise preconditioning improves behavioral functions following transient cerebral ischemia induced by 4-vessel occlusion (4-vo) in rats. Arch. Iran. Med. 2013, 16, 697–704. [Google Scholar] [PubMed]
- Fukai, T.; Siegfried, M.R.; Ushio-Fukai, M.; Cheng, Y.; Kojda, G.; Harrison, D.G. Regulation of the vascular extracellular superoxide dismutase by nitric oxide and exercise training. J. Clin. Investig. 2000, 105, 1631–1639. [Google Scholar] [CrossRef] [PubMed]
- Hambrecht, R.; Wolf, A.; Gielen, S.; Linke, A.; Hofer, J.; Erbs, S.; Schoene, N.; Schuler, G. Effect of exercise on coronary endothelial function in patients with coronary artery disease. N. Engl. J. Med. 2000, 342, 454–460. [Google Scholar] [CrossRef] [PubMed]
- Hornig, B.; Maier, V.; Drexler, H. Physical training improves endothelial function in patients with chronic heart failure. Circulation 1996, 93, 210–214. [Google Scholar] [CrossRef] [PubMed]
- Sessa, W.C.; Pritchard, K.; Seyedi, N.; Wang, J.; Hintze, T.H. Chronic exercise in dogs increases coronary vascular nitric oxide production and endothelial cell nitric oxide synthase gene expression. Circ. Res. 1994, 74, 349–353. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.T.; Chang, Y.C.; Tu, Y.F.; Huang, C.C. VEGF-a/VEGFR-2 signaling leading to camp response element-binding protein phosphorylation is a shared pathway underlying the protective effect of preconditioning on neurons and endothelial cells. J. Neurosci. 2009, 29, 4356–4368. [Google Scholar] [CrossRef] [PubMed]
- Hayes, K.; Sprague, S.; Guo, M.; Davis, W.; Friedman, A.; Kumar, A.; Jimenez, D.F.; Ding, Y. Forced, not voluntary, exercise effectively induces neuroprotection in stroke. Acta Neuropathol. 2008, 115, 289–296. [Google Scholar] [CrossRef] [PubMed]
- Lezi, E.; Lu, J.; Selfridge, J.E.; Burns, J.M.; Swerdlow, R.H. Lactate administration reproduces specific brain and liver exercise-related changes. J. Neurochem. 2013, 127, 91–100. [Google Scholar] [CrossRef]
- Lauritzen, K.H.; Morland, C.; Puchades, M.; Holm-Hansen, S.; Hagelin, E.M.; Lauritzen, F.; Attramadal, H.; Storm-Mathisen, J.; Gjedde, A.; Bergersen, L.H. Lactate receptor sites link neurotransmission, neurovascular coupling, and brain energy metabolism. Cereb. Cortex 2014, 24, 2784–2795. [Google Scholar] [CrossRef] [PubMed]
- Slevin, M.; Krupinski, J.; Slowik, A.; Kumar, P.; Szczudlik, A.; Gaffney, J. Serial measurement of vascular endothelial growth factor and transforming growth factor-β1 in serum of patients with acute ischemic stroke. Stroke 2000, 31, 1863–1870. [Google Scholar] [CrossRef] [PubMed]
- Matsuo, R.; Ago, T.; Kamouchi, M.; Kuroda, J.; Kuwashiro, T.; Hata, J.; Sugimori, H.; Fukuda, K.; Gotoh, S.; Makihara, N.; et al. Clinical significance of plasma VEGF value in ischemic stroke—Research for biomarkers in ischemic stroke (rebios) study. BMC Neurol. 2013, 13, 32. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.C.; Lee, K.Y.; Kim, Y.J.; Kim, S.H.; Koh, S.H.; Lee, Y.J. Serum VEGF levels in acute ischaemic strokes are correlated with long-term prognosis. Eur. J. Neurol. 2010, 17, 45–51. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
VEGF-A-mediated vascular sprouting. VEGFR-2 is expressed on endothelial cells (shown in gray) including proliferating cells, where it binds VEGF-A, which then induces sprouting. One form of VEGFR-1 is expressed on mature endothelial cells, while another form is secreted (sVEGFR-1). Both forms of VEGFR-1 bind VEGF-A, hence preventing the binding of VEGF-A to VEGFR-2 on non-sprouting parts of the blood vessel (decoy function). This is important to guide the growing vessel in the right direction and prevent the sprouting of neighboring cells.
Figure 1.
VEGF-A-mediated vascular sprouting. VEGFR-2 is expressed on endothelial cells (shown in gray) including proliferating cells, where it binds VEGF-A, which then induces sprouting. One form of VEGFR-1 is expressed on mature endothelial cells, while another form is secreted (sVEGFR-1). Both forms of VEGFR-1 bind VEGF-A, hence preventing the binding of VEGF-A to VEGFR-2 on non-sprouting parts of the blood vessel (decoy function). This is important to guide the growing vessel in the right direction and prevent the sprouting of neighboring cells.

Figure 2.
VEGF-A mediated disruption of the blood brain barrier (BBB). VEGF-A binds to VEGFR-2 on endothelial cells (shown in gray) of the BBB leading to the activation of the VEGFR-2–Src–Rac1–PAK pathway. Activated PAK phosphorylates (red dot) the internal tail of the cell-adhesion molecule VE-cadherin (blue), leading to its internalization and subsequently to the disruption of the intercellular junctions of the BBB. Internalized VE-cadherin is then either recycled to the membrane or degraded. The green cell exemplifies a systemic immune cell that is allowed to enter the brain through the fenestrated vessel wall along with other molecules that are normally prevented from entering when the BBB is intact. An intact adherent junction between two endothelial cells in the absence of a VEGF-A signal is shown to the left.
Figure 2.
VEGF-A mediated disruption of the blood brain barrier (BBB). VEGF-A binds to VEGFR-2 on endothelial cells (shown in gray) of the BBB leading to the activation of the VEGFR-2–Src–Rac1–PAK pathway. Activated PAK phosphorylates (red dot) the internal tail of the cell-adhesion molecule VE-cadherin (blue), leading to its internalization and subsequently to the disruption of the intercellular junctions of the BBB. Internalized VE-cadherin is then either recycled to the membrane or degraded. The green cell exemplifies a systemic immune cell that is allowed to enter the brain through the fenestrated vessel wall along with other molecules that are normally prevented from entering when the BBB is intact. An intact adherent junction between two endothelial cells in the absence of a VEGF-A signal is shown to the left.

Figure 3.
Effects of VEGF-A in cerebral stroke–timeline. Before stroke onset, the upregulation of VEGF-A (large arrow labelled VEGF-A), preconditioning or exercise decreases the risk of stroke as well as the outcome after stroke. The latter is, at least partly, due to the increased formation of collateral (angiogenic effects) and direct neuroprotective effects of VEGF-A. In the acute phase (0–24 h after stroke onset), the systemic administration of VEGF-A at levels leading to angiogenesis, or the intrinsic upregulation of VEGF-A lead to a leaky BBB and corresponding detrimental effects. The application of low non-angiogenic doses (e.g., via a cerebral artery) as well as the intraventricular or topical application of VEGF-A have a neuroprotective effect, even in the acute phase. In the later phase (>24 h) after stroke, increased levels of VEGF-A decrease stroke-induced neural damage.
Figure 3.
Effects of VEGF-A in cerebral stroke–timeline. Before stroke onset, the upregulation of VEGF-A (large arrow labelled VEGF-A), preconditioning or exercise decreases the risk of stroke as well as the outcome after stroke. The latter is, at least partly, due to the increased formation of collateral (angiogenic effects) and direct neuroprotective effects of VEGF-A. In the acute phase (0–24 h after stroke onset), the systemic administration of VEGF-A at levels leading to angiogenesis, or the intrinsic upregulation of VEGF-A lead to a leaky BBB and corresponding detrimental effects. The application of low non-angiogenic doses (e.g., via a cerebral artery) as well as the intraventricular or topical application of VEGF-A have a neuroprotective effect, even in the acute phase. In the later phase (>24 h) after stroke, increased levels of VEGF-A decrease stroke-induced neural damage.

© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Geiseler, S.J.; Morland, C. The Janus Face of VEGF in Stroke. Int. J. Mol. Sci. 2018, 19, 1362. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms19051362
AMA Style
Geiseler SJ, Morland C. The Janus Face of VEGF in Stroke. International Journal of Molecular Sciences. 2018; 19(5):1362. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms19051362
Chicago/Turabian StyleGeiseler, Samuel J., and Cecilie Morland. 2018. "The Janus Face of VEGF in Stroke" International Journal of Molecular Sciences 19, no. 5: 1362. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms19051362
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.