Pharmacological Targeting of Cell Cycle, Apoptotic and Cell Adhesion Signaling Pathways Implicated in Chemoresistance of Cancer Cells

 , , and
, , and
Abstract
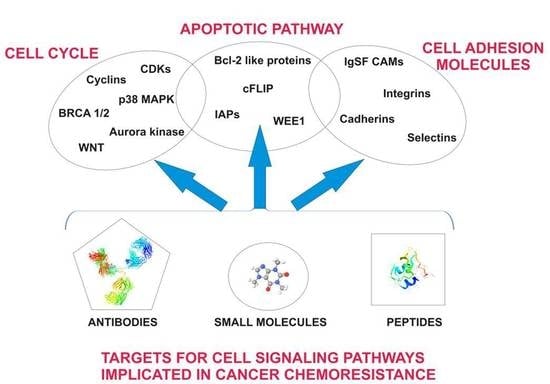
:
1. Introduction
1.1. Cell Cycle
1.2. Apoptosis
1.3. Cell Adhesion
2. Cell Cycle as a Target for Overcoming Chemoresistance during Cancer Therapy
2.1. Cyclins and Cyclin Dependent Kinases
2.2. The DNA Damage Responsive p53 Pathway
2.3. Targetting Mutated p53
2.4. Aurora Kinase Signaling
2.5. BRCA1/2
2.6. Wingless (WNT) Signaling
2.7. The p38 MAP Kinase Pathway
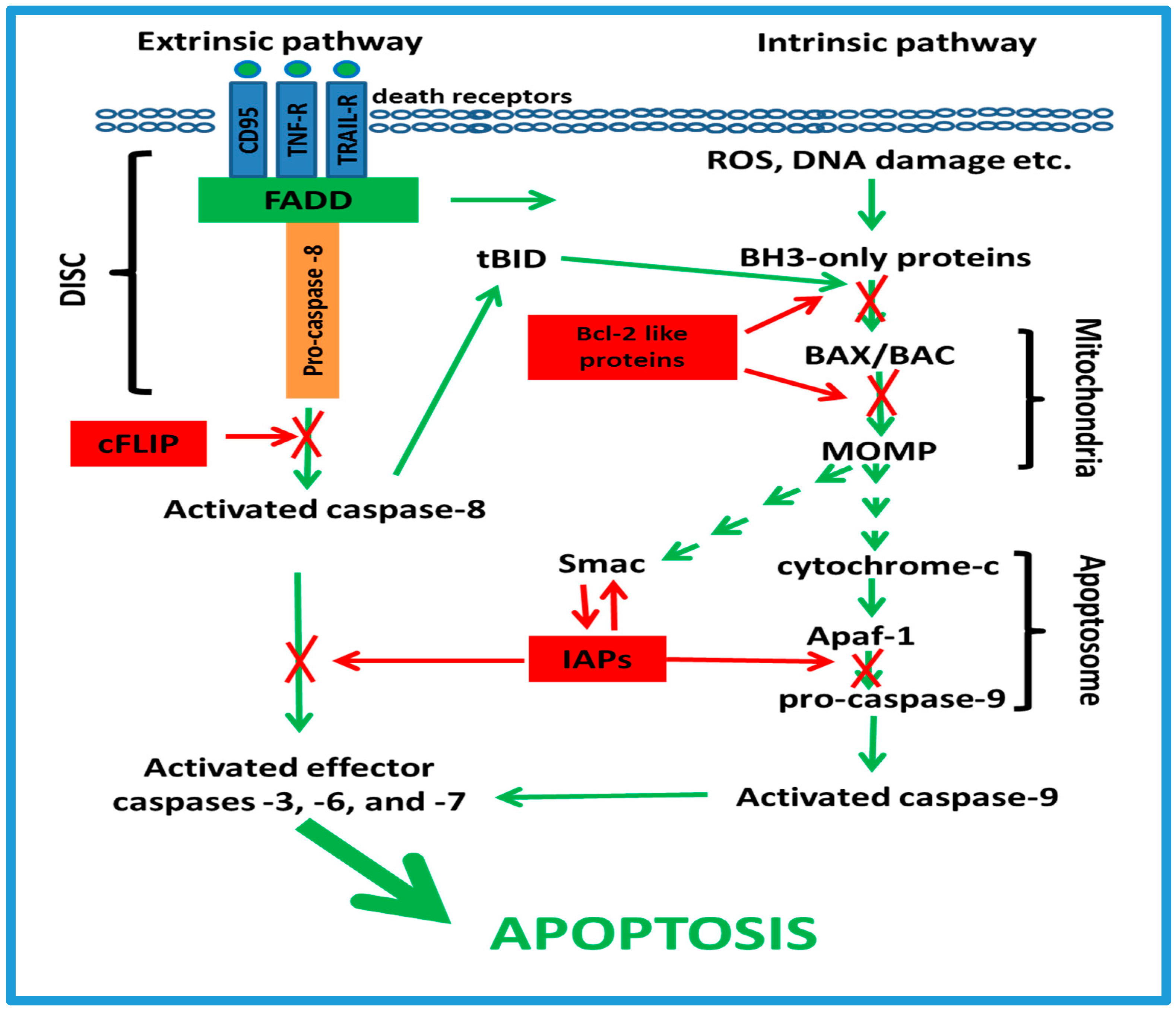
3. Anti-Apoptotic Mechanisms in Resistance to Chemotherapy
3.1. Apoptotic Cell-Signaling Pathways
3.2. The cFLIP Proteins
3.3. The Bcl-2-Like Proteins
3.4. The IAP Family
4. Transforming Growth Factors
5. Role of Cell Adhesion Molecules (CAMs) in Chemoresistance
6. Conclusions
Acknowledgments
Conflicts of Interest
References
- Kelland, L. The resurgence of platinum-based cancer chemotherapy. Nat. Rev. Cancer 2007, 7, 573–584. [Google Scholar] [CrossRef] [PubMed]
- Socinski, M.A. Cytotoxic chemotherapy in advanced non-small cell lung cancer: A review of standard treatment paradigms. Clin. Cancer Res. 2004, 10, 4210s–4214s. [Google Scholar] [CrossRef] [PubMed]
- Bartkova, J.; Rezaei, N.; Liontos, M.; Karakaidos, P.; Kletsas, D.; Issaeva, N.; Vassiliou, L.V.; Kolettas, E.; Niforou, K.; Zoumpourlis, V.C.; et al. Oncogene-induced senescence is part of the tumorigenesis barrier imposed by DNA damage checkpoints. Nature 2006, 444, 633–637. [Google Scholar] [CrossRef] [PubMed]
- Ciccia, A.; Elledge, S.J. The DNA damage response: Making it safe to play with knives. Mol. Cell 2010, 40, 179–204. [Google Scholar] [CrossRef] [PubMed]
- Jackson, S.P.; Bartek, J. The DNA-damage response in human biology and disease. Nature 2009, 461, 1071–1078. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grossi, V.; Peserico, A.; Tezil, T.; Simone, C. p38α MAPK pathway: A key factor in colorectal cancer therapy and chemoresistance. World J. Gastroenterol. 2014, 20, 9744–9758. [Google Scholar] [CrossRef] [PubMed]
- Dominguez-Brauer, C.; Thu, K.L.; Mason, J.M.; Blaser, H.; Bray, M.R.; Mak, T.W. Targeting Mitosis in Cancer: Emerging Strategies. Mol. Cell 2015, 60, 524–536. [Google Scholar] [CrossRef] [PubMed]
- Cicenas, J.; Kalyan, K.; Sorokinas, A.; Jatulyte, A.; Valiunas, D.; Kaupinis, A.; Valius, M. Highlights of the Latest Advances in Research on CDK Inhibitors. Cancers 2014, 6, 2224–2242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yim, H. Current clinical trials with polo-like kinase 1 inhibitors in solid tumors. Anticancer Drugs 2013, 24, 999–1006. [Google Scholar] [CrossRef] [PubMed]
- Kerr, J.F.; Wyllie, A.H.; Currie, A.R. Apoptosis: A basic biological phenomenon with wide-ranging implications in tissue kinetics. Br. J. Cancer 1972, 26, 239–257. [Google Scholar] [CrossRef] [PubMed]
- Ubah, O.C.; Wallace, H.M. Cancer therapy: Targeting mitochondria and other sub-cellular organelles. Curr. Pharm. Des. 2014, 20, 201–222. [Google Scholar] [CrossRef] [PubMed]
- Wong, R.S.Y. Apoptosis in cancer: From pathogenesis to treatment. J. Exp. Clin. Cancer Res. 2011, 30, 87. [Google Scholar] [CrossRef] [PubMed]
- Moh, M.C.; Shen, S. The roles of cell adhesion molecules in tumor suppression and cell migration: A new paradox. Cell Adhes. Migr. 2009, 3, 334–336. [Google Scholar] [CrossRef]
- Albelda, S.M.; Buck, C.A. Integrins and other cell adhesion molecules. FASEB J. 1990, 4, 2868–2880. [Google Scholar] [CrossRef] [PubMed]
- Cohen, M.B.; Griebling, T.L.; Ahaghotu, C.A.; Rokhlin, O.W.; Ross, J.S. Cellular adhesion molecules in urologic malignancies. Am. J. Clin. Pathol. 1997, 107, 56–63. [Google Scholar] [CrossRef] [PubMed]
- Schmidmaier, R.; Baumann, P. ANTI-ADHESION evolves to a promising therapeutic concept in oncology. Curr. Med. Chem. 2008, 15, 978–990. [Google Scholar] [CrossRef] [PubMed]
- Schmidmaier, R.; Baumann, P.; Bumeder, I.; Meinhardt, G.; Straka, C.; Emmerich, B. First clinical experience with simvastatin to overcome drug resistance in refractory multiple myeloma. Eur. J. Haematol. 2007, 79, 240–243. [Google Scholar] [CrossRef] [PubMed]
- Durand, R.E.; Sutherland, R.M. Effects of intercellular contact on repair of radiation damage. Exp. Cell Res. 1972, 71, 75–80. [Google Scholar] [CrossRef]
- Sherr, C.J.; Roberts, J.M. CDK inhibitors: Positive and negative regulators of G1-phase progression. Genes Dev. 1999, 13, 1501–1512. [Google Scholar] [CrossRef] [PubMed]
- Canavese, M.; Santo, L.; Raje, N. Cyclin dependent kinases in cancer. Cancer Biol. Ther. 2012, 13, 451–457. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bagella, L.; Sun, A.; Tonini, T.; Abbadessa, G.; Cottone, G.; Paggi, M.G.; De Luca, A.; Claudio, P.P.; Giordano, A. A small molecule based on the pRb2/p130 spacer domain leads to inhibition of cdk2 activity, cell cycle arrest and tumor growth reduction in vivo. Oncogene 2007, 26, 1829–1839. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, G.K. CDK inhibitors: Cell cycle arrest versus apoptosis. Cell Cycle 2002, 1, 122–123. [Google Scholar] [CrossRef] [PubMed]
- Cirillo, D.; Pentimalli, F.; Giordano, A. Peptides or small molecules? Different approaches to develop more effective CDK inhibitors. Curr. Med. Chem. 2011, 18, 2854–2866. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.H.; Kim, S.U.; Shin, D.Y.; Choi, K.S. Roscovitine sensitizes glioma cells to TRAIL-mediated apoptosis by downregulation of survivin and XIAP. Oncogene 2004, 23, 446–456. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lubanska, D.; Porter, L. Revisiting CDK Inhibitors for Treatment of Glioblastoma Multiforme. Drugs R D 2017, 17, 255–263. [Google Scholar] [CrossRef] [PubMed]
- Bose, P.; Simmons, G.L.; Grant, S. Cyclin-dependent kinase inhibitor therapy for hematologic malignancies. Expert Opin. Investig. Drugs 2013, 22, 723–738. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hayashi, T.; Adachi, K.; Ohba, S.; Hirose, Y. The Cdk inhibitor flavopiridol enhances temozolomide-induced cytotoxicity in human glioma cells. J. Neurooncol. 2013, 115, 169–178. [Google Scholar] [CrossRef] [PubMed]
- Haider, C.; Grubinger, M.; Reznickova, E.; Weiss, T.S.; Rotheneder, H.; Miklos, W.; Berger, W.; Jorda, R.; Zatloukal, M.; Gucky, T.; et al. Novel inhibitors of cyclin-dependent kinases combat hepatocellular carcinoma without inducing chemoresistance. Mol. Cancer Ther. 2013, 12, 1947–1957. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Zhou, Y.; Li, Y.; Zhou, J.; Wu, Y.; Cui, Y.; Yang, G.; Hong, Y. Mutations of p53 and KRAS activate NF-kappaB to promote chemoresistance and tumorigenesis via dysregulation of cell cycle and suppression of apoptosis in lung cancer cells. Cancer Lett. 2015, 357, 520–526. [Google Scholar] [CrossRef] [PubMed]
- Woo, M.G.; Xue, K.; Liu, J.; McBride, H.; Tsang, B.K. Calpain-mediated processing of p53-associated parkin-like cytoplasmic protein (PARC) affects chemosensitivity of human ovarian cancer cells by promoting p53 subcellular trafficking. J. Biol. Chem. 2012, 287, 3963–3975. [Google Scholar] [CrossRef] [PubMed]
- Mirzayans, R.; Andrais, B.; Scott, A.; Murray, D. New insights into p53 signaling and cancer cell response to DNA damage: Implications for cancer therapy. J. Biomed. Biotechnol. 2012, 2012, 170325. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.; Piwnica-Worms, H. ATR-mediated checkpoint pathways regulate phosphorylation and activation of human Chk1. Mol. Cell. Biol. 2001, 21, 4129–4139. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.; Watkins, J.L.; Piwnica-Worms, H. Disruption of the checkpoint kinase 1/cell division cycle 25A pathway abrogates ionizing radiation-induced S and G2 checkpoints. Proc. Natl. Acad. Sci. USA 2002, 99, 14795–14800. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, C.; Li, L.; Guan, X.; Xiong, L.; Miao, X. Mutant p53 Gain of Function and Chemoresistance: The Role of Mutant p53 in Response to Clinical Chemotherapy. Chemotherapy 2017, 62, 43–53. [Google Scholar] [CrossRef] [PubMed]
- Lehmann, B.D.; Pietenpol, J.A. Targeting mutant p53 in human tumors. J. Clin. Oncol. 2012, 30, 3648–3650. [Google Scholar] [CrossRef] [PubMed]
- Fan, S.; Smith, M.L.; Rivet, D.J., 2nd; Duba, D.; Zhan, Q.; Kohn, K.W.; Fornace, A.J., Jr.; O'Connor, P.M. Disruption of p53 function sensitizes breast cancer MCF-7 cells to cisplatin and pentoxifylline. Cancer Res. 1995, 55, 1649–1654. [Google Scholar] [PubMed]
- Maréchal, A.; Zou, L. DNA Damage Sensing by the ATM and ATR Kinases. Cold Spring Harb. Perspect. Biol. 2013, 5, a012716. [Google Scholar] [CrossRef] [PubMed]
- Chin, K.V.; Ueda, K.; Pastan, I.; Gottesman, M.M. Modulation of activity of the promoter of the human MDR1 gene by Ras and p53. Science 1992, 255, 459–462. [Google Scholar] [CrossRef] [PubMed]
- Ma, C.X.; Janetka, J.W.; Piwnica-Worms, H. Death by releasing the breaks: CHK1 inhibitors as cancer therapeutics. Trends Mol. Med. 2011, 17, 88–96. [Google Scholar] [CrossRef] [PubMed]
- Visconti, R.; Della Monica, R.; Grieco, D. Cell cycle checkpoint in cancer: A therapeutically targetable double-edged sword. J. Exp. Clin. Cancer Res. 2016, 35, 153. [Google Scholar] [CrossRef] [PubMed]
- Dixon, H.; Norbury, C.J. Therapeutic exploitation of checkpoint defects in cancer cells lacking p53 function. Cell Cycle 2002, 1, 362–368. [Google Scholar] [CrossRef] [PubMed]
- Iwata, T.; Uchino, T.; Koyama, A.; Johmura, Y.; Koyama, K.; Saito, T.; Ishiguro, S.; Arikawa, T.; Komatsu, S.; Miyachi, M.; et al. The G2 checkpoint inhibitor CBP-93872 increases the sensitivity of colorectal and pancreatic cancer cells to chemotherapy. PLoS ONE 2017, 12, e0178221. [Google Scholar] [CrossRef] [PubMed]
- Harada, N.; Watanabe, Y.; Yoshimura, Y.; Sakumoto, H.; Makishima, F.; Tsuchiya, M.; Nakanishi, K.; Nakanishi, M.; Aoki, Y. Identification of a checkpoint modulator with synthetic lethality to p53 mutants. Anticancer Drugs 2011, 22, 986–994. [Google Scholar] [CrossRef] [PubMed]
- Hirokawa, T.; Shiotani, B.; Shimada, M.; Murata, K.; Johmura, Y.; Haruta, M.; Tahara, H.; Takeyama, H.; Nakanishi, M. CBP-93872 inhibits NBS1-mediated ATR activation, abrogating maintenance of the DNA double-strand break-specific G2 checkpoint. Cancer Res. 2014, 74, 3880–3889. [Google Scholar] [CrossRef] [PubMed]
- Blandino, G.; Di Agostino, S. New therapeutic strategies to treat human cancers expressing mutant p53 proteins. J. Exp. Clin. Cancer Res. 2018, 37, 30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hirai, H.; Iwasawa, Y.; Okada, M.; Arai, T.; Nishibata, T.; Kobayashi, M.; Kimura, T.; Kaneko, N.; Ohtani, J.; Yamanaka, K.; et al. Small-molecule inhibition of Wee1 kinase by MK-1775 selectively sensitizes p53-deficient tumor cells to DNA-damaging agents. Mol. Cancer Ther. 2009, 8, 2992–3000. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- U.S. National Library of Medicine. Clinical Trials. 2018. Available online: https://clinicaltrials.gov/ct2/show/NCT01357161 (accessed on 31 May 2018).
- Duffy, M.J.; Synnott, N.C.; Crown, J. Mutant p53 as a target for cancer treatment. Eur. J. Cancer 2017, 83, 258–265. [Google Scholar] [CrossRef] [PubMed]
- Haupt, S.; Raghu, D.; Haupt, Y. Mutant p53 Drives Cancer by Subverting Multiple Tumor Suppression Pathways. Front. Oncol. 2016, 6, 12. [Google Scholar] [CrossRef] [PubMed]
- Willis, A.; Jung, E.J.; Wakefield, T.; Chen, X. Mutant p53 exerts a dominant negative effect by preventing wild-type p53 from binding to the promoter of its target genes. Oncogene 2004, 23, 2330–2338. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vijayakumaran, R.; Tan, K.H.; Miranda, P.J.; Haupt, S.; Haupt, Y. Regulation of Mutant p53 Protein Expression. Front. Oncol. 2015, 5, 284. [Google Scholar] [CrossRef] [PubMed]
- Xue, W.; Zender, L.; Miething, C.; Dickins, R.A.; Hernando, E.; Krizhanovsky, V.; Cordon-Cardo, C.; Lowe, S.W. Senescence and tumour clearance is triggered by p53 restoration in murine liver carcinomas. Nature 2007, 445, 656–660. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muller, P.A.; Vousden, K.H. p53 mutations in cancer. Nat. Cell Biol. 2013, 15, 2–8. [Google Scholar] [CrossRef] [PubMed]
- Parrales, A.; Ranjan, A.; Iyer, S.V.; Padhye, S.; Weir, S.J.; Roy, A.; Iwakuma, T. DNAJA1 controls the fate of misfolded mutant p53 through the mevalonate pathway. Nat. Cell Biol. 2016, 18, 1233–1243. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nordstrom, T.; Clements, M.; Karlsson, R.; Adolfsson, J.; Gronberg, H. The risk of prostate cancer for men on aspirin, statin or antidiabetic medications. Eur. J. Cancer 2015, 51, 725–733. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Suh, Y.A.; Fuller, M.Y.; Jackson, J.G.; Xiong, S.; Terzian, T.; Quintas-Cardama, A.; Bankson, J.A.; El-Naggar, A.K.; Lozano, G. Restoring expression of wild-type p53 suppresses tumor growth but does not cause tumor regression in mice with a p53 missense mutation. J. Clin. Investig. 2011, 121, 893–904. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, T.; Lee, K.; Rehman, A.; Daoud, S.S. PRIMA-1 induces apoptosis by inhibiting JNK signaling but promoting the activation of Bax. Biochem. Biophys. Res. Commun. 2007, 352, 203–212. [Google Scholar] [CrossRef] [PubMed]
- Lambert, J.M.; Moshfegh, A.; Hainaut, P.; Wiman, K.G.; Bykov, V.J. Mutant p53 reactivation by PRIMA-1MET induces multiple signaling pathways converging on apoptosis. Oncogene 2010, 29, 1329–1338. [Google Scholar] [CrossRef] [PubMed]
- Zandi, R.; Selivanova, G.; Christensen, C.L.; Gerds, T.A.; Willumsen, B.M.; Poulsen, H.S. PRIMA-1Met/APR-246 induces apoptosis and tumor growth delay in small cell lung cancer expressing mutant p53. Clin. Cancer Res. 2011, 17, 2830–2841. [Google Scholar] [CrossRef] [PubMed]
- Saha, M.N.; Jiang, H.; Yang, Y.; Reece, D.; Chang, H. PRIMA-1Met/APR-246 displays high antitumor activity in multiple myeloma by induction of p73 and Noxa. Mol. Cancer Ther. 2013, 12, 2331–2341. [Google Scholar] [CrossRef] [PubMed]
- Liang, Y.; Besch-Williford, C.; Hyder, S.M. PRIMA-1 inhibits growth of breast cancer cells by re-activating mutant p53 protein. Int. J. Oncol. 2009, 35, 1015–1023. [Google Scholar] [PubMed]
- Walerych, D.; Lisek, K.; Sommaggio, R.; Piazza, S.; Ciani, Y.; Dalla, E.; Rajkowska, K.; Gaweda-Walerych, K.; Ingallina, E.; Tonelli, C.; et al. Proteasome machinery is instrumental in a common gain-of-function program of the p53 missense mutants in cancer. Nat. Cell Biol. 2016, 18, 897–909. [Google Scholar] [CrossRef] [PubMed]
- Garufi, A.; Pistritto, G.; Cirone, M.; D’Orazi, G. Reactivation of mutant p53 by capsaicin, the major constituent of peppers. J. Exp. Clin. Cancer Res. 2016, 35, 136. [Google Scholar] [CrossRef] [PubMed]
- Cordani, M.; Oppici, E.; Dando, I.; Butturini, E.; Dalla Pozza, E.; Nadal-Serrano, M.; Oliver, J.; Roca, P.; Mariotto, S.; Cellini, B.; et al. Mutant p53 proteins counteract autophagic mechanism sensitizing cancer cells to mTOR inhibition. Mol. Oncol. 2016, 10, 1008–1029. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aggarwal, M.; Saxena, R.; Sinclair, E.; Fu, Y.; Jacobs, A.; Dyba, M.; Wang, X.; Cruz, I.; Berry, D.; Kallakury, B.; et al. Reactivation of mutant p53 by a dietary-related compound phenethyl isothiocyanate inhibits tumor growth. Cell Death Differ. 2016, 23, 1615–1627. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Agostino, S.; Cortese, G.; Monti, O.; Dell’Orso, S.; Sacchi, A.; Eisenstein, M.; Citro, G.; Strano, S.; Blandino, G. The disruption of the protein complex mutantp53/p73 increases selectively the response of tumor cells to anticancer drugs. Cell Cycle 2008, 7, 3440–3447. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, X.; Wilcken, R.; Joerger, A.C.; Chuckowree, I.S.; Amin, J.; Spencer, J.; Fersht, A.R. Small molecule induced reactivation of mutant p53 in cancer cells. Nucleic Acids Res. 2013, 41, 6034–6044. [Google Scholar] [CrossRef] [PubMed]
- Goldenson, B.; Crispino, J.D. The aurora kinases in cell cycle and leukemia. Oncogene 2015, 34, 537–545. [Google Scholar] [CrossRef] [PubMed]
- Nikonova, A.S.; Astsaturov, I.; Serebriiskii, I.G.; Dunbrack, R.L.; Golemis, E.A. Aurora-A kinase (AURKA) in normal and pathological cell growth. Cell. Mol. Life Sci. 2013, 70, 661–687. [Google Scholar] [CrossRef] [PubMed]
- Guan, Z.; Wang, X.R.; Zhu, X.F.; Huang, X.F.; Xu, J.; Wang, L.H.; Wan, X.B.; Long, Z.J.; Liu, J.N.; Feng, G.K.; et al. Aurora-A, a negative prognostic marker, increases migration and decreases radiosensitivity in cancer cells. Cancer Res. 2007, 67, 10436–10444. [Google Scholar] [CrossRef] [PubMed]
- Oh, E.T.; Byun, M.S.; Lee, H.; Park, M.T.; Jue, D.M.; Lee, C.W.; Lim, B.U.; Park, H.J. Aurora-a contributes to radioresistance by increasing NF-kappaB DNA binding. Radiat. Res. 2010, 174, 265–273. [Google Scholar] [CrossRef] [PubMed]
- Thollet, A.; Vendrell, J.A.; Payen, L.; Ghayad, S.E.; Ben Larbi, S.; Grisard, E.; Collins, C.; Villedieu, M.; Cohen, P.A. ZNF217 confers resistance to the pro-apoptotic signals of paclitaxel and aberrant expression of Aurora-A in breast cancer cells. Mol. Cancer 2010, 9, 291. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, K.; Chen, J.; Chen, D.; Huang, J.; Feng, B.; Han, S.; Chen, Y.; Song, H.; De, W.; Zhu, Z.; et al. Aurora-A promotes chemoresistance in hepatocelluar carcinoma by targeting NF-kappaB/microRNA-21/PTEN signaling pathway. Oncotarget 2014, 5, 12916–12935. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, H.; He, L.; Kruk, P.; Nicosia, S.V.; Cheng, J.Q. Aurora-A induces cell survival and chemoresistance by activation of Akt through a p53-dependent manner in ovarian cancer cells. Int. J. Cancer 2006, 119, 2304–2312. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, H.; Wang, Y.; Wang, Z.; Meng, J.; Qi, Z.; Yang, G. Aurora-A controls cancer cell radio- and chemoresistance via ATM/Chk2-mediated DNA repair networks. Biochim. Biophys. Acta 2014, 1843, 934–944. [Google Scholar] [CrossRef] [PubMed]
- Kelly, K.R.; Nawrocki, S.T.; Espitia, C.M.; Zhang, M.; Yang, J.J.; Padmanabhan, S.; Ecsedy, J.; Giles, F.J.; Carew, J.S. Targeting Aurora A kinase activity with the investigational agent alisertib increases the efficacy of cytarabine through a FOXO-dependent mechanism. Int. J. Cancer 2012, 131, 2693–2703. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kelly, K.R.; Ecsedy, J.; Medina, E.; Mahalingam, D.; Padmanabhan, S.; Nawrocki, S.T.; Giles, F.J.; Carew, J.S. The novel Aurora A kinase inhibitor MLN8237 is active in resistant chronic myeloid leukaemia and significantly increases the efficacy of nilotinib. J. Cell. Mol. Med. 2011, 15, 2057–2070. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chefetz, I.; Holmberg, J.C.; Alvero, A.B.; Visintin, I.; Mor, G. Inhibition of Aurora-A kinase induces cell cycle arrest in epithelial ovarian cancer stem cells by affecting NFkB pathway. Cell Cycle 2011, 10, 2206–2214. [Google Scholar] [CrossRef] [PubMed]
- Narod, S.A.; Foulkes, W.D. BRCA1 and BRCA2: 1994 and beyond. Nat. Rev. Cancer 2004, 4, 665–676. [Google Scholar] [CrossRef] [PubMed]
- Kadouri, L.; Hubert, A.; Rotenberg, Y.; Hamburger, T.; Sagi, M.; Nechushtan, C.; Abeliovich, D.; Peretz, T. Cancer risks in carriers of the BRCA1/2 Ashkenazi founder mutations. J. Med. Genet. 2007, 44, 467–471. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Venkitaraman, A.R. Tracing the network connecting BRCA and Fanconi anaemia proteins. Nat. Rev. Cancer 2004, 4, 266–276. [Google Scholar] [CrossRef] [PubMed]
- D'Andrea, A.D.; Grompe, M. The Fanconi anaemia/BRCA pathway. Nat. Rev. Cancer 2003, 3, 23–34. [Google Scholar] [CrossRef] [PubMed]
- Farmer, H.; McCabe, N.; Lord, C.J.; Tutt, A.N.; Johnson, D.A.; Richardson, T.B.; Santarosa, M.; Dillon, K.J.; Hickson, I.; Knights, C.; et al. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature 2005, 434, 917–921. [Google Scholar] [CrossRef] [PubMed]
- Bryant, H.E.; Schultz, N.; Thomas, H.D.; Parker, K.M.; Flower, D.; Lopez, E.; Kyle, S.; Meuth, M.; Curtin, N.J.; Helleday, T. Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nature 2005, 434, 913–917. [Google Scholar] [CrossRef] [PubMed]
- Sakai, W.; Swisher, E.M.; Jacquemont, C.; Chandramohan, K.V.; Couch, F.J.; Langdon, S.P.; Wurz, K.; Higgins, J.; Villegas, E.; Taniguchi, T. Functional restoration of BRCA2 protein by secondary BRCA2 mutations in BRCA2-mutated ovarian carcinoma. Cancer Res. 2009, 69, 6381–6386. [Google Scholar] [CrossRef] [PubMed]
- Dhillon, K.K.; Swisher, E.M.; Taniguchi, T. Secondary mutations of BRCA1/2 and drug resistance. Cancer Sci. 2011, 102, 663–669. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wiltshire, T.; Senft, J.; Wang, Y.; Konat, G.W.; Wenger, S.L.; Reed, E.; Wang, W. BRCA1 contributes to cell cycle arrest and chemoresistance in response to the anticancer agent irofulven. Mol. Pharmacol. 2007, 71, 1051–1060. [Google Scholar] [CrossRef] [PubMed]
- Huang, F.; Goyal, N.; Sullivan, K.; Hanamshet, K.; Patel, M.; Mazina, O.M.; Wang, C.X.; An, W.F.; Spoonamore, J.; Metkar, S.; et al. Targeting BRCA1- and BRCA2-deficient cells with RAD52 small molecule inhibitors. Nucleic Acids Res. 2016, 44, 4189–4199. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dziadkowiec, K.N.; Gąsiorowska, E.; Nowak-Markwitz, E.; Jankowska, A. PARP inhibitors: Review of mechanisms of action and BRCA1/2 mutation targeting. Prz. Menopauzalny 2016, 15, 215–219. [Google Scholar] [CrossRef] [PubMed]
- Wei, W.; Sun, H.H.; Li, N.; Li, H.Y.; Li, X.; Li, Q.; Shen, X.H. WNT5A modulates cell cycle progression and contributes to the chemoresistance in pancreatic cancer cells. Hepatobiliary Pancreat. Dis. Int. 2014, 13, 529–538. [Google Scholar] [CrossRef]
- Asem, M.S.; Buechler, S.; Wates, R.B.; Miller, D.L.; Stack, M.S. Wnt5a Signaling in Cancer. Cancers 2016, 8, 79. [Google Scholar] [CrossRef] [PubMed]
- Medrek, C.; Landberg, G.; Andersson, T.; Leandersson, K. Wnt-5a-CKIα signaling promotes β-catenin/E-cadherin complex formation and intercellular adhesion in human breast epithelial cells. J. Biol. Chem. 2009, 284, 10968–10979. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, H.; Oue, N.; Sato, A.; Hasegawa, Y.; Matsubara, A.; Yasui, W.; Kikuchi, A. Wnt5a signaling is involved in the aggressiveness of prostate cancer and expression of metalloproteinase. Oncogene 2010, 29, 2036–2046. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Da Forno, P.D.; Pringle, J.H.; Hutchinson, P.; Osborn, J.; Huang, Q.; Potter, L.; Hancox, R.A.; Fletcher, A.; Saldanha, G.S. WNT5A expression increases during melanoma progression and correlates with outcome. Clin. Cancer Res. 2008, 14, 5825–5832. [Google Scholar] [CrossRef] [PubMed]
- Anastas, J.N.; Kulikauskas, R.M.; Tamir, T.; Rizos, H.; Long, G.V.; von Euw, E.M.; Yang, P.T.; Chen, H.W.; Haydu, L.; Toroni, R.A.; et al. WNT5A enhances resistance of melanoma cells to targeted BRAF inhibitors. J. Clin. Investig. 2014, 124, 2877–2890. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dissanayake, S.K.; Weeraratna, A.T. Detecting PKC phosphorylation as part of the Wnt/calcium pathway in cutaneous melanoma. Methods Mol. Biol. 2008, 468, 157–172. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Zhang, K.; Wu, J.; Shi, J.; Xue, J.; Li, J.; Chen, J.; Zhu, Y.; Wei, J.; He, J.; et al. Wnt5a Increases Properties of Lung Cancer Stem Cells and Resistance to Cisplatin through Activation of Wnt5a/PKC Signaling Pathway. Stem Cells Int. 2016, 2016, 1690896. [Google Scholar] [CrossRef] [PubMed]
- Griesmann, H.; Ripka, S.; Pralle, M.; Ellenrieder, V.; Baumgart, S.; Buchholz, M.; Pilarsky, C.; Aust, D.; Gress, T.M.; Michl, P. WNT5A-NFAT signaling mediates resistance to apoptosis in pancreatic cancer. Neoplasia 2013, 15, 11–22. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Prieto, R.; Rojas, J.M.; Taya, Y.; Gutkind, J.S. A role for the p38 mitogen-acitvated protein kinase pathway in the transcriptional activation of p53 on genotoxic stress by chemotherapeutic agents. Cancer Res. 2000, 60, 2464–2472. [Google Scholar] [PubMed]
- Cai, B.; Chang, S.H.; Becker, E.B.; Bonni, A.; Xia, Z. p38 MAP kinase mediates apoptosis through phosphorylation of BimEL at Ser-65. J. Biol. Chem. 2006, 281, 25215–25222. [Google Scholar] [CrossRef] [PubMed]
- Manke, I.A.; Nguyen, A.; Lim, D.; Stewart, M.Q.; Elia, A.E.; Yaffe, M.B. MAPKAP kinase-2 is a cell cycle checkpoint kinase that regulates the G2/M transition and S phase progression in response to UV irradiation. Mol. Cell 2005, 17, 37–48. [Google Scholar] [CrossRef] [PubMed]
- Reinhardt, H.C.; Aslanian, A.S.; Lees, J.A.; Yaffe, M.B. p53-deficient cells rely on ATM- and ATR-mediated checkpoint signaling through the p38MAPK/MK2 pathway for survival after DNA damage. Cancer Cell 2007, 11, 175–189. [Google Scholar] [CrossRef] [PubMed]
- Pan, S.T.; Li, Z.L.; He, Z.X.; Qiu, J.X.; Zhou, S.F. Molecular mechanisms for tumour resistance to chemotherapy. Clin. Exp. Pharmacol. Physiol. 2016, 43, 723–737. [Google Scholar] [CrossRef] [PubMed]
- Igea, A.; Nebreda, A.R. The Stress Kinase p38α as a Target for Cancer Therapy. Cancer Res. 2015, 75, 3997–4002. [Google Scholar] [CrossRef] [PubMed]
- Mikhailov, A.; Shinohara, M.; Rieder, C.L. Topoisomerase II and histone deacetylase inhibitors delay the G2/M transition by triggering the p38 MAPK checkpoint pathway. J. Cell Biol. 2004, 166, 517–526. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mikhailov, A.; Shinohara, M.; Rieder, C.L. The p38-mediated stress-activated checkpoint. A rapid response system for delaying progression through antephase and entry into mitosis. Cell Cycle 2005, 4, 57–62. [Google Scholar] [CrossRef] [PubMed]
- Bucher, N.; Britten, C.D. G2 checkpoint abrogation and checkpoint kinase-1 targeting in the treatment of cancer. Br. J. Cancer 2008, 98, 523–528. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morandell, S.; Reinhardt, H.C.; Cannell, I.G.; Kim, J.S.; Ruf, D.M.; Mitra, T.; Couvillon, A.D.; Jacks, T.; Yaffe, M.B. A reversible gene-targeting strategy identifies synthetic lethal interactions between MK2 and p53 in the DNA damage response in vivo. Cell Rep. 2013, 5, 868–877. [Google Scholar] [CrossRef] [PubMed]
- Tan, W.; Yu, H.G.; Luo, H.S. Inhibition of the p38 MAPK pathway sensitizes human gastric cells to doxorubicin treatment in vitro and in vivo. Mol. Med. Rep. 2014, 10, 3275–3281. [Google Scholar] [CrossRef] [PubMed]
- Paillas, S.; Boissiere, F.; Bibeau, F.; Denouel, A.; Mollevi, C.; Causse, A.; Denis, V.; Vezzio-Vie, N.; Marzi, L.; Cortijo, C.; et al. Targeting the p38 MAPK pathway inhibits irinotecan resistance in colon adenocarcinoma. Cancer Res. 2011, 71, 1041–1049. [Google Scholar] [CrossRef] [PubMed]
- Kopper, F.; Binkowski, A.M.; Bierwirth, C.; Dobbelstein, M. The MAPK-activated protein kinase 2 mediates gemcitabine sensitivity in pancreatic cancer cells. Cell Cycle 2014, 13, 884–889. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gu, C.; Cheng, H.; Yang, H.; Bian, Y.; Wang, Y.; Zhang, Y.; Pisano, M.; Hu, G.; Yang, Y. MK2 is a therapeutic target for high-risk multiple myeloma. Haematologica 2018, 103. [Google Scholar] [CrossRef] [PubMed]
- Walczak, H. Death receptor-ligand systems in cancer, cell death, and inflammation. Cold Spring Harb. Perspect. Biol. 2013, 5, a008698. [Google Scholar] [CrossRef] [PubMed]
- Kurokawa, M.; Kornbluth, S. Caspases and kinases in a death grip. Cell 2009, 138, 838–854. [Google Scholar] [CrossRef] [PubMed]
- Shamas-Din, A.; Brahmbhatt, H.; Leber, B.; Andrews, D.W. BH3-only proteins: Orchestrators of apoptosis. Biochim. Biophys. Acta 2011, 1813, 508–520. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Zhu, H.; Xu, C.J.; Yuan, J. Cleavage of BID by caspase 8 mediates the mitochondrial damage in the Fas pathway of apoptosis. Cell 1998, 94, 491–501. [Google Scholar] [CrossRef]
- LaCasse, E.C.; Mahoney, D.J.; Cheung, H.H.; Plenchette, S.; Baird, S.; Korneluk, R.G. IAP-targeted therapies for cancer. Oncogene 2008, 27, 6252–6275. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sadarangani, A.; Kato, S.; Espinoza, N.; Lange, S.; Llados, C.; Espinosa, M.; Villalon, M.; Lipkowitz, S.; Cuello, M.; Owen, G.I. TRAIL mediates apoptosis in cancerous but not normal primary cultured cells of the human reproductive tract. Apoptosis 2007, 12, 73–85. [Google Scholar] [CrossRef] [PubMed]
- Trivedi, R.; Mishra, D.P. Trailing TRAIL Resistance: Novel Targets for TRAIL Sensitization in Cancer Cells. Front. Oncol. 2015, 5, 69. [Google Scholar] [CrossRef] [PubMed]
- Twomey, J.D.; Kim, S.-R.; Zhao, L.; Bozza, W.P.; Zhang, B. Spatial dynamics of TRAIL death receptors in cancer cells. Drug Resist. Updates 2015, 19, 13–21. [Google Scholar] [CrossRef] [PubMed]
- Yoon, M.J.; Kang, Y.J.; Kim, I.Y.; Kim, E.H.; Lee, J.A.; Lim, J.H.; Kwon, T.K.; Choi, K.S. Monensin, a polyether ionophore antibiotic, overcomes TRAIL resistance in glioma cells via endoplasmic reticulum stress, DR5 upregulation and c-FLIP downregulation. Carcinogenesis 2013, 34, 1918–1928. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trivedi, R.; Maurya, R.; Mishra, D.P. Medicarpin, a legume phytoalexin sensitizes myeloid leukemia cells to TRAIL-induced apoptosis through the induction of DR5 and activation of the ROS-JNK-CHOP pathway. Cell Death Dis. 2014, 5, e1465. [Google Scholar] [CrossRef] [PubMed]
- Murai, M.; Inoue, T.; Suzuki-Karasaki, M.; Ochiai, T.; Ra, C.; Nishida, S.; Suzuki-Karasaki, Y. Diallyl trisulfide sensitizes human melanoma cells to TRAIL-induced cell death by promoting endoplasmic reticulum-mediated apoptosis. Int. J. Oncol. 2012, 41, 2029–2037. [Google Scholar] [CrossRef] [PubMed]
- Jiang, C.C.; Chen, L.H.; Gillespie, S.; Kiejda, K.A.; Mhaidat, N.; Wang, Y.F.; Thorne, R.; Zhang, X.D.; Hersey, P. Tunicamycin sensitizes human melanoma cells to tumor necrosis factor-related apoptosis-inducing ligand-induced apoptosis by up-regulation of TRAIL-R2 via the unfolded protein response. Cancer Res. 2007, 67, 5880–5888. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.F.; Cao, J.G.; Tian, L.; Liu, F. 5,7-Dimethoxyflavone sensitizes TRAIL-induced apoptosis through DR5 upregulation in hepatocellular carcinoma cells. Cancer Chemother. Pharmacol. 2012, 69, 195–206. [Google Scholar] [CrossRef] [PubMed]
- Kandasamy, K.; Kraft, A.S. Proteasome inhibitor PS-341 (VELCADE) induces stabilization of the TRAIL receptor DR5 mRNA through the 3'-untranslated region. Mol. Cancer Ther. 2008, 7, 1091–1100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, Y.; Lakshmikanthan, V.; Lewis, R.W.; Kumar, M.V. Sensitization of TRAIL-resistant cells by inhibition of heat shock protein 90 with low-dose geldanamycin. Mol. Cancer Ther. 2006, 5, 170–178. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siegelin, M.D.; Habel, A.; Gaiser, T. 17-AAG sensitized malignant glioma cells to death-receptor mediated apoptosis. Neurobiol. Dis. 2009, 33, 243–249. [Google Scholar] [CrossRef] [PubMed]
- Mellier, G.; Liu, D.; Bellot, G.; Holme, A.L.; Pervaiz, S. Small molecule sensitization to TRAIL is mediated via nuclear localization, phosphorylation and inhibition of chaperone activity of Hsp27. Cell Death Dis. 2013, 4, e890. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Lu, Y.; Shen, H.-M. Targeting p53 as a therapeutic strategy in sensitizing TRAIL-induced apoptosis in cancer cells. Cancer Lett. 2012, 314, 8–23. [Google Scholar] [CrossRef] [PubMed]
- Hou, W.; Han, J.; Lu, C.; Goldstein, L.A.; Rabinowich, H. Enhancement of tumor-TRAIL susceptibility by modulation of autophagy. Autophagy 2008, 4, 940–943. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hernandez, A.; Wang, Q.D.; Schwartz, S.A.; Evers, B.M. Sensitization of human colon cancer cells to TRAIL-mediated apoptosis. J. Gastrointest. Surg. 2001, 5, 56–65. [Google Scholar] [CrossRef]
- Kim, H.B.; Kim, M.J.; Lee, S.H.; Lee, J.W.; Bae, J.H.; Kim, D.W.; Dao, T.T.; Oh, W.K.; Kang, C.D.; Kim, S.H. Amurensin G, a novel SIRT1 inhibitor, sensitizes TRAIL-resistant human leukemic K562 cells to TRAIL-induced apoptosis. Biochem. Pharmacol. 2012, 84, 402–410. [Google Scholar] [CrossRef] [PubMed]
- Ueffing, N.; Keil, E.; Freund, C.; Kühne, R.; Schulze-Osthoff, K.; Schmitz, I. Mutational analyses of c-FLIPR, the only murine short FLIP isoform, reveal requirements for DISC recruitment. Cell Death Differ. 2008, 15, 773–782. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thome, M.; Schneider, P.; Hofmann, K.; Fickenscher, H.; Meinl, E.; Neipel, F.; Mattmann, C.; Burns, K.; Bodmer, J.L.; Schroter, M.; et al. Viral FLICE-inhibitory proteins (FLIPs) prevent apoptosis induced by death receptors. Nature 1997, 386, 517–521. [Google Scholar] [CrossRef] [PubMed]
- Kataoka, T.; Ito, M.; Budd, R.C.; Tschopp, J.; Nagai, K. Expression level of c-FLIP versus Fas determines susceptibility to Fas ligand-induced cell death in murine thymoma EL-4 cells. Exp. Cell Res. 2002, 273, 256–264. [Google Scholar] [CrossRef] [PubMed]
- Wilson, T.R.; McLaughlin, K.M.; McEwan, M.; Sakai, H.; Rogers, K.M.; Redmond, K.M.; Johnston, P.G.; Longley, D.B. c-FLIP: A key regulator of colorectal cancer cell death. Cancer Res. 2007, 67, 5754–5762. [Google Scholar] [CrossRef] [PubMed]
- El-Gazzar, A.; Wittinger, M.; Perco, P.; Anees, M.; Horvat, R.; Mikulits, W.; Grunt, T.W.; Mayer, B.; Krainer, M. The role of c-FLIP(L) in ovarian cancer: Chaperoning tumor cells from immunosurveillance and increasing their invasive potential. Gynecol. Oncol. 2010, 117, 451–459. [Google Scholar] [CrossRef] [PubMed]
- Haag, C.; Stadel, D.; Zhou, S.; Bachem, M.G.; Moller, P.; Debatin, K.M.; Fulda, S. Identification of c-FLIP(L) and c-FLIP(S) as critical regulators of death receptor-induced apoptosis in pancreatic cancer cells. Gut 2011, 60, 225–237. [Google Scholar] [CrossRef] [PubMed]
- Ullenhag, G.J.; Mukherjee, A.; Watson, N.F.; Al-Attar, A.H.; Scholefield, J.H.; Durrant, L.G. Overexpression of FLIPL is an independent marker of poor prognosis in colorectal cancer patients. Clin. Cancer Res. 2007, 13, 5070–5075. [Google Scholar] [CrossRef] [PubMed]
- Shirley, S.; Micheau, O. Targeting c-FLIP in cancer. Cancer Lett. 2013, 332, 141–150. [Google Scholar] [CrossRef] [PubMed]
- Wilson, C.; Wilson, T.; Johnston, P.G.; Longley, D.B.; Waugh, D.J. Interleukin-8 signaling attenuates TRAIL- and chemotherapy-induced apoptosis through transcriptional regulation of c-FLIP in prostate cancer cells. Mol. Cancer Ther. 2008, 7, 2649–2661. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seo, S.U.; Cho, H.K.; Min, K.-J.; Woo, S.M.; Kim, S.; Park, J.-W.; Kim, S.H.; Choi, Y.H.; Keum, Y.S.; Hyun, J.W.; et al. Thioridazine enhances sensitivity to carboplatin in human head and neck cancer cells through downregulation of c-FLIP and Mcl-1 expression. Cell Death Dis. 2017, 8, e2599. [Google Scholar] [CrossRef] [PubMed]
- Kauh, J.; Fan, S.; Xia, M.; Yue, P.; Yang, L.; Khuri, F.R.; Sun, S.Y. c-FLIP degradation mediates sensitization of pancreatic cancer cells to TRAIL-induced apoptosis by the histone deacetylase inhibitor LBH589. PLoS ONE 2010, 5, e10376. [Google Scholar] [CrossRef] [PubMed]
- Henrich, C.J.; Brooks, A.D.; Erickson, K.L.; Thomas, C.L.; Bokesch, H.R.; Tewary, P.; Thompson, C.R.; Pompei, R.J.; Gustafson, K.R.; McMahon, J.B.; et al. Withanolide E sensitizes renal carcinoma cells to TRAIL-induced apoptosis by increasing cFLIP degradation. Cell Death Dis. 2015, 6, e1666. [Google Scholar] [CrossRef] [PubMed]
- Kolar, Z.; Murray, P.G.; Scott, K.; Harrison, A.; Vojtesek, B.; Dusek, J. Relation of Bcl-2 expression to androgen receptor, p21WAF1/CIP1, and cyclin D1 status in prostate cancer. Mol. Pathol. 2000, 53, 15–18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, J.; Jin, S.; Abraham, V.; Huang, X.; Liu, B.; Mitten, M.J.; Nimmer, P.; Lin, X.; Smith, M.; Shen, Y.; et al. The Bcl-2/Bcl-X(L)/Bcl-w inhibitor, navitoclax, enhances the activity of chemotherapeutic agents in vitro and in vivo. Mol. Cancer Ther. 2011, 10, 2340–2349. [Google Scholar] [CrossRef] [PubMed]
- Scherr, A.L.; Gdynia, G.; Salou, M.; Radhakrishnan, P.; Duglova, K.; Heller, A.; Keim, S.; Kautz, N.; Jassowicz, A.; Elssner, C.; et al. Bcl-xL is an oncogenic driver in colorectal cancer. Cell Death Dis. 2016, 7, e2342. [Google Scholar] [CrossRef] [PubMed]
- Kotschy, A.; Szlavik, Z.; Murray, J.; Davidson, J.; Maragno, A.L.; Le Toumelin-Braizat, G.; Chanrion, M.; Kelly, G.L.; Gong, J.N.; Moujalled, D.M.; et al. The MCL1 inhibitor S63845 is tolerable and effective in diverse cancer models. Nature 2016, 538, 477–482. [Google Scholar] [CrossRef] [PubMed]
- Bruncko, M.; Wang, L.; Sheppard, G.S.; Phillips, D.C.; Tahir, S.K.; Xue, J.; Erickson, S.; Fidanze, S.; Fry, E.; Hasvold, L.; et al. Structure-guided design of a series of MCL-1 inhibitors with high affinity and selectivity. J. Med. Chem. 2015, 58, 2180–2194. [Google Scholar] [CrossRef] [PubMed]
- Bittker, J.A.; Weiwer, M.; Wei, G.; Germain, A.; Brown, E.; Dandapani, S.; Munoz, B.; Palmer, M.; Golub, T.; Schreiber, S.L. Discovery of inhibitors of anti-apoptotic protein A1. In Probe Reports from the NIH Molecular Libraries Program; National Center for Biotechnology Information (US): Bethesda, MD, USA, 2010. [Google Scholar]
- Pluta, P.; Jeziorski, A.; Cebula-Obrzut, A.P.; Wierzbowska, A.; Piekarski, J.; Smolewski, P. Expression of IAP family proteins and its clinical importance in breast cancer patients. Neoplasma 2015, 62, 666–673. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Endo, T.; Abe, S.; Seidlar, H.B.; Nagaoka, S.; Takemura, T.; Utsuyama, M.; Kitagawa, M.; Hirokawa, K. Expression of IAP family proteins in colon cancers from patients with different age groups. Cancer Immunol. Immunother. 2004, 53, 770–776. [Google Scholar] [CrossRef] [PubMed]
- Nemoto, T.; Kitagawa, M.; Hasegawa, M.; Ikeda, S.; Akashi, T.; Takizawa, T.; Hirokawa, K.; Koike, M. Expression of IAP family proteins in esophageal cancer. Exp. Mol. Pathol. 2004, 76, 253–259. [Google Scholar] [CrossRef] [PubMed]
- Espinosa, M.; Cantu, D.; Herrera, N.; Lopez, C.M.; De la Garza, J.G.; Maldonado, V.; Melendez-Zajgla, J. Inhibitors of apoptosis proteins in human cervical cancer. BMC Cancer 2006, 6, 45. [Google Scholar] [CrossRef] [PubMed]
- Moon, J.-H.; Shin, J.-S.; Hong, S.-W.; Jung, S.-A.; Hwang, I.-Y.; Kim, J.H.; Choi, E.K.; Ha, S.-H.; Kim, J.-S.; Kim, K.-M.; et al. A novel small-molecule IAP antagonist, AZD5582, draws Mcl-1 down-regulation for induction of apoptosis through targeting of cIAP1 and XIAP in human pancreatic cancer. Oncotarget 2015, 6, 26895–26908. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- LaCasse, E.C.; Cherton-Horvat, G.G.; Hewitt, K.E.; Jerome, L.J.; Morris, S.J.; Kandimalla, E.R.; Yu, D.; Wang, H.; Wang, W.; Zhang, R.; et al. Preclinical Characterization of AEG35156/GEM 640, a Second-Generation Antisense Oligonucleotide Targeting X-Linked Inhibitor of Apoptosis. Clin. Cancer Res. 2006, 12, 5231–5241. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dean, E.; Jodrell, D.; Connolly, K.; Danson, S.; Jolivet, J.; Durkin, J.; Morris, S.; Jowle, D.; Ward, T.; Cummings, J.; et al. Phase I trial of AEG35156 administered as a 7-day and 3-day continuous intravenous infusion in patients with advanced refractory cancer. J. Clin. Oncol 2009, 27, 1660–1666. [Google Scholar] [CrossRef] [PubMed]
- Gyuraszova, K.; Mikes, J.; Halaburkova, A.; Jendzelovsky, R.; Fedorocko, P. YM155, a small molecule inhibitor of survivin expression, sensitizes cancer cells to hypericin-mediated photodynamic therapy. Photochem. Photobiol. Sci. 2016, 15, 812–821. [Google Scholar] [CrossRef] [PubMed]
- Foster, F.; Owens, T.; Tanianis-Hughes, J.; Clarke, R.; Brennan, K.; Bundred, N.; Streuli, C. Targeting inhibitor of apoptosis proteins in combination with ErbB antagonists in breast cancer. Breast Cancer Res. 2009, 11, R41. [Google Scholar] [CrossRef] [PubMed]
- Fandy, T.E.; Shankar, S.; Srivastava, R.K. Smac/DIABLO enhances the therapeutic potential of chemotherapeutic drugs and irradiation, and sensitizes TRAIL-resistant breast cancer cells. Mol. Cancer 2008, 7, 60. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ziegler, D.S.; Wright, R.D.; Kesari, S.; Lemieux, M.E.; Tran, M.A.; Jain, M.; Zawel, L.; Kung, A.L. Resistance of human glioblastoma multiforme cells to growth factor inhibitors is overcome by blockade of inhibitor of apoptosis proteins. J. Clin. Investig. 2008, 118, 3109–3122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weisberg, E.; Kung, A.L.; Wright, R.D.; Moreno, D.; Catley, L.; Ray, A.; Zawel, L.; Tran, M.; Cools, J.; Gilliland, G.; et al. Potentiation of antileukemic therapies by Smac mimetic, LBW242: Effects on mutant FLT3-expressing cells. Mol. Cancer Ther. 2007, 6, 1951–1961. [Google Scholar] [CrossRef] [PubMed]
- Weisberg, E.; Ray, A.; Barrett, R.; Nelson, E.; Christie, A.L.; Porter, D.; Straub, C.; Zawel, L.; Daley, J.F.; Lazo-Kallanian, S.; et al. Smac mimetics: Implications for enhancement of targeted therapies in leukemia. Leukemia 2010, 24, 2100–2109. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.-A.; Kim, S.-W.; Nam, J.; Sung, E.-G.; Song, I.-H.; Kim, J.-Y.; Kwon, T.K.; Lee, T.-J. Inhibition of c-FLIP(L) expression by miRNA-708 increases the sensitivity of renal cancer cells to anti-cancer drugs. Oncotarget 2016, 7, 31832–31846. [Google Scholar] [CrossRef] [PubMed]
- Herman, M.D.; Nyman, T.; Welin, M.; Lehtio, L.; Flodin, S.; Tresaugues, L.; Kotenyova, T.; Flores, A.; Nordlund, P. Completing the family portrait of the anti-apoptotic Bcl-2 proteins: Crystal structure of human Bfl-1 in complex with Bim. FEBS Lett. 2008, 582, 3590–3594. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deveraux, Q.L.; Takahashi, R.; Salvesen, G.S.; Reed, J.C. X-linked IAP is a direct inhibitor of cell-death proteases. Nature 1997, 388, 300–304. [Google Scholar] [CrossRef] [PubMed]
- Bai, L.; Smith, D.C.; Wang, S. Small-Molecule SMAC Mimetics as New Cancer Therapeutics. Pharmacol. Ther. 2014, 144, 82–95. [Google Scholar] [CrossRef] [PubMed]
- Galbán, S.; Hwang, C.; Rumble, J.M.; Oetjen, K.A.; Wright, C.W.; Boudreault, A.; Durkin, J.; Gillard, J.W.; Jaquith, J.B.; Morris, S.J.; et al. Cytoprotective effects of iaps revealed by a small molecule antagonist. Biochem. J. 2009, 417, 765–771. [Google Scholar] [CrossRef] [PubMed]
- Marusawa, H.; Matsuzawa, S.; Welsh, K.; Zou, H.; Armstrong, R.; Tamm, I.; Reed, J.C. HBXIP functions as a cofactor of survivin in apoptosis suppression. EMBO J. 2003, 22, 2729–2740. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, Z.; Yao, X.; Wu, M. Direct interaction between survivin and Smac/DIABLO is essential for the anti-apoptotic activity of survivin during taxol-induced apoptosis. J. Biol. Chem. 2003, 278, 23130–23140. [Google Scholar] [CrossRef] [PubMed]
- Partridge, M.; Green, M.R.; Langdon, J.D.; Feldmann, M. Production of TGF-α and TGF-β by cultured keratinocytes, skin and oral squamous cell carcinomas--potential autocrine regulation of normal and malignant epithelial cell proliferation. Br. J. Cancer 1989, 60, 542–548. [Google Scholar] [CrossRef] [PubMed]
- Imamura, T.; Hikita, A.; Inoue, Y. The roles of TGF-β signaling in carcinogenesis and breast cancer metastasis. Breast Cancer 2012, 19, 118–124. [Google Scholar] [CrossRef] [PubMed]
- Du, B.; Shim, J.S. Targeting Epithelial-Mesenchymal Transition (EMT) to Overcome Drug Resistance in Cancer. Molecules 2016, 21. [Google Scholar] [CrossRef] [PubMed]
- Colak, S.; Ten Dijke, P. Targeting TGF-β Signaling in Cancer. Trends Cancer 2017, 3, 56–71. [Google Scholar] [CrossRef] [PubMed]
- Roy, L.O.; Poirier, M.B.; Fortin, D. Differential Expression and Clinical Significance of Transforming Growth Factor-β Isoforms in GBM Tumors. Int. J. Mol. Sci. 2018, 19. [Google Scholar] [CrossRef] [PubMed]
- Mao, L.; Li, Y.; Zhao, J.; Li, Q.; Yang, B.; Wang, Y.; Zhu, Z.; Sun, H.; Zhai, Z. Transforming growth factor-β1 contributes to oxaliplatin resistance in colorectal cancer via epithelial to mesenchymal transition. Oncol. Lett. 2017, 14, 647–654. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.; Liu, H.; Yu, J.; Yu, H. Chemoresistance to doxorubicin induces epithelial-mesenchymal transition via upregulation of transforming growth factor β signaling in HCT116 colon cancer cells. Mol. Med. Rep. 2015, 12, 192–198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saxena, M.; Stephens, M.A.; Pathak, H.; Rangarajan, A. Transcription factors that mediate epithelial-mesenchymal transition lead to multidrug resistance by upregulating ABC transporters. Cell Death Dis. 2011, 2, e179. [Google Scholar] [CrossRef] [PubMed]
- Bhola, N.E.; Balko, J.M.; Dugger, T.C.; Kuba, M.G.; Sanchez, V.; Sanders, M.; Stanford, J.; Cook, R.S.; Arteaga, C.L. TGF-β inhibition enhances chemotherapy action against triple-negative breast cancer. J. Clin. Investig. 2013, 123, 1348–1358. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, C.Y.; Son, J.Y.; Jin, C.H.; Nam, J.S.; Kim, D.K.; Sheen, Y.Y. EW-7195, a novel inhibitor of ALK5 kinase inhibits EMT and breast cancer metastasis to lung. Eur. J. Cancer 2011, 47, 2642–2653. [Google Scholar] [CrossRef] [PubMed]
- Park, C.Y.; Kim, D.K.; Sheen, Y.Y. EW-7203, a novel small molecule inhibitor of transforming growth factor-β (TGF-β) type I receptor/activin receptor-like kinase-5, blocks TGF-β1-mediated epithelial-to-mesenchymal transition in mammary epithelial cells. Cancer Sci. 2011, 102, 1889–1896. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, C.Y.; Min, K.N.; Son, J.Y.; Park, S.Y.; Nam, J.S.; Kim, D.K.; Sheen, Y.Y. An novel inhibitor of TGF-β type I receptor, IN-1130, blocks breast cancer lung metastasis through inhibition of epithelial-mesenchymal transition. Cancer Lett. 2014, 351, 72–80. [Google Scholar] [CrossRef] [PubMed]
- Serova, M.; Tijeras-Raballand, A.; Dos Santos, C.; Albuquerque, M.; Paradis, V.; Neuzillet, C.; Benhadji, K.A.; Raymond, E.; Faivre, S.; de Gramont, A. Effects of TGF-β signalling inhibition with galunisertib (LY2157299) in hepatocellular carcinoma models and in ex vivo whole tumor tissue samples from patients. Oncotarget 2015, 6, 21614–21627. [Google Scholar] [CrossRef] [PubMed]
- Shain, K.H.; Dalton, W.S. Cell adhesion is a key determinant in de novo multidrug resistance (MDR): New targets for the prevention of acquired MDR. Mol. Cancer Ther. 2001, 1, 69–78. [Google Scholar] [PubMed]
- Dickreuter, E.; Cordes, N. The cancer cell adhesion resistome: Mechanisms, targeting and translational approaches. Biol. Chem. 2017, 398, 721–735. [Google Scholar] [CrossRef] [PubMed]
- Hazlehurst, L.A.; Dalton, W.S. Mechanisms associated with cell adhesion mediated drug resistance (CAM-DR) in hematopoietic malignancies. Cancer Metastasis Rev. 2001, 20, 43–50. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, T.; Kato, Y.; Fuji, H.; Horiuchi, T.; Chiba, Y.; Tanaka, K. E-cadherin-dependent intercellular adhesion enhances chemoresistance. Int. J. Mol. Med. 2003, 12, 693–700. [Google Scholar] [CrossRef] [PubMed]
- Schmidmaier, R.; Morsdorf, K.; Baumann, P.; Emmerich, B.; Meinhardt, G. Evidence for cell adhesion-mediated drug resistance of multiple myeloma cells in vivo. Int. J. Biol. Mark. 2006, 21, 218–222. [Google Scholar] [CrossRef]
- Bewick, M.A.; Lafrenie, R.M. Adhesion dependent signalling in the tumour microenvironment: The future of drug targetting. Curr. Pharm. Des. 2006, 12, 2833–2848. [Google Scholar] [CrossRef] [PubMed]
- Makrilia, N.; Kollias, A.; Manolopoulos, L.; Syrigos, K. Cell adhesion molecules: Role and clinical significance in cancer. Cancer Investig. 2009, 27, 1023–1037. [Google Scholar] [CrossRef] [PubMed]
- Okegawa, T.; Pong, R.C.; Li, Y.; Hsieh, J.T. The role of cell adhesion molecule in cancer progression and its application in cancer therapy. Acta Biochim. Pol. 2004, 51, 445–457. [Google Scholar] [PubMed]
- Trzpis, M.; McLaughlin, P.M.J.; de Leij, L.M.F.H.; Harmsen, M.C. Epithelial Cell Adhesion Molecule: More than a Carcinoma Marker and Adhesion Molecule. Am. J. Pathol. 2007, 171, 386–395. [Google Scholar] [CrossRef] [PubMed]
- Bellone, S.; Siegel, E.R.; Cocco, E.; Cargnelutti, M.; Silasi, D.A.; Azodi, M.; Schwartz, P.E.; Rutherford, T.J.; Pecorelli, S.; Santin, A.D. Overexpression of epithelial cell adhesion molecule in primary, metastatic, and recurrent/chemotherapy-resistant epithelial ovarian cancer: Implications for epithelial cell adhesion molecule-specific immunotherapy. Int. J. Gynecol. Cancer 2009, 19, 860–866. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Wang, L.; Mizokami, A.; Shi, J.; Zou, C.; Dai, J.; Keller, E.T.; Lu, Y.; Zhang, J. Down-regulation of E-cadherin enhances prostate cancer chemoresistance via Notch signaling. Chin. J. Cancer 2017, 36, 35. [Google Scholar] [CrossRef] [PubMed]
- Thiery, J.P.; Acloque, H.; Huang, R.Y.; Nieto, M.A. Epithelial-mesenchymal transitions in development and disease. Cell 2009, 139, 871–890. [Google Scholar] [CrossRef] [PubMed]
- Marin-Aguilera, M.; Codony-Servat, J.; Reig, O.; Lozano, J.J.; Fernandez, P.L.; Pereira, M.V.; Jimenez, N.; Donovan, M.; Puig, P.; Mengual, L.; et al. Epithelial-to-mesenchymal transition mediates docetaxel resistance and high risk of relapse in prostate cancer. Mol. Cancer Ther. 2014, 13, 1270–1284. [Google Scholar] [CrossRef] [PubMed]
- Shah, M.Y.; Martinez-Garcia, E.; Phillip, J.M.; Chambliss, A.B.; Popovic, R.; Ezponda, T.; Small, E.C.; Will, C.; Phillip, M.P.; Neri, P.; et al. MMSET/WHSC1 enhances DNA damage repair leading to an increase in resistance to chemotherapeutic agents. Oncogene 2016, 35, 5905–5915. [Google Scholar] [CrossRef] [PubMed]
- Furukawa, Y.; Kikuchi, J. Epigenetic mechanisms of cell adhesion-mediated drug resistance in multiple myeloma. Int. J. Hematol. 2016, 104, 281–292. [Google Scholar] [CrossRef] [PubMed]
- Jones, C.B.; McIntosh, J.; Huang, H.; Graytock, A.; Hoyt, D.G. Regulation of bleomycin-induced DNA breakage and chromatin structure in lung endothelial cells by integrins and poly(ADP-ribose) polymerase. Mol. Pharmacol. 2001, 59, 69–75. [Google Scholar] [CrossRef] [PubMed]
- Oloumi, A.; MacPhail, S.H.; Johnston, P.J.; Banath, J.P.; Olive, P.L. Changes in subcellular distribution of topoisomerase IIα correlate with etoposide resistance in multicell spheroids and xenograft tumors. Cancer Res. 2000, 60, 5747–5753. [Google Scholar] [PubMed]
- Damiano, J.S.; Hazlehurst, L.A.; Dalton, W.S. Cell adhesion-mediated drug resistance (CAM-DR) protects the K562 chronic myelogenous leukemia cell line from apoptosis induced by BCR/ABL inhibition, cytotoxic drugs, and gamma-irradiation. Leukemia 2001, 15, 1232–1239. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.; Van der Sluis, P.C.; Boulware, D.; Hazlehurst, L.A.; Dalton, W.S. The FA/BRCA pathway is involved in melphalan-induced DNA interstrand cross-link repair and accounts for melphalan resistance in multiple myeloma cells. Blood 2005, 106, 698–705. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hazlehurst, L.A.; Argilagos, R.F.; Emmons, M.; Boulware, D.; Beam, C.A.; Sullivan, D.M.; Dalton, W.S. Cell adhesion to fibronectin (CAM-DR) influences acquired mitoxantrone resistance in U937 cells. Cancer Res. 2006, 66, 2338–2345. [Google Scholar] [CrossRef] [PubMed]
- Aslan, B.; Monroig, P.; Hsu, M.C.; Pena, G.A.; Rodriguez-Aguayo, C.; Gonzalez-Villasana, V.; Rupaimoole, R.; Nagaraja, A.S.; Mangala, S.; Han, H.D.; et al. The ZNF304-integrin axis protects against anoikis in cancer. Nat. Commun. 2015, 6, 7351. [Google Scholar] [CrossRef] [PubMed]
- Simpson, C.D.; Anyiwe, K.; Schimmer, A.D. Anoikis resistance and tumor metastasis. Cancer Lett. 2008, 272, 177–185. [Google Scholar] [CrossRef] [PubMed]
- Uekita, T.; Tanaka, M.; Takigahira, M.; Miyazawa, Y.; Nakanishi, Y.; Kanai, Y.; Yanagihara, K.; Sakai, R. CUB-domain-containing protein 1 regulates peritoneal dissemination of gastric scirrhous carcinoma. Am. J. Pathol. 2008, 172, 1729–1739. [Google Scholar] [CrossRef] [PubMed]
- Shain, K.H.; Landowski, T.H.; Dalton, W.S. Adhesion-mediated intracellular redistribution of c-Fas-associated death domain-like IL-1-converting enzyme-like inhibitory protein-long confers resistance to CD95-induced apoptosis in hematopoietic cancer cell lines. J. Immunol. 2002, 168, 2544–2553. [Google Scholar] [CrossRef] [PubMed]
- Zahreddine, H.; Borden, K.L.B. Mechanisms and insights into drug resistance in cancer. Front. Pharmacol. 2013, 4, 28. [Google Scholar] [CrossRef] [PubMed]
- Jiang, W.G. E-cadherin and its associated protein catenins, cancer invasion and metastasis. Br. J. Surg. 1996, 83, 437–446. [Google Scholar] [CrossRef] [PubMed]
- Johnson, J.P. Cell adhesion molecules in the development and progression of malignant melanoma. Cancer Metastasis Rev. 1999, 18, 345–357. [Google Scholar] [CrossRef] [PubMed]
- Hazan, R.B.; Qiao, R.; Keren, R.; Badano, I.; Suyama, K. Cadherin switch in tumor progression. Ann. N. Y. Acad. Sci. 2004, 1014, 155–163. [Google Scholar] [CrossRef] [PubMed]
- Sheridan, C.; Kishimoto, H.; Fuchs, R.K.; Mehrotra, S.; Bhat-Nakshatri, P.; Turner, C.H.; Goulet, R., Jr.; Badve, S.; Nakshatri, H. CD44+/CD24- breast cancer cells exhibit enhanced invasive properties: An early step necessary for metastasis. Breast Cancer Res. 2006, 8, R59. [Google Scholar] [CrossRef] [PubMed]
- Zang, Z.J.; Cutcutache, I.; Poon, S.L.; Zhang, S.L.; McPherson, J.R.; Tao, J.; Rajasegaran, V.; Heng, H.L.; Deng, N.; Gan, A.; et al. Exome sequencing of gastric adenocarcinoma identifies recurrent somatic mutations in cell adhesion and chromatin remodeling genes. Nat. Genet. 2012, 44, 570–574. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Gao, Z.; Li, F.; Li, X.; Sun, Y.; Wang, M.; Li, D.; Wang, R.; Fang, R.; Pan, Y.; et al. Whole Exome Sequencing Identifies Frequent Somatic Mutations in Cell-Cell Adhesion Genes in Chinese Patients with Lung Squamous Cell Carcinoma. Sci. Rep. 2015, 5, 14237. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cifola, I.; Lionetti, M.; Pinatel, E.; Todoerti, K.; Mangano, E.; Pietrelli, A.; Fabris, S.; Mosca, L.; Simeon, V.; Petrucci, M.T.; et al. Whole-exome sequencing of primary plasma cell leukemia discloses heterogeneous mutational patterns. Oncotarget 2015, 6, 17543–17558. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neri, P.; Bahlis, N.J. Targeting of adhesion molecules as a therapeutic strategy in multiple myeloma. Curr. Cancer Drug Targets 2012, 12, 776–796. [Google Scholar] [CrossRef] [PubMed]
- Tolomelli, A.; Galletti, P.; Baiula, M.; Giacomini, D. Can Integrin Agonists Have Cards to Play against Cancer? A Literature Survey of Small Molecules Integrin Activators. Cancers 2017, 9, 78. [Google Scholar] [CrossRef] [PubMed]
- Seguin, L.; Desgrosellier, J.S.; Weis, S.M.; Cheresh, D.A. Integrins and cancer: Regulators of cancer stemness, metastasis, and drug resistance. Trends Cell Biol. 2015, 25, 234–240. [Google Scholar] [CrossRef] [PubMed]
- Damiano, J.S. Integrins as novel drug targets for overcoming innate drug resistance. Curr. Cancer Drug Targets 2002, 2, 37–43. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, Y.T.; Jiang, E.; Shishido, S.N.; Kim, H.N.; Pham, J.; Khazal, S.; Osborne, A.; Esguerra, Z.A.; Kwok, E.; Jang, J.; et al. Effects of the small-molecule inhibitor of integrin α4, TBC3486, on pre-B-ALL cells. Leukemia 2014, 28, 2101–2104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hsieh, Y.-T.; Gang, E.J.; Bonig, H.; Biediger, R.J.; Vanderslice, P.; Kim, Y.-M. The Small Molecule Inhibitor of VLA4 TBC3486 Sensitizes Resistant ALL to Chemotherapy. Blood 2012, 120, 1500. [Google Scholar]
- Mori, Y.; Shimizu, N.; Dallas, M.; Niewolna, M.; Story, B.; Williams, P.J.; Mundy, G.R.; Yoneda, T. Anti-α4 integrin antibody suppresses the development of multiple myeloma and associated osteoclastic osteolysis. Blood 2004, 104, 2149–2154. [Google Scholar] [CrossRef] [PubMed]
- Silginer, M.; Weller, M.; Ziegler, U.; Roth, P. Integrin inhibition promotes atypical anoikis in glioma cells. Cell Death Dis. 2014, 5, e1012. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakahara, S.; Miyoshi, E.; Noda, K.; Ihara, S.; Gu, J.; Honke, K.; Inohara, H.; Kubo, T.; Taniguchi, N. Involvement of oligosaccharide changes in α5β1 integrin in a cisplatin-resistant human squamous cell carcinoma cell line. Mol. Cancer Ther. 2003, 2, 1207–1214. [Google Scholar] [PubMed]
- Damiano, J.S.; Cress, A.E.; Hazlehurst, L.A.; Shtil, A.A.; Dalton, W.S. Cell adhesion mediated drug resistance (CAM-DR): Role of integrins and resistance to apoptosis in human myeloma cell lines. Blood 1999, 93, 1658–1667. [Google Scholar] [PubMed]
- Kobune, M.; Chiba, H.; Kato, J.; Kato, K.; Nakamura, K.; Kawano, Y.; Takada, K.; Takimoto, R.; Takayama, T.; Hamada, H.; et al. Wnt3/RhoA/ROCK signaling pathway is involved in adhesion-mediated drug resistance of multiple myeloma in an autocrine mechanism. Mol. Cancer Ther. 2007, 6, 1774–1784. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmidmaier, R.; Baumann, P.; Simsek, M.; Dayyani, F.; Emmerich, B.; Meinhardt, G. The HMG-CoA reductase inhibitor simvastatin overcomes cell adhesion–mediated drug resistance in multiple myeloma by geranylgeranylation of Rho protein and activation of Rho kinase. Blood 2004, 104, 1825–1832. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sartori, A.; Portioli, E.; Battistini, L.; Calorini, L.; Pupi, A.; Vacondio, F.; Arosio, D.; Bianchini, F.; Zanardi, F. Synthesis of Novel c(AmpRGD)-Sunitinib Dual Conjugates as Molecular Tools Targeting the αvβ3 Integrin/VEGFR2 Couple and Impairing Tumor-Associated Angiogenesis. J. Med. Chem. 2017, 60, 248–262. [Google Scholar] [CrossRef] [PubMed]
- Ley, K.; Rivera-Nieves, J.; Sandborn, W.J.; Shattil, S. Integrin-based therapeutics: Biological basis, clinical use and new drugs. Nat. Rev. Drug Discov. 2016, 15, 173–183. [Google Scholar] [CrossRef] [PubMed]
- Scaringi, C.; Minniti, G.; Caporello, P.; Enrici, R.M. Integrin inhibitor cilengitide for the treatment of glioblastoma: A brief overview of current clinical results. Anticancer Res. 2012, 32, 4213–4223. [Google Scholar] [PubMed]
- Stupp, R.; Hegi, M.E.; Gorlia, T.; Erridge, S.C.; Perry, J.; Hong, Y.K.; Aldape, K.D.; Lhermitte, B.; Pietsch, T.; Grujicic, D.; et al. Cilengitide combined with standard treatment for patients with newly diagnosed glioblastoma with methylated MGMT promoter (CENTRIC EORTC 26071-22072 study): A multicentre, randomised, open-label, phase 3 trial. Lancet Oncol. 2014, 15, 1100–1108. [Google Scholar] [CrossRef]
- Vansteenkiste, J.; Barlesi, F.; Waller, C.F.; Bennouna, J.; Gridelli, C.; Goekkurt, E.; Verhoeven, D.; Szczesna, A.; Feurer, M.; Milanowski, J.; et al. Cilengitide combined with cetuximab and platinum-based chemotherapy as first-line treatment in advanced non-small-cell lung cancer (NSCLC) patients: Results of an open-label, randomized, controlled phase II study (CERTO). Ann. Oncol. 2015, 26, 1734–1740. [Google Scholar] [CrossRef] [PubMed]
- Vermorken, J.B.; Peyrade, F.; Krauss, J.; Mesia, R.; Remenar, E.; Gauler, T.C.; Keilholz, U.; Delord, J.P.; Schafhausen, P.; Erfan, J.; et al. Cisplatin, 5-fluorouracil, and cetuximab (PFE) with or without cilengitide in recurrent/metastatic squamous cell carcinoma of the head and neck: Results of the randomized phase I/II ADVANTAGE trial (phase II part). Ann. Oncol. 2014, 25, 682–688. [Google Scholar] [CrossRef] [PubMed]
- Khasraw, M.; Lee, A.; McCowatt, S.; Kerestes, Z.; Buyse, M.E.; Back, M.; Kichenadasse, G.; Ackland, S.; Wheeler, H. Cilengitide with metronomic temozolomide, procarbazine, and standard radiotherapy in patients with glioblastoma and unmethylated MGMT gene promoter in ExCentric, an open-label phase II trial. J. Neuro Oncol. 2016, 128, 163–171. [Google Scholar] [CrossRef] [PubMed]
- Mas-Moruno, C.; Rechenmacher, F.; Kessler, H. Cilengitide: The first anti-angiogenic small molecule drug candidate design, synthesis and clinical evaluation. Anticancer Agents Med. Chem. 2010, 10, 753–768. [Google Scholar] [CrossRef] [PubMed]
- Podar, K.; Zimmerhackl, A.; Fulciniti, M.; Tonon, G.; Hainz, U.; Tai, Y.T.; Vallet, S.; Halama, N.; Jager, D.; Olson, D.L.; et al. The selective adhesion molecule inhibitor Natalizumab decreases multiple myeloma cell growth in the bone marrow microenvironment: Therapeutic implications. Br. J. Haematol. 2011, 155, 438–448. [Google Scholar] [CrossRef] [PubMed]
- Weitz-Schmidt, G.; Welzenbach, K.; Brinkmann, V.; Kamata, T.; Kallen, J.; Bruns, C.; Cottens, S.; Takada, Y.; Hommel, U. Statins selectively inhibit leukocyte function antigen-1 by binding to a novel regulatory integrin site. Nat. Med. 2001, 7, 687. [Google Scholar] [CrossRef] [PubMed]
- Rezaie-Majd, A.; Prager, G.W.; Bucek, R.A.; Schernthaner, G.H.; Maca, T.; Kress, H.-G.; Valent, P.; Binder, B.R.; Minar, E.; Baghestanian, M. Simvastatin Reduces the Expression of Adhesion Molecules in Circulating Monocytes From Hypercholesterolemic Patients. Arterioscler. Thromb. Vasc. Biol. 2003, 23, 397–403. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hazlehurst, L.A.; Enkemann, S.A.; Beam, C.A.; Argilagos, R.F.; Painter, J.; Shain, K.H.; Saporta, S.; Boulware, D.; Moscinski, L.; Alsina, M.; et al. Genotypic and Phenotypic Comparisons of de Novo and Acquired Melphalan Resistance in an Isogenic Multiple Myeloma Cell Line Model. Cancer Res. 2003, 63, 7900–7906. [Google Scholar] [PubMed]
- Wagner, B.J.; Lob, S.; Lindau, D.; Horzer, H.; Guckel, B.; Klein, G.; Glatzle, J.; Rammensee, H.G.; Brucher, B.L.; Konigsrainer, A. Simvastatin reduces tumor cell adhesion to human peritoneal mesothelial cells by decreased expression of VCAM-1 and β1 integrin. Int. J. Oncol. 2011, 39, 1593–1600. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Venkataramani, V.; Küffer, S.; Cheung, K.C.P.; Jiang, X.; Trümper, L.H.P.; Wulf, G.G.; Ströbel, P. CD31 Expression Determines Redox Status and Chemoresistance in Human Angiosarcomas. Clin. Cancer Res. 2018, 24, 460–473. [Google Scholar] [CrossRef] [PubMed]
- Podar, K.; Tonon, G.; Sattler, M.; Tai, Y.-T.; LeGouill, S.; Yasui, H.; Ishitsuka, K.; Kumar, S.; Kumar, R.; Pandite, L.N.; et al. The small-molecule VEGF receptor inhibitor pazopanib (GW786034B) targets both tumor and endothelial cells in multiple myeloma. Proc. Natl. Acad. Sci. USA 2006, 103, 19478–19483. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Du Bois, A.; Floquet, A.; Kim, J.W.; Rau, J.; Del Campo, J.M.; Friedlander, M.; Pignata, S.; Fujiwara, K.; Vergote, I.; Colombo, N.; et al. Randomized, double-blind, phase III trial of pazopanib versus placebo in women who have not progressed after first-line chemotherapy for advanced epithelial ovarian, fallopian tube, or primary peritoneal cancer (AEOC): Results of an international Intergroup trial (AGO-OVAR16). J. Clin. Oncol. 2013, 31, LBA5503. [Google Scholar] [CrossRef]
- Puhr, M.; Hoefer, J.; Schäfer, G.; Erb, H.H.H.; Oh, S.J.; Klocker, H.; Heidegger, I.; Neuwirt, H.; Culig, Z. Epithelial-to-Mesenchymal Transition Leads to Docetaxel Resistance in Prostate Cancer and Is Mediated by Reduced Expression of miR-200c and miR-205. Am. J. Pathol. 2012, 181, 2188–2201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shou, J.; Ross, S.; Koeppen, H.; de Sauvage, F.J.; Gao, W.Q. Dynamics of notch expression during murine prostate development and tumorigenesis. Cancer Res. 2001, 61, 7291–7297. [Google Scholar] [PubMed]
- Olsauskas-Kuprys, R.; Zlobin, A.; Osipo, C. Gamma secretase inhibitors of Notch signaling. OncoTargets Ther. 2013, 6, 943–955. [Google Scholar] [CrossRef]
- Schott, A.F.; Landis, M.D.; Dontu, G.; Griffith, K.A.; Layman, R.M.; Krop, I.; Paskett, L.A.; Wong, H.; Dobrolecki, L.E.; Lewis, M.T.; et al. Preclinical and clinical studies of gamma secretase inhibitors with docetaxel on human breast tumors. Clin. Cancer Res. 2013, 19, 1512–1524. [Google Scholar] [CrossRef] [PubMed]
- Brooks, S.A.; Lomax-Browne, H.J.; Carter, T.M.; Kinch, C.E.; Hall, D.M. Molecular interactions in cancer cell metastasis. Acta Histochem. 2010, 112, 3–25. [Google Scholar] [CrossRef] [PubMed]
- Woodward, J. Crossing the endothelium: E-selectin regulates tumor cell migration under flow conditions. Cell Adhes. Migr. 2008, 2, 151–152. [Google Scholar] [CrossRef]
- Bevilacqua, M.P.; Nelson, R.M. Selectins. J. Clin. Investig. 1993, 91, 379–387. [Google Scholar] [CrossRef] [PubMed]
- Azab, A.K.; Quang, P.; Azab, F.; Pitsillides, C.; Thompson, B.; Chonghaile, T.; Patton, J.T.; Maiso, P.; Monrose, V.; Sacco, A.; et al. P-selectin glycoprotein ligand regulates the interaction of multiple myeloma cells with the bone marrow microenvironment. Blood 2012, 119, 1468–1478. [Google Scholar] [CrossRef] [PubMed]
- Yue, Z.; Wang, A.; Zhu, Z.; Tao, L.; Li, Y.; Zhou, L.; Chen, W.; Lu, Y. Holothurian glycosaminoglycan inhibits metastasis via inhibition of P-selectin in B16F10 melanoma cells. Mol. Cell Biochem. 2015, 410, 143–154. [Google Scholar] [CrossRef] [PubMed]
- Muz, B.; Azab, F.; de la Puente, P.; Rollins, S.; Alvarez, R.; Kawar, Z.; Azab, A.K. Inhibition of P-Selectin and PSGL-1 Using Humanized Monoclonal Antibodies Increases the Sensitivity of Multiple Myeloma Cells to Bortezomib. BioMed Res. Int. 2015, 2015, 417586. [Google Scholar] [CrossRef] [PubMed]
- Natoni, A.; Smith, T.A.G.; Keane, N.; Locatelli-Hoops, S.C.; Oliva, I.; Fogler, W.E.; Magnani, J.L.; Dwyer, M. E-Selectin Ligand Expression Increases with Progression of Myeloma and Induces Drug Resistance in a Murine Transplant Model, Which Is Overcome By the Glycomimetic E-Selectin Antagonist, GMI-1271. Blood 2015, 126, 1805. [Google Scholar]
- Borsig, L.; Wong, R.; Hynes, R.O.; Varki, N.M.; Varki, A. Synergistic effects of L- and P-selectin in facilitating tumor metastasis can involve non-mucin ligands and implicate leukocytes as enhancers of metastasis. Proc. Natl. Acad. Sci USA 2002, 99, 2193–2198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- DeAngelo, D.J.; Jonas, B.A.; Becker, P.S.; O’Dwyer, M.; Advani, A.S.; Marlton, P.; Magnani, J.; Thackray, H.M.; Liesveld, J. GMI-1271, a novel E-selectin antagonist, combined with induction chemotherapy in elderly patients with untreated AML. J. Clin. Oncol. 2017, 35, 2560. [Google Scholar] [CrossRef]
- Bjorklund, C.C.; Baladandayuthapani, V.; Lin, H.Y.; Jones, R.J.; Kuiatse, I.; Wang, H.; Yang, J.; Shah, J.J.; Thomas, S.K.; Wang, M.; et al. Evidence of a Role for CD44 and Cell Adhesion in Mediating Resistance to Lenalidomide in Multiple Myeloma: Therapeutic Implications. Leukemia 2014, 28, 373–383. [Google Scholar] [CrossRef] [PubMed]
- Negi, L.M.; Talegaonkar, S.; Jaggi, M.; Ahmad, F.J.; Iqbal, Z.; Khar, R.K. Role of CD44 in tumour progression and strategies for targeting. J. Drug Target. 2012, 20, 561–573. [Google Scholar] [CrossRef] [PubMed]
- Zheng, F.-M.; Long, Z.-J.; Hou, Z.-J.; Luo, Y.; Xu, L.-Z.; Xia, J.-L.; Lai, X.-J.; Liu, J.-W.; Wang, X.; Kamran, M.; et al. A Novel Small Molecule Aurora Kinase Inhibitor Attenuates Breast Tumor–Initiating Cells and Overcomes Drug Resistance. Mol. Cancer Ther. 2014, 13, 1991–2003. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
| Target Proteins | Anti-Apoptotic Mechanisms | Inhibitors | Type of Tumor | |
|---|---|---|---|---|
| The cellular FLICE-like inhibitory protein (cFLIP) | Competitive interference with caspase-8 recruitment to DISC | Cycloheximide [136] | murine thymoma EL-4 cells | |
| miRNA-708 [165] | renal cancer cells | |||
| CXCR2 antagonist Z10397767 [142] | prostate cancer cells | |||
| Thioridazine [143] | human head and neck cancer cells | |||
| Histone deacetylase inhibitor LBH589 [144] | pancreatic cancer cells | |||
| Withanolide E and analogues [145] | renal carcinoma | |||
| Bcl-2-like proteins | Bcl-2 Bcl-XL Bcl-W | Inhibition of pore-forming Bax/Bak in mitochondria | Small-molecule ABT-737 [148] | human colorectal cancer |
| Small-molecule ABT-263 (navitoclax) [147] | small cell lung cancer leukemia lymphoma hematologic malignances | |||
| Mcl-1 | Antagonizes Bax and Bak activation | Small-molecule S63845 [149] | myeloma leukaemia lymphoma | |
| Bfl-1/A1 | Binds to BH3-only proteins [166] | 4-chloro-1-methyl-3-nitroquinolin-2-one [151] | MEF and melanoma cell lines primed with various A1 constructs | |
| IAPs | BIRC4 (X-linked IAP/XIAP/hILP) BIRC2 (cellular IAP1/cIAP1/HIAP2) BIRC3 (cellular IAP2/cIAP2/HIAP1) | Prevent downstream proteolytic processing of pro-caspase-3, -6 and -7 [117,167] | AZD5582 [156] | pancreatic cancer |
| Oligonucleotide AEG-35156 [157] | Panc-1 pancreatic carcinoma cells, xenograft models of prostate, colon, and lung cancer, lymphoma, melanoma, breast cancer, | |||
| Smac mimetics [160,161,162,163,164,168]. | breast cancer multiple myeloma human glioblastoma non-small cell lung cancer leukemia | |||
| Small-molecule AEG40730 [169] | HCT116 Cell Line human colon carcinoma | |||
| BIRC5 (Survivin) | Binds to pro-caspase-9, preventing its recruitment to Apaf1 [170] Inhibits SMAC [171] | Small-molecule YM155 [159] | colorectal and lung adenocarcinoma | |
| Serine/threonine protein kinases | WEE1 | Dysregulates CDK1 and CDK2 | MK1775 (AZD 1775) | ovarian cancer advanced gastric adenocarcinoma metastatic solid tumors [47] |
| Type of CAM | Type of Tumor | Chemotherapy Drugs | Signaling Pathway | Preclinical Anti CAM-DR Treatment | Clinical |
|---|---|---|---|---|---|
| Integrin α4 | acute lymphoblastic leukemia | vincristine | Direct inhibition | Integrin α4 inhibitor small molecule TBC3486 [221,222] | - |
| multiple myeloma | melphalan | Direct inhibition | Anti-integrin α4 antibody [223] | - | |
| Integrin α4 | multiple myeloma | bortezomib | Direct inhibition | inhibitor Natalizumab, a recombinant humanized IgG4 monoclonal antibody, which binds integrin α4 [237] | Natalizumab—Phase I/II (NCT00675428)—multiple myeloma (terminated) |
| Integrin α4 | Glioma cells | temozolomide | Direct inhibition | EMD-121974 (Cilengitide), a synthetic Arg-Gly-Asp-motif peptide-α4 integrin inhibitor [224] | Cilengitide with temozolomide—Phase III (NCT00689221)—newly diagnosed glioblastoma [232] |
| Cilengitide combined with cetuximab and platinum-based chemotherapy (NCT00842712)—Phase II-non-small-cell lung cancer [233] Cilengitide with Cisplatin, 5-fluorouracil, and cetuximab (NCT00705016)—Phase I/II—squamous cell carcinoma of the head and neck [234] | |||||
| Cilengitide with metronomic temozolomide, procarbazine, and standard radiotherapy (NCT01124240)—Phase II [235] | |||||
| Integrin α5β1 | squamous cell carcinoma | cisplatin | Direct inhibition | Anti-α5β1 Integrin Neutral Antibody [225] | - |
| VLA-4 (integrin α4β1) and VLA-5 (integrin α5β1) | myeloma | doxorubicin and melphalan | Direct inhibition | Anti-VLA-4 VLA-5 antibody [226] | - |
| VLA-4 (integrin α4β1) and LFA-1 (integrin αLβ2) | multiple myeloma | melphalan, treosulfan, doxorubicin, dexamethasone, and bortezomib | HMG-CoA/GG-PP/Rho/Rho-kinase | Anti LFA-1 and VLA-4 antibodies. Geranylgeranyl transferase (GGTase) inhibitor GGTI-298 and Rho kinase specific inhibitors Y-27632. The HMG-CoA reductase inhibitor simvastatin [238,239,240]. | Simvastatin with bortezomib, bendamustin dexamethasone—Phase II (NCT00399867) [227,228]—in Patients with Refractory Multiple Myeloma [17] |
| CD31/PECAM-1 | Angiosarcoma | Doxorubicin | YAP | YAP inhibitors (Pazopanib) [242] | Pazopanib—Phase III NCT00866697- Ovarian, Fallopian Tube or Primary Peritoneal Adenocarcinoma [244] |
| E-cadherin | Prostate cancer | Paclitaxel | Notch | The γ-secretase inhibitor (GSI, a Notch inhibitor) [195] | The γ-secretase inhibitor MK-0752—Phase II-NCT00645333 Breast cancer |
| PSGL-1/P-selectin | In macrophage for macrophage-mediated myeloma drug resistance | Bortezomib | Direct inhibition | The pan-selectin inhibitor GMI-1070 [252] | - |
| P-selectin | Melanoma | - | Direct inhibition | P-selectin inhibitor- Holothurian glycosaminoglycan [253] | - |
| Multiple myeloma | Bortezomib | Direct inhibition | Humanized Monoclonal Antibodies [254] | - | |
| E-selectin | Multiple myeloma | Bortezomib | Direct inhibition | E-selectin inhibitor GMI-1271 [255] | E-selectin inhibitor GMI-1271-with mitoxantrone, etoposide and cytarabine Phase I/II NCT02306291-acute myeloid leukemia [257] |
| CD44 | Breast cancer | epirubicin | Aurora kinase | Aurora kinase inhibitor AKI603 [260] | - |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alimbetov, D.; Askarova, S.; Umbayev, B.; Davis, T.; Kipling, D. Pharmacological Targeting of Cell Cycle, Apoptotic and Cell Adhesion Signaling Pathways Implicated in Chemoresistance of Cancer Cells. Int. J. Mol. Sci. 2018, 19, 1690. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms19061690
Alimbetov D, Askarova S, Umbayev B, Davis T, Kipling D. Pharmacological Targeting of Cell Cycle, Apoptotic and Cell Adhesion Signaling Pathways Implicated in Chemoresistance of Cancer Cells. International Journal of Molecular Sciences. 2018; 19(6):1690. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms19061690
Chicago/Turabian StyleAlimbetov, Dauren, Sholpan Askarova, Bauyrzhan Umbayev, Terence Davis, and David Kipling. 2018. "Pharmacological Targeting of Cell Cycle, Apoptotic and Cell Adhesion Signaling Pathways Implicated in Chemoresistance of Cancer Cells" International Journal of Molecular Sciences 19, no. 6: 1690. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms19061690