ALUminating the Path of Atherosclerosis Progression: Chaos Theory Suggests a Role for Alu Repeats in the Development of Atherosclerotic Vascular Disease



Abstract
:
{kind=link}
{kind=link}
{kind=link}
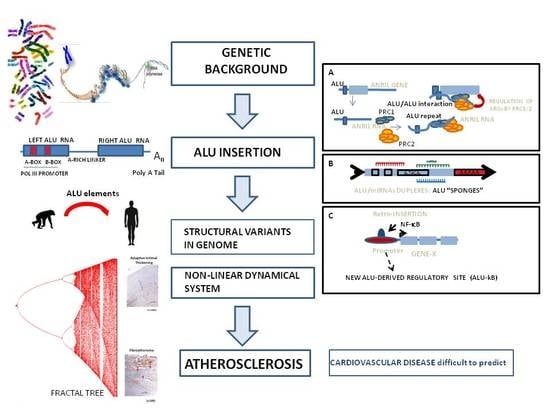
1. Atherosclerosis Is a Complex Vascular Disease with Distinctive Traits of Nonlinear Behavior
2. Multifractal and Chaos-Theory Analysis of the Human Genome Highlights the Involvement of Alu Elements in the Development of Complex, Nonlinear Human Diseases
2.1. Chaos Theory Provides Tools for the Analysis of Global Genomic Signatures
2.2. Mathematical Analysis of the Human Genome Highlights Features of Nonlinear Correlations in the Alu Family of Genomic Elements
3. The Family of Alu Repeated Elements and Their Impact on the Mechanisms Regulating Gene Expression
3.1. The Human Genome Is Mostly Composed of Transcribed, Non-Protein-Coding (ncRNA) Genes
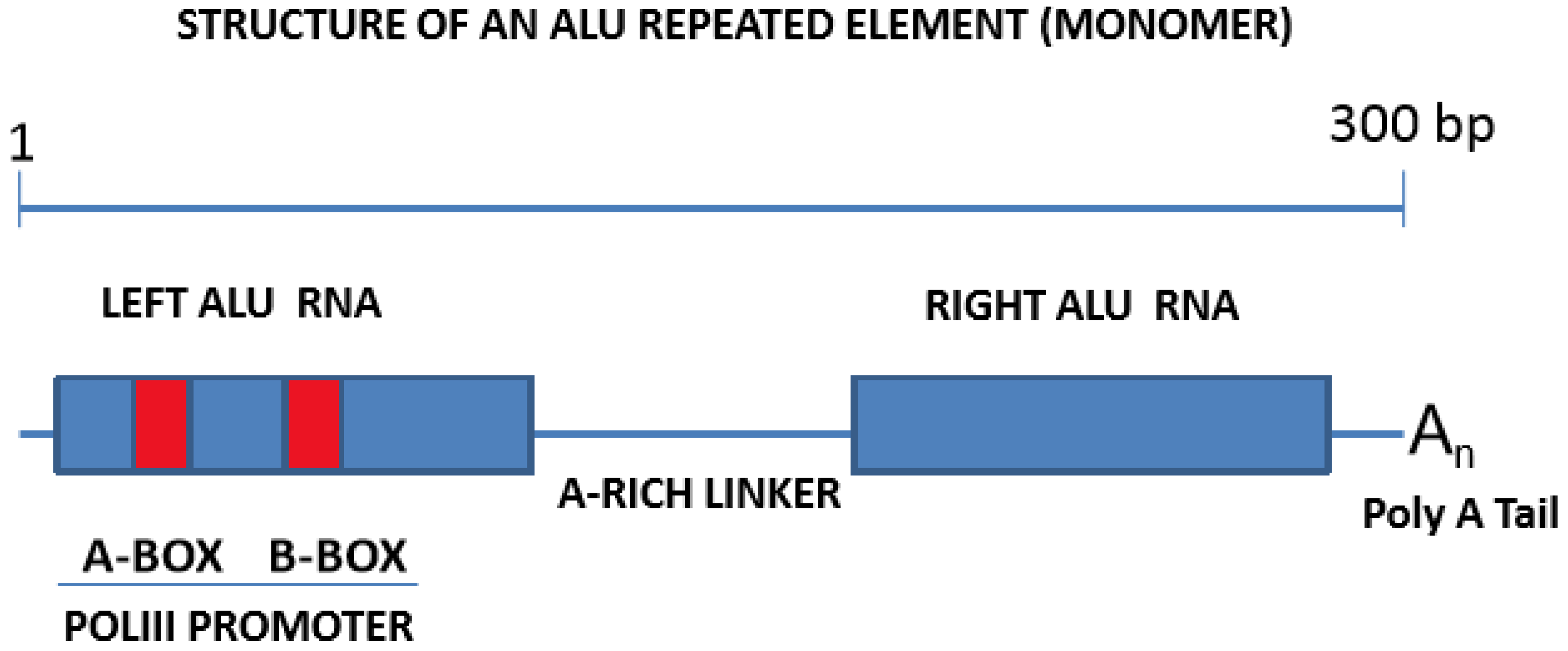
3.2. Alu Repeats: A Family of Highly Succesful Genomic Invaders
3.3. Mature Alu-RNAs Include Free Alu Elements Transcribed by RNA Polymerase III, or mRNA-Embedded Alu Elements Transcribed by RNA Polymerase II
3.4. Genomic Alu Elements Are Involved in Transcriptional Regulation and Have an Impact on Human Disease
4. Anril: A Long Noncoding RNA Harboring a Risk Factor for Atherosclerosis
5. Alu Elements May Play Multiple Roles in the Progression of Atherosclerosis
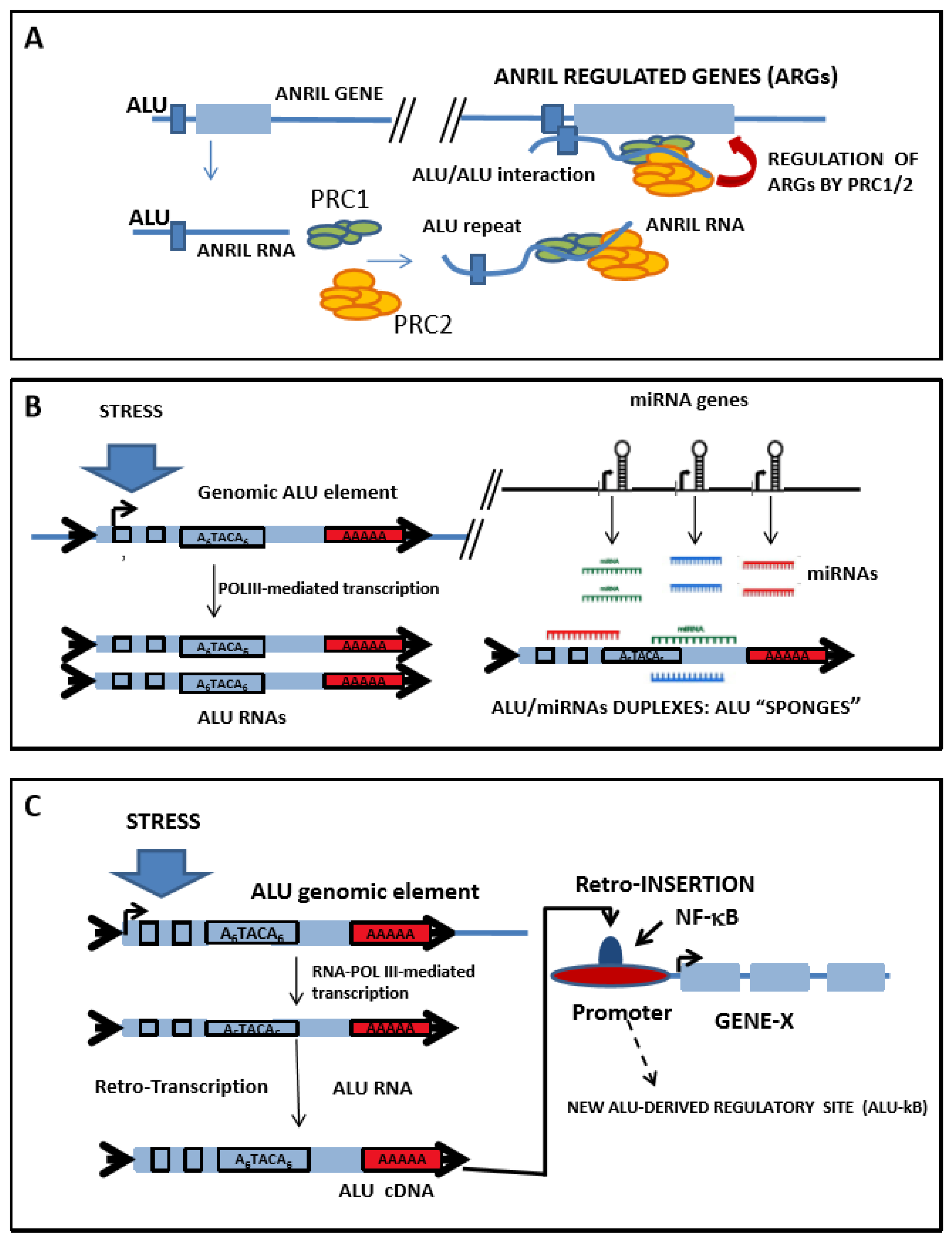
5.1. Role of Alu Elements in the Regulation of ANRIL Function
5.2. Interaction between Alu-RNAs and miRNAs Creates Complex Regulatory Networks
5.3. Alu Elements Are Common Binding Sites for Transcription Factors, Such as NF-κB, and May Impact Gene Expression of the Inflammatory Response
5.4. A Polymorphic Alu Insertion Controls the Renin-Angiotensin System
6. Concluding Remarks
Author Contributions
Acknowledgments
Conflicts of Interest
Abbreviations
| asRNA | antisense transcripts from coding regions |
| ASVD | arteriosclerotic vascular disease |
| ATH | Atherosclerosis |
| CAC | coronary artery calcification |
| CAD | coronary artery disease |
| circRNA | circular noncoding RNA |
| eQTL | expression quantitative trait locus |
| ECM | extracellular matrix |
| EMT | epithelial–mesenchymal transition |
| FH | familial hypercholesterolemia |
| GWAS | genome-wide association studies |
| HIV | human immunodeficiency virus |
| LINE | long interspersed nuclear element |
| ncRNA | noncoding RNA |
| lncRNA | long noncoding RNA |
| lincRNA | long intergenic noncoding RNA |
| MACE | major adverse cardiovascular event |
| miRNA | microRNA |
| piRNA | piwi-interacting RNA |
| PRC | polycomb repressive complex |
| PRR | pattern recognition receptor |
| RAAS | renin–angiotensin–aldosterone system |
| ROS | reactive oxygen species |
| SMC | smooth muscle cells |
| SNP | single-nucleotide polymorphism |
| SINE | short interspersed nuclear element |
| SRP | signal recognition particle |
| TSS | transcription start site |
| UTR | untranslated region |
| VEC | vascular endothelial cell |
| Ang II | angiotensin II |
| ACE | angiotensin-converting enzyme |
| ANRIL/CDKN2B-AS1 | antisense noncoding RNA in the INK4 locus, CDKN2B antisense 1 |
| BCR | B-cell receptor |
| CBX7 | chromobox protein homolog 7 |
| CDK | cyclin-dependent kinase |
| FKBP9 | FK506 binding protein 9 |
| GTSE1 | G2 and S phase-expressed protein 1 |
| HDL | high-density lipoprotein |
| HGH | human growth hormone |
| IKK | IκB kinase |
| INK | inhibitor of CDK4 |
| LDL | low-density lipoprotein |
| MDM2 | MDM2 proto-oncogene |
| MDM4 | MDM4 p53 regulator |
| MMP-9 | matrix metalloproteinase 9 |
| NEMO | NF-κB essential modulator |
| NIK | NF-κB-inducing kinase |
| NF1 | neurofibromatosis 1 gene |
| NF-κB | activated nuclear factor kappa B |
| NR2C1 | nuclear receptor subfamily 2 group C member 1 |
| oxLDL | oxidized LDL |
| PODXL | podocalyxin |
| RAD1 | checkpoint DNA exonuclease |
| RANK | receptor activator of NF-κB |
| STAT1 | signal transducer and activator of transcription 1 |
| SUZ12 | suppressor of zest 12 (subunit of polycomb repressive complex 2) |
| TCR | T-cell receptor |
| TNFR | tumor necrosis factor receptor |
| UBE2I | ubiquitin-conjugating enzyme E2 |
| VEGF | vascular endothelial growth factor |
References
- Libby, P. Inflammation in atherosclerosis. Nature 2002, 420, 868–874. [Google Scholar] [CrossRef] [PubMed]
- Chalmers, A.D.; Bursill, C.A.; Myerscough, M.R. Nonlinear dynamics of early atherosclerotic plaque formation may determine the efficacy of high density lipoproteins (HDL) in plaque regression. PLoS ONE 2017, 12, e0187674. [Google Scholar] [CrossRef] [PubMed]
- Ramsey, S.A.; Gold, E.S.; Aderem, A. A system biology approach to understanding atherosclerosis. EMBO Mol. Med. 2010, 2, 79–89. [Google Scholar] [CrossRef] [PubMed]
- Hao, W.; Friedman, A. The LDL-HDL profile determines the risk of atherosclerosis: A mathematical model. PLoS ONE 2014, 9, e90497. [Google Scholar] [CrossRef] [PubMed]
- Holzer, M.; Trieb, M.; Konya, V.; Wadsack, C.; Heinemann, A.; Marsche, G. Aging affects high-density lipoprotein composition and function. Biochim. Biophys. Acta 2013, 1831, 1442–1448. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.Y.; Howarth, S.P.; Tang, T.; Gillard, J.H. How critical is fibrous cap thickness to carotid plaque stability? A flow-plaque interaction model. Stroke 2006, 37, 1195–1199. [Google Scholar] [CrossRef] [PubMed]
- El Khatib, N.; Génieys, S.; Kazmierczak, B.; Volpert, V. Mathematical modelling of atherosclerosis as an inflammatory disease. Philos. Trans. A Math. Phys. Eng. Sci. 2009, 367, 4877–4886. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.; Sevag Packard, R.R.; Hsiai, T.K. Blood Flow Modulation of Vascular Dynamics. Curr. Opin. Lipidol. 2015, 26, 376–383. [Google Scholar] [CrossRef] [PubMed]
- Goldberger, A.L. Non-linear dynamics for clinicians: Chaos theory, fractals, and complexity at the bedside. Lancet 1996, 347, 1312–1314. [Google Scholar] [CrossRef]
- Bruschke, A.V.; Kramer, J.R.; Bal, E.T.; Haque, I.U.; Detrano, R.C.; Goormastic, M. The dynamics of progression of coronary atherosclerosis studied in 168 medically treated patients who underwent coronary arteriography three times. Am. Heart J. 1989, 117, 296–305. [Google Scholar] [CrossRef]
- Veress, A.I.; Vince, D.G.; Anderson, P.M.; Cornhill, J.F.; Herderick, E.E.; Klingensmith, J.D.; Kuban, B.D.; Greenberg, N.L.; Thomas, J.D. Vascular mechanics of the coronary artery. Z. Kardiol. 2000, 89 (Suppl. 2), 92–100. [Google Scholar] [CrossRef] [PubMed]
- Kamenskiy, A.V.; Dzenis, Y.A.; MacTaggart, J.N.; Lynch, T.G.; Jaffar Kazmi, S.A.; Pipinos, I.I. Nonlinear mechanical behavior of the human common, external, and internal carotid arteries in vivo. J. Surg. Res. 2012, 176, 329–336. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Han, H.C. Mechanical buckling of artery under pulsatile pressure. J. Biomech. 2012, 45, 1192–1198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Z.Y.; Gillard, J.H. Simulation of the interaction between blood flow and atherosclerotic plaque. In Proceedings of the 29th Annual International Conference of the IEEE Engineering in Medicine and Biology Society, Lyon, France, 22–26 August 2007; pp. 1699–1702. [Google Scholar]
- Nguyen, C.M.; Levy, A.J. The mechanics of atherosclerotic plaque rupture by inclusion/matrix interfacial decohesion. J. Biomech. 2010, 43, 2702–2708. [Google Scholar] [CrossRef] [PubMed]
- Xiao, Y.; Huang, Y.; Li, M.; Xu, R.; Xiao, S. Nonlinear analysis of correlations in Alu repeat sequences in DNA. Phys. Rev. E Stat. Nonlinear Soft Matter Phys. 2003, 68 Pt 1, 061913. [Google Scholar] [CrossRef]
- Häsler, J.; Samuelsson, T.; Strub, K. Useful “junk”: Alu RNAs in the human transcriptome. Cell. Mol. Life Sci. 2007, 64, 1793–1800. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.L.; Yang, L. ALUternative Regulation for Gene Expression. Trends Cell Biol. 2017, 27, 480–490. [Google Scholar] [CrossRef] [PubMed]
- Conti, A.; Carnevali, D.; Bollati, V.; Fustinoni, S.; Pellegrini, M.; Dieci, G. Identification of RNA polymerase III-transcribed Alu loci by computational screening of RNA-Seq data. Nucleic Acids Res. 2015, 43, 817–835. [Google Scholar] [CrossRef] [PubMed]
- Lazaros, L.; Kitsou, C.; Kostoulas, C.; Bellou, S.; Hatzi, E.; Ladias, P.; Stefos, T.; Markoula, S.; Galani, V.; Vartholomatos, G.; et al. Retrotransposon expression and incorporation of cloned human and mouse retroelements in human spermatozoa. Fertil. Steril. 2017, 107, 821–830. [Google Scholar] [CrossRef] [PubMed]
- Navarro, E.; Espinosa, L.; Adell, T.; Torà, M.; Berrozpe, G.; Real, F.X. Expressed sequence tag (EST) phenotyping of HT-29 cells: Cloning of ser/thr protein kinase EMK1, kinesin KIF3B, and of transcripts that include Alu repeated elements. Biochim. Biophys. Acta 1999, 1450, 254–264. [Google Scholar] [CrossRef]
- Kim, S.; Cho, C.S.; Han, K.; Lee, J. Structural Variation of Alu Element and Human Disease. Genom. Inform. 2016, 14, 70–77. [Google Scholar] [CrossRef] [PubMed]
- Chopra-Tandon, N.; Wu, H.; Arcaro, K.F.; Sturgeon, S.R. Relationships between Global DNA Methylation in Circulating White Blood Cells and Breast Cancer Risk Factors. J. Cancer Epidemiol. 2017, 2017, 2705860. [Google Scholar] [CrossRef] [PubMed]
- Jeffrey, H.J. Chaos game representation of gene structure. Nucleic Acids Res. 1990, 18, 2163–2170. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arakawa, K.; Oshita, K.; Tomita, M. A web server for interactive and zoomable Chaos Game Representation images. Source Code Biol. Med. 2009, 4, 6. [Google Scholar] [CrossRef] [PubMed]
- Almeida, J.S. Sequence analysis by iterated maps, a review. Brief. Bioinform. 2014, 15, 369–375. [Google Scholar] [CrossRef] [PubMed]
- Oliver, J.L.; Bernaola-Galván, P.; Guerrero-García, J.; Román-Roldán, R. Entropic profiles of DNA sequences through chaos-game-derived images. J. Theor. Biol. 1993, 160, 457–470. [Google Scholar] [CrossRef] [PubMed]
- Messaoudi, I.; Elloumi-Oueslati, A.; Lachiri, Z. Building Specific Signals from Frequency Chaos Game and Revealing Periodicities Using a Smoothed Fourier Analysis. IEEE/ACM Trans. Comput. Biol. Bioinform. 2014, 11, 863–877. [Google Scholar] [CrossRef] [PubMed]
- Joseph, J.; Sasikumar, R. Chaos game representation for comparison of whole genomes. BMC Bioinform. 2006, 7, 243. [Google Scholar] [CrossRef] [PubMed]
- Anh, V.V.; Lau, K.S.; Yu, Z.G. Recognition of an organism from fragments of its complete genome. Phys. Rev. E Stat. Nonlinear Soft Matter Phys. 2002, 66 Pt 1, 031910. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, Z.G.; Anh, V.; Lau, K.S. Measure representation and multifractal analysis of complete genomes. Phys. Rev. E Stat. Nonlinear Soft Matter Phys. 2001, 64 Pt 1, 031903. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, L.Q.; Yu, Z.G.; Deng, J.Q.; Anh, V.; Long, S.C. A fractal method to distinguish coding and non-coding sequences in a complete genome based on a number sequence representation. J. Theor. Biol. 2005, 232, 559–567. [Google Scholar] [CrossRef] [PubMed]
- Fu, W.; Wang, Y.; Lu, D. Multifractal Analysis of Genomic Sequences CGR Images. In Proceedings of the 27th Annual International Conference of the Engineering in Medicine and Biology Society, Shanghai, China, 17–18 January 2005; Volume 5, pp. 4783–4786. [Google Scholar]
- Provata, A.; Oikonomou, T. Power law exponents characterizing human DNA. Phys. Rev. E Stat. Nonlinear Soft Matter Phys. 2007, 75 Pt 2, 056102. [Google Scholar] [CrossRef] [PubMed]
- Stan, C.; Cristescu, M.T.; Luiza, B.I.; Cristescu, C.P. Investigation on series of length of coding and non-coding DNA sequences of bacteria using multifractal detrended cross-correlation analysis. J. Theor. Biol. 2013, 321, 54–62. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.Y.; Zhou, Y.; Yu, Z.G.; Anh, V.; Zhou, L.Q. Human Pol II promoter recognition based on primary sequences and free energy of dinucleotides. BMC Bioinform. 2008, 9, 113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vélez, P.E.; Garreta, L.E.; Martínez, E.; Díaz, N.; Amador, S.; Tischer, I.; Gutiérrez, J.M.; Moreno, P.A. The Caenorhabditis elegans genome: A multifractal analysis. Genet. Mol. Res. 2010, 9, 949–965. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Audit, B.; Vaillant, C.; Arnéodo, A.; d’Aubenton-Carafa, Y.; Thermes, C. Wavelet Analysis of DNA Bending Profiles reveals Structural Constraints on the Evolution of Genomic Sequences. J. Biol. Phys. 2004, 30, 33–81. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Swain, T. Fast comparison of microbial genomes using the chaos game representation for metagenomic applications. Procedia Comput. Sci. 2013, 18, 1372–1380. [Google Scholar] [CrossRef] [Green Version]
- Mandal, S.; Roychowdhury, T.; Chirom, K.; Bhattacharya, A.; Brojen Singh, R.K. Complex multifractal nature in Mycobacterium tuberculosis genome. Sci. Rep. 2017, 7, 46395. [Google Scholar] [CrossRef] [PubMed]
- Yu, Z.G.; Anh, V.; Lau, K.S. Multifractal and correlation analyses of protein sequences from complete genomes. Phys. Rev. E Stat. Nonlinear Soft Matter Phys. 2003, 68 Pt 1, 021913. [Google Scholar] [CrossRef] [PubMed]
- Nie, G.; Li, Y.; Wang, F.; Wang, S.; Hu, X. A novel fractal approach for predicting G-protein-coupled receptors and their subfamilies with support vector machines. Biomed. Mater. Eng. 2015, 26 (Suppl. 1), S1829–S1836. [Google Scholar] [CrossRef] [PubMed]
- Moreno, P.A.; Vélez, P.E.; Martínez, E.; Garreta, L.E.; Díaz, N.; Amador, S.; Tischer, I.; Gutiérrez, J.M.; Naik, A.K.; Tobar, F.; et al. The human genome: A multifractal analysis. BMC Genom. 2011, 12, 506. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peng, C.K.; Buldyrev, S.V.; Goldberger, A.L.; Havlin, S.; Sciortino, F.; Simons, M.; Stanley, H.E. Long-range correlations in nucleotide sequences. Nature 1992, 356, 168–170. [Google Scholar] [CrossRef] [PubMed]
- Ossadnik, S.M.; Buldyrev, S.V.; Goldberger, A.L.; Havlin, S.; Mantegna, R.N.; Peng, C.K.; Simons, M.; Stanley, H.E. Correlation approach to identify coding regions in DNA sequences. Biophys. J. 1994, 67, 64–70. [Google Scholar] [CrossRef] [Green Version]
- Havlin, S.; Buldyrev, S.V.; Goldberger, A.L.; Mantegna, R.N.; Peng, C.K.; Simons, M.; Stanley, H.E. Statistical and linguistic features of DNA sequences. Fractals 1995, 3, 269–284. [Google Scholar] [CrossRef] [PubMed]
- Sellis, D.; Provata, A.; Almirantis, Y. Alu and LINE1 distributions in the human chromosomes: Evidence of global genomic organization expressed in the form of power laws. Mol. Biol. Evol. 2007, 24, 2385–2399. [Google Scholar] [CrossRef] [PubMed]
- Holste, D.; Grosse, I.; Beirer, S.; Schieg, P.; Herzel, H. Repeats and correlations in human DNA sequences. Phys. Rev. E Stat. Nonlinear Soft Matter Phys. 2003, 67 Pt 1, 061913. [Google Scholar] [CrossRef] [PubMed]
- Maddox, J. Long-range correlations within DNA. Nature 1992, 358, 103. [Google Scholar] [CrossRef] [PubMed]
- Labuda, D.; Striker, G. Sequence conservation in Alu evolution. Nucleic Acids Res. 1989, 17, 2477–2491. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sinnett, D.; Richer, C.; Deragon, J.M.; Labuda, D. Alu RNA secondary structure consists of two independent 7 SL RNA-like folding units. J. Biol. Chem. 1991, 266, 8675–8678. [Google Scholar] [PubMed]
- Podgornaya, O.I.; Ostromyshenskii, D.I.; Enukashvily, N.I. Who Needs This Junk, or Genomic Dark Matter. Biochemistry 2018, 83, 450–466. [Google Scholar] [CrossRef] [PubMed]
- Shabalina, S.A.; Spiridonov, N.A. The mammalian transcriptome and the function of non-coding DNA sequences. Genome Biol. 2004, 5, 105. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Samuels, D.C.; Zhao, S.; Xiang, Y.; Zhao, Y.Y.; Guo, Y. Current Research on Non-Coding Ribonucleic Acid (RNA). Genes 2017, 8, 366. [Google Scholar] [CrossRef] [PubMed]
- Haque, S.; Harries, L.W. Circular RNAs (circRNAs) in Health and Disease. Genes 2017, 8, 353. [Google Scholar] [CrossRef] [PubMed]
- Derrien, T.; Johnson, R.; Bussotti, G.; Tanzer, A.; Djebali, S.; Tilgner, H.; Guernec, G.; Martin, D.; Merkel, A.; Knowles, D.G.; et al. The GENCODE v7 catalog of human long noncoding RNAs: Analysis of their gene structure, evolution, and expression. Genome Res. 2012, 22, 1775–1789. [Google Scholar] [CrossRef] [PubMed]
- Bartel, D.P. Metazoan MicroRNAs. Cell 2018, 173, 20–51. [Google Scholar] [CrossRef] [PubMed]
- Ponnusamy, M.; Yan, K.W.; Liu, C.Y.; Li, P.F.; Wang, K. PIWI family emerging as a decisive factor of cell fate: An overview. Eur. J. Cell Biol. 2017, 96, 746–757. [Google Scholar] [CrossRef] [PubMed]
- Platt, R.N.; Vandewege, M.W.; Ray, D.A. Mammalian transposable elements and their impacts on genome evolution. Chromosome Res. 2018, 26, 25–43. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lander, E.S.; Linton, L.M.; Birren, B.; Nusbaum, C.; Zody, M.C.; Baldwin, J.; Devon, K.; Dewar, K.; Doyle, M.; FitzHugh, W.; et al. Initial sequencing and analysis of the human genome. Nature 2001, 409, 860–921. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Chen, J.G.; Zhao, Q. Regulatory roles of Alu transcript on gene expression. Exp. Cell Res. 2015, 338, 113–118. [Google Scholar] [CrossRef] [PubMed]
- Schmid, C.W.; Jelinek, W.R. The Alu family of dispersed repetitive sequences. Science 1982, 216, 1065–1070. [Google Scholar] [CrossRef] [PubMed]
- Daniel, C.; Behm, M.; Öhman, M. The role of Alu elements in the cis-regulation of RNA processing. Cell. Mol. Life Sci. 2015, 72, 4063–4076. [Google Scholar] [CrossRef] [PubMed]
- Batzer, M.A.; Deininger, P.L.; Hellmann-Blumberg, U.; Jurka, J.; Labuda, D.; Rubin, C.M.; Schmid, C.W.; Zietkiewicz, E.; Zuckerkandl, E. Standardized nomenclature for Alu repeats. J. Mol. Evol. 1996, 42, 3–6. [Google Scholar] [CrossRef] [PubMed]
- Mighell, A.J.; Markham, A.F.; Robinson, P.A. Alu sequences. FEBS Lett. 1997, 417, 1–5. [Google Scholar] [CrossRef] [Green Version]
- Wallace, N.; Wagstaff, B.J.; Deininger, P.L.; Roy-Engel, A.M. LINE-1 ORF1 protein enhances Alu SINE retrotransposition. Gene 2008, 419, 1–6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cordaux, R.; Batzer, M.A. The impact of retrotransposons on human genome evolution. Nat. Rev. Genet. 2009, 10, 691–703. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sela, N.; Mersch, B.; Gal-Mark, N.; Lev-Maor, G.; Hotz-Wagenblatt, A.; Ast, G. Comparative analysis of transposed element insertion within human and mouse genomes reveals Alu’s unique role in shaping the human transcriptome. Genome Biol. 2007, 8, R127. [Google Scholar] [CrossRef] [PubMed]
- Häsler, J.; Strub, K. Alu elements as regulators of gene expression. Nucleic Acids Res. 2006, 34, 5491–5497. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chenais, B. Transposable elements in cancer and other human diseases. Curr. Cancer Drug Targets 2015, 15, 227–242. [Google Scholar] [CrossRef] [PubMed]
- Russanova, V.R.; Driscoll, C.T.; Howard, B.H. Adenovirus type 2 preferentially stimulates polymerase III transcription of Alu elements by relieving repression: A potential role for chromatin. Mol. Cell. Biol. 1995, 15, 4282–4290. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.M.; Chu, W.M.; Choudary, P.V.; Schmid, C.W. Cell stress and translational inhibitors transiently increase the abundance of mammalian SINE transcripts. Nucleic Acids Res. 1995, 23, 1758–1765. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, C.; Rubin, C.M.; Schmid, C.W. Genome-wide chromatin remodeling modulates the Alu heat shock response. Gene 2001, 276, 127–133. [Google Scholar] [CrossRef]
- Moolhuijzen, P.; Kulski, J.K.; Dunn, D.S.; Schibeci, D.; Barrero, R.; Gojobori, T.; Bellgard, M. The transcript repeat element: The human Alu sequence as a component of gene networks influencing cancer. Funct. Integr. Genom. 2010, 10, 307–319. [Google Scholar] [CrossRef] [PubMed]
- Ichiyanagi, K. Regulating Pol III transcription to change Pol II transcriptome. Cell Cycle 2014, 13, 3625–3626. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pandey, R.; Mukerji, M. From ‘JUNK’ to just unexplored noncoding knowledge: The case of transcribed Alus. Brief. Funct. Genomics 2011, 10, 294–311. [Google Scholar] [CrossRef] [PubMed]
- Holdt, L.M.; Hoffmann, S.; Sass, K.; Langenberger, D.; Scholz, M.; Krohn, K.; Finstermeier, K.; Stahringer, A.; Wilfert, W.; Beutner, F.; et al. Alu elements in ANRIL non-coding RNA at chromosome 9p21 modulate atherogenic cell functions through trans-regulation of gene networks. PLoS Genet. 2013, 9, e1003588. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.H.; Frankel, F.R. The induction of ALU-sequence transcripts by glucocorticoid in rat liver cells. J. Steroid Biochem. 1986, 25, 201–207. [Google Scholar] [PubMed]
- Koga, Y.; Lindstrom, E.; Fenyo, E.M.; Wigzell, H.; Mak, T. High levels of heterodisper RNA accumulate in T-cell infected with human immunodeficiency virus and in normal thymocytes. Proc. Natl. Acad. Sci. USA 1988, 85, 4521–4525. [Google Scholar] [CrossRef] [PubMed]
- Panning, B.; Smiley, J.R. Activation of RNA polymerase III transcription of human Alu repetitive elements by adenovirus type 5: Requirement for the E1b 58-kilodalton protein and the products of E4 open reading frames 3 and 6. Mol. Cell. Biol. 1993, 13, 3231–3244. [Google Scholar] [CrossRef] [PubMed]
- Panning, B.; Smiley, J.R. Activation of RNA polymerase III transcription of human Alu elements by herpes simplex virus. Virology 1994, 202, 408–417. [Google Scholar] [CrossRef] [PubMed]
- Gu, Z.; Jin, K.; Crabbe, M.J.; Zhang, Y.; Liu, X.; Huang, Y.; Hua, M.; Nan, P.; Zhang, Z.; Zhong, Y. Enrichment analysis of Alu elements with different spatial chromatin proximity in the human genome. Protein Cell 2016, 7, 250–266. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hambor, J.E.; Mennone, J.; Coon, M.E.; Hanke, J.H.; Kavathas, P. Identification and characterization of an Alu-containing, T-cell-specific enhancer located in the last intron of the human CD8 alpha gene. Mol. Cell. Biol. 1993, 13, 7056–7070. [Google Scholar] [CrossRef] [PubMed]
- Hanke, J.H.; Hambor, J.E.; Kavathas, P. Repetitive Alu elements form a cruciform structure that regulates the function of the human CD8 alpha T cell-specific enhancer. J. Mol. Biol. 1995, 246, 63–73. [Google Scholar] [CrossRef] [PubMed]
- Trujillo, M.A.; Sakagashira, M.; Eberhardt, N.L. The human growth hormone gene contains a silencer embedded within an Alu repeat in the 3’-flanking region. Mol. Endocrinol. 2006, 20, 2559–2575. [Google Scholar] [CrossRef] [PubMed]
- Ichiyanagi, K. Transposable elements in eukaryotic genomes: Epigenetic regulation by the host and functionalization for the host. Genes Genet. Syst. 2013, 88, 1. [Google Scholar] [CrossRef] [PubMed]
- Bouttier, M.; Laperriere, D.; Memari, B.; Mangiapane, J.; Fiore, A.; Mitchell, E.; Verway, M.; Behr, M.A.; Sladek, R.; Barreiro, L.B.; et al. Alu repeats as transcriptional regulatory platforms in macrophage responses to M. tuberculosis infection. Nucleic Acids Res. 2016, 44, 10571–10587. [Google Scholar] [CrossRef] [PubMed]
- Zuckerkandl, E.; Latter, G.; Jurka, J. Maintenance of function without selection: Alu sequences as “cheap genes”. J. Mol. Evol. 1989, 29, 504–512. [Google Scholar] [CrossRef] [PubMed]
- Ade, C.; Roy-Engel, A.M.; Deininger, P.L. Alu elements: An intrinsic source of human genome instability. Curr. Opin. Virol. 2013, 3, 639–645. [Google Scholar] [CrossRef] [PubMed]
- Payer, L.M.; Steranka, J.P.; Yang, W.R.; Kryatova, M.; Medabalimi, S.; Ardeljan, D.; Liu, C.; Boeke, J.D.; Avramopoulos, D.; Burns, K.H. Structural variants caused by Alu insertions are associated with risks for many human diseases. Proc. Natl. Acad. Sci. USA 2017, 114, E3984–E3992. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Huan, W.; Zuo, H.; Zhao, L.; Huang, C.; Liu, X.; Hou, S.; Qi, J.; Shi, W. Alu methylation serves as a biomarker for non-invasive diagnosis of glioma. Oncotarget 2016, 7, 26099–26106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kolomietz, E.; Meyn, M.S.; Pandita, A.; Squire, J.A. The role of Alu repeat clusters as mediators of recurrent chromosomal aberrations in tumors. Genes Chromosomes Cancer 2002, 35, 97–112. [Google Scholar] [CrossRef] [PubMed]
- Wallace, M.R.; Andersen, L.B.; Saulino, A.M.; Gregory, P.E.; Glover, T.W.; Collins, F.S. A de novo Alu insertion results in neurofibromatosis type 1. Nature 1991, 353, 864–866. [Google Scholar] [CrossRef] [PubMed]
- Chae, J.J.; Park, Y.B.; Kim, S.H.; Hong, S.S.; Song, G.J.; Han, K.H.; Namkoong, Y.; Kim, H.S.; Lee, C.C. Two partial deletion mutations involving the same Alu sequence within intron 8 of the LDL receptor gene in Korean patients with familial hypercholesterolemia. Hum. Genet. 1997, 99, 155–163. [Google Scholar] [CrossRef] [PubMed]
- Ricci, V.; Regis, S.; di Duca, M.; Filocamo, M. An Alu-mediated rearrangement as cause of exon skipping in Hunter disease. Hum. Genet. 2003, 112, 419–425. [Google Scholar] [PubMed]
- Sen, S.K.; Han, K.; Wang, J.; Lee, J.; Wang, H.; Callinan, P.A.; Dyer, M.; Cordaux, R.; Liang, P.; Batzer, M.A. Human genomic deletions mediated by recombination between Alu elements. Am. J. Hum. Genet. 2006, 79, 41–53. [Google Scholar] [CrossRef] [PubMed]
- McPherson, R.; Tybjaerg-Hansen, A. Genetics of Coronary Artery Disease. Circ. Res. 2016, 118, 564–578. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lloyd-Jones, D.M.; Nam, B.H.; D’Agostino, R.B.; Levy, D.; Murabito, J.M.; Wang, T.J.; Wilson, P.W.; O’Donnell, C.J. Parental cardiovascular disease as a risk factor for cardiovascular disease in middle-aged adults: A prospective study of parents and offspring. JAMA 2004, 291, 2204–2211. [Google Scholar] [CrossRef] [PubMed]
- Peyser, P.A.; Bielak, L.F.; Chu, J.S.; Turner, S.T.; Ellsworth, D.L.; Boerwinkle, E.; Sheedy, P.F. Heritability of coronary artery calcium quantity measured by electron beam computed tomography in asymptomatic adults. Circulation 2002, 106, 304–308. [Google Scholar] [CrossRef] [PubMed]
- McPherson, R.; Pertsemlidis, A.; Kavaslar, N.; Stewart, A.; Roberts, R.; Cox, D.R.; Hinds, D.A.; Pennacchio, L.A.; Tybjaerg-Hansen, A.; Folsom, A.R.; et al. A common allele on chromosome 9 associated with coronary heart disease. Science 2007, 316, 1488–1491. [Google Scholar] [CrossRef] [PubMed]
- Shen, G.Q.; Li, L.; Rao, S.; Abdullah, K.G.; Ban, J.M.; Lee, B.S.; Park, J.E.; Wang, Q.K. Four SNPs on chromosome 9p21 in a South Korean population implicate a genetic locus that confers high cross-race risk for development of coronary artery disease. Arterioscler. Thromb. Vasc. Biol. 2008, 28, 360–365. [Google Scholar] [CrossRef] [PubMed]
- Shen, G.Q.; Rao, S.; Martinelli, N.; Li, L.; Olivieri, O.; Corrocher, R.; Abdullah, K.G.; Hazen, S.L.; Smith, J.; Barnard, J.; et al. Association between four SNPs on chromosome 9p21 and myocardial infarction is replicated in an Italian population. J. Hum. Genet. 2008, 53, 144–150. [Google Scholar] [CrossRef] [PubMed]
- Hinohara, K.; Nakajima, T.; Takahashi, M.; Hohda, S.; Sasaoka, T.; Nakahara, K.; Chida, K.; Sawabe, M.; Arimura, T.; Sato, A.; et al. Replication of the association between a chromosome 9p21 polymorphism and coronary artery disease in Japanese and Korean populations. J. Hum. Genet. 2008, 53, 357–359. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abdullah, K.G.; Li, L.; Shen, G.Q.; Hu, Y.; Yang, Y.; MacKinlay, K.G.; Topol, E.J.; Wang, Q.K. Four SNPS on chromosome 9p21 confer risk to premature, familial CAD and MI in an American Caucasian population (GeneQuest). Ann. Hum. Genet. 2008, 72 Pt 5, 654–657. [Google Scholar] [CrossRef] [PubMed]
- Ding, H.; Xu, Y.; Wang, X.; Wang, Q.; Zhang, L.; Tu, Y.; Yan, J.; Wang, W.; Hui, R.; Wang, C.Y.; et al. 9p21 is a shared susceptibility locus strongly for coronary artery disease and weakly for ischemic stroke in Chinese Han population. Circ. Cardiovasc. Genet. 2009, 2, 338–346. [Google Scholar] [CrossRef] [PubMed]
- Kumar, J.; Yumnam, S.; Basu, T.; Ghosh, A.; Garg, G.; Karthikeyan, G.; Sengupta, S. Association of polymorphisms in 9p21 region with CAD in North Indian population: Replication of SNPs identified through GWAS. Clin. Genet. 2011, 79, 588–593. [Google Scholar] [CrossRef] [PubMed]
- Kral, B.G.; Mathias, R.A.; Suktitipat, B.; Ruczinski, I.; Vaidya, D.; Yanek, L.R.; Quyyumi, A.A.; Patel, R.S.; Zafari, A.M.; Vaccarino, V.; et al. A common variant in the CDKN2B gene on chromosome 9p21 protects against coronary artery disease in Americans of African ancestry. J. Hum. Genet. 2011, 56, 224–229. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Munir, M.S.; Wang, Z.; Alahdab, F.; Steffen, M.W.; Erwin, P.J.; Kullo, I.J.; Murad, M.H. The association of 9p21-3 locus with coronary atherosclerosis: A systematic review and meta-analysis. BMC Med. Genet. 2014, 15, 66. [Google Scholar] [CrossRef] [PubMed]
- Pott, J.; Burkhardt, R.; Beutner, F.; Horn, K.; Teren, A.; Kirsten, H.; Holdt, L.M.; Schuler, G.; Teupser, D.; Loeffler, M.; et al. Genome-wide meta-analysis identifies novel loci of plaque burden in carotid artery. Atherosclerosis 2017, 259, 32–40. [Google Scholar] [CrossRef] [PubMed]
- Chan, K.; Patel, R.S.; Newcombe, P.; Nelson, C.P.; Qasim, A.; Epstein, S.E.; Burnett, S.; Vaccarino, V.L.; Zafari, A.M.; Shah, S.H.; et al. Association between the chromosome 9p21 locus and angiographic coronary artery disease burden: A collaborative meta-analysis. J. Am. Coll. Cardiol. 2013, 61, 957–970. [Google Scholar] [CrossRef] [PubMed]
- Preuss, M.; König, I.R.; Thompson, J.R.; Erdmann, J.; Absher, D.; Assimes, T.L.; Blankenberg, S.; Boerwinkle, E.; Chen, L.; Cupples, L.A.; et al. Design of the Coronary ARtery DIsease Genome-Wide Replication and Meta-Analysis (CARDIoGRAM) Study: A Genome-wide association meta-analysis involving more than 22000 cases and 60000 controls. Circ. Cardiovasc. Genet. 2010, 3, 475–483. [Google Scholar] [CrossRef] [PubMed]
- Aarabi, G.; Zeller, T.; Seedorf, H.; Reissmann, D.R.; Heydecke, G.; Schaefer, A.S.; Seedorf, U. Genetic Susceptibility Contributing to Periodontal and Cardiovascular Disease. J. Dent. Res. 2017, 96, 610–617. [Google Scholar] [CrossRef] [PubMed]
- Kessler, T.; Vilne, B.; Schunkert, H. The impact of genome-wide association studies on the pathophysiology and therapy of cardiovascular disease. EMBO Mol. Med. 2016, 8, 688–701. [Google Scholar] [CrossRef] [PubMed]
- Helgadottir, A.; Thorleifsson, G.; Manolescu, A.; Gretarsdottir, S.; Blondal, T.; Jonasdottir, A.; Sigurdsson, A.; Baker, A.; Palsson, A.; Masson, G.; et al. A common variant on chromosome 9p21 affects the risk of myocardial infarction. Science 2007, 316, 1491–1493. [Google Scholar] [CrossRef] [PubMed]
- Tajbakhsh, A.; Khorrami, M.S.; Hassanian, S.M.; Aghasizade, M.; Pasdar, A.; Maftouh, M.; Tabatabai, E.; Parizadeh, S.M.; Fazeli, M.; Ferns, G.A.; et al. The 9p21 Locus and its Potential Role in Atherosclerosis Susceptibility; Molecular Mechanisms and Clinical Implications. Curr. Pharm. Des. 2016, 22, 5730–5737. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.S.; Smith, J.A.; Bielak, L.F.; Wu, C.Y.; Sun, Y.V.; Sheedy, P.F.; Turner, S.T.; Peyser, P.A.; Kardia, S.L. The relationship between diastolic blood pressure and coronary artery calcification is dependent on single nucleotide polymorphisms on chromosome 9p21.3. BMC Med. Genet. 2014, 15, 89. [Google Scholar] [CrossRef] [PubMed]
- Arslan, S.; Berkan, Ö.; Lalem, T.; Özbilüm, N.; Göksel, S.; Korkmaz, Ö.; Çetin, N.; Devaux, Y.; Cardiolinc™ Network. Long non-coding RNAs in the atherosclerotic plaque. Atherosclerosis 2017, 266, 176–181. [Google Scholar] [CrossRef] [PubMed]
- Pasmant, E.; Laurendeau, I.; Héron, D.; Vidaud, M.; Vidaud, D.; Bièche, I. Characterization of a germ-line deletion, including the entire INK4/ARF locus, in a melanoma-neural system tumor family: Identification of ANRIL, an antisense noncoding RNA whose expression coclusters with ARF. Cancer Res. 2007, 67, 3963–3969. [Google Scholar] [CrossRef] [PubMed]
- Bunch, H. Gene regulation of mammalian long non-coding RNA. Mol. Genet. Genom. 2017. [Google Scholar] [CrossRef] [PubMed]
- Aguilo, F.; Di Cecilia, S.; Walsh, M.J. Long Non-coding RNA ANRIL and Polycomb in Human Cancers and Cardiovascular Disease. Curr. Top. Microbiol. Immunol. 2016, 394, 29–39. [Google Scholar] [PubMed]
- Xu, S.T.; Xu, J.H.; Zheng, Z.R.; Zhao, Q.Q.; Zeng, X.S.; Cheng, S.X.; Liang, Y.H.; Hu, Q.F. Long non-coding RNA ANRIL promotes carcinogenesis via sponging miR-199a in triple-negative breast cancer. Biomed. Pharmacother. 2017, 96, 14–21. [Google Scholar] [CrossRef] [PubMed]
- Li, R.; Yin, F.; Guo, Y.Y.; Zhao, K.C.; Ruan, Q.; Qi, Y.M. Knockdown of ANRIL aggravates H2O2-induced injury in PC-12 cells by targeting microRNA-125a. Biomed. Pharmacother. 2017, 92, 952–961. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.J.; Wang, D.D.; Du, C.X.; Wang, Y. Long Noncoding RNA ANRIL Promotes Cervical Cancer Development by Acting as a Sponge of miR-186. Oncol. Res. 2017. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Wang, X.; Chen, X. Potential Role of Long Non-Coding RNA ANRIL in Pediatric Medulloblastoma Through Promotion on Proliferation and Migration by Targeting miR-323. J. Cell. Biochem. 2017, 118, 4735–4744. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Zhang, J.Q.; Chen, J.Z.; Chen, H.X.; Qiu, F.N.; Yan, M.L.; Chen, Y.L.; Peng, C.H.; Tian, Y.F.; Wang, Y.D. The over expression of long non-coding RNA ANRIL promotes epithelial-mesenchymal transition by activating the ATM-E2F1 signaling pathway in pancreatic cancer: An in vivo and in vitro study. Int. J. Biol. Macromol. 2017, 102, 718–728. [Google Scholar] [CrossRef] [PubMed]
- Thomas, A.A.; Feng, B.; Chakrabarti, S. ANRIL: A Regulator of VEGF in Diabetic Retinopathy. Investig. Ophthalmol. Vis. Sci. 2017, 58, 470–480. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Sun, G.; Zhang, H.; Tian, J.; Li, Y. Long non-coding RNA ANRIL indicates a poor prognosis of cervical cancer and promotes carcinogenesis via PI3K/Akt pathways. Biomed. Pharmacother. 2017, 85, 511–516. [Google Scholar] [CrossRef] [PubMed]
- Royds, J.A.; Pilbrow, A.P.; Ahn, A.; Morrin, H.R.; Frampton, C.; Russell, I.A.; Moravec, C.S.; Sweet, W.E.; Tang, W.H.; Currie, M.J.; et al. The rs11515 Polymorphism Is More Frequent and Associated with Aggressive Breast Tumors with Increased ANRIL and Decreased p16 (INK4a) Expression. Front. Oncol. 2015, 5, 306. [Google Scholar] [PubMed]
- Chen, D.; Zhang, Z.; Mao, C.; Zhou, Y.; Yu, L.; Yin, Y.; Wu, S.; Mou, X.; Zhu, Y. ANRIL inhibits p15(INK4b) through the TGFβ1 signaling pathway in human esophageal squamous cell carcinoma. Cell. Immunol. 2014, 289, 91–96. [Google Scholar] [CrossRef] [PubMed]
- Burd, C.E.; Jeck, W.R.; Liu, Y.; Sanoff, H.K.; Wang, Z.; Sharpless, N.E. Expression of linear and novel circular forms of an INK4/ARF-associated non-coding RNA correlates with atherosclerosis risk. PLoS Genet. 2010, 6, e1001233. [Google Scholar] [CrossRef] [PubMed]
- Assimes, T.L.; Knowles, J.W.; Basu, A.; Iribarren, C.; Southwick, A.; Tang, H.; Absher, D.; Li, J.; Fair, J.M.; Rubin, G.D.; et al. Susceptibility locus for clinical and subclinical coronary artery disease at chromosome 9p21 in the multi-ethnic ADVANCE study. Hum. Mol. Genet. 2008, 17, 2320–2328. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McLaren, J.E.; Ramji, D.P. Interferon gamma: A master regulator of atherosclerosis. Cytokine Growth Factor Rev. 2009, 20, 125–135. [Google Scholar] [CrossRef] [PubMed]
- Hannou, S.A.; Wouters, K.; Paumelle, R.; Staels, B. Functional genomics of the CDKN2A/B locus in cardiovascular and metabolic disease: What have we learned from GWASs? Trends Endocrinol. Metab. 2015, 26, 176–184. [Google Scholar] [CrossRef] [PubMed]
- Arbiol-Roca, A.; Padró-Miquel, A.; Hueso, M.; Navarro, E.; Alía-Ramos, P.; González-Álvarez, M.T.; Rama, I.; Torras, J.; Grinyó, J.M.; Cruzado, J.M.; et al. Association of ANRIL gene polymorphisms with major adverse cardiovascular events in hemodialysis patients. Clin. Chim. Acta 2017, 466, 61–67. [Google Scholar] [CrossRef] [PubMed]
- Feinberg, M.W.; Moore, K.J. MicroRNA Regulation of Atherosclerosis. Circ. Res. 2016, 118, 703–720. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andreou, I.; Sun, X.; Stone, P.H.; Edelman, E.R.; Feinberg, M.W. miRNAs in atherosclerotic plaque initiation, progression, and rupture. Trends Mol. Med. 2015, 21, 307–318. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aryal, B.; Rotllan, N.; Fernández-Hernando, C. Noncoding RNAs and atherosclerosis. Curr. Atheroscler. Rep. 2014, 16, 407. [Google Scholar] [CrossRef] [PubMed]
- Urbich, C.; Kuehbacher, A.; Dimmeler, S. Role of microRNAs in vascular diseases, inflammation, and angiogenesis. Cardiovasc. Res. 2008, 79, 581–588. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feng, J.; Li, A.; Deng, J.; Yang, Y.; Dang, L.; Ye, Y.; Li, Y.; Zhang, W. miR21 attenuates lipopolysaccharide-induced lipid accumulation and inflammatory response: Potential role in cerebrovascular disease. Lipids Health Dis. 2014, 13, 27. [Google Scholar] [CrossRef] [PubMed]
- Haver, V.G.; Slart, R.H.; Zeebregts, C.J.; Peppelenbosch, M.P.; Tio, R.A. Rupture of vulnerable atherosclerotic plaques: MicroRNAs conducting the orchestra? Trends Cardiovasc. Med. 2010, 20, 65–71. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Yao, N.; Zhang, J.; Liu, Z. MicroRNA-125b is involved in atherosclerosis obliterans in vitro by targeting podocalyxin. Mol. Med. Rep. 2015, 12, 561–568. [Google Scholar] [CrossRef] [PubMed]
- Hueso, M.; De Ramon, L.; Navarro, E.; Ripoll, E.; Cruzado, J.M.; Grinyo, J.M.; Torras, J. Silencing of CD40 in vivo reduces progression of experimental atherogenesis through an NF-κB/miR-125b axis and reveals new potential mediators in the pathogenesis of atherosclerosis. Atherosclerosis 2016, 255, 80–89. [Google Scholar] [CrossRef] [PubMed]
- Holdt, L.M.; Teupser, D. From genotype to phenotype in human atherosclerosis recent findings. Curr. Opin. Lipidol. 2013, 24, 410–418. [Google Scholar] [CrossRef] [PubMed]
- Visel, A.; Zhu, Y.; May, D.; Afzal, V.; Gong, E.; Attanasio, C.; Blow, M.J.; Cohen, J.C.; Rubin, E.M.; Pennacchio, L.A. Targeted deletion of the 9p21 non-coding coronary artery disease risk interval in mice. Nature 2010, 464, 409–412. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, S.; Gu, W.; Li, Y.; Zhu, H. ANRIL/CDKN2B-AS shows two-stage clade-specific evolution and becomes conserved after transposon insertions in simians. BMC Evol. Biol. 2013, 13, 247. [Google Scholar] [CrossRef] [PubMed]
- Tsirigos, A.; Rigoutsos, I. Alu and b1 repeats have been selectively retained in the upstream and intronic regions of genes of specific functional classes. PLoS Comput. Biol. 2009, 5, e1000610. [Google Scholar] [CrossRef] [PubMed]
- Burenina, O.Y.; Oretskaya, T.S.; Kubareva, E.A. Non-coding RNAs as Transcriptional Regulators in Eukaryotes. Acta Nat. 2017, 9, 13–25. [Google Scholar]
- Spengler, R.M.; Oakley, C.K.; Davidson, B.L. Functional microRNAs and target sites are created by lineage-specific transposition. Hum. Mol. Genet. 2014, 23, 1783–1793. [Google Scholar] [CrossRef] [PubMed]
- Gu, T.J.; Yi, X.; Zhao, X.W.; Zhao, Y.; Yin, J.Q. Alu-directed transcriptional regulation of some novel miRNAs. BMC Genom. 2009, 10, 563. [Google Scholar] [CrossRef] [PubMed]
- Monteys, A.M.; Spengler, R.M.; Wan, J.; Tecedor, L.; Lennox, K.A.; Xing, Y.; Davidson, B.L. Structure and activity of putative intronic miRNA promoters. RNA 2010, 16, 495–505. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Borchert, G.M.; Lanier, W.; Davidson, B.L. RNA polymerase III transcribes human microRNAs. Nat. Struct. Mol. Biol. 2006, 13, 1097–1101. [Google Scholar] [CrossRef] [PubMed]
- Saito, Y.; Suzuki, H.; Tsugawa, H.; Nakagawa, I.; Matsuzaki, J.; Kanai, Y.; Hibi, T. Chromatin remodeling at Alu repeats by epigenetic treatment activates silenced microRNA-512-5p with downregulation of Mcl-1 in human gastric cancer cells. Oncogene 2009, 28, 2738–2744. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oei, S.L.; Babich, V.S.; Kazakov, V.I.; Usmanova, N.M.; Kropotov, A.V.; Tomilin, N.V. Clusters of regulatory signals for RNA polymerase II transcription associated with Alu family repeats and CpG islands in human promoters. Genomics 2004, 83, 873–882. [Google Scholar] [CrossRef] [PubMed]
- Smalheiser, N.R.; Torvik, V.I. Alu elements within human mRNAs are probable microRNA targets. Trends Genet. 2006, 22, 532–536. [Google Scholar] [CrossRef] [PubMed]
- Daskalova, E.; Baev, V.; Rusinov, V.; Minkov, I. 3’UTR-located ALU elements: Donors of potential miRNA target sites and mediators of network miRNA-based regulatory interactions. Evol. Bioinform. Online 2007, 2, 103–120. [Google Scholar] [CrossRef] [PubMed]
- Lehnert, S.; van Loo, P.; Thilakarathne, P.J.; Marynen, P.; Verbeke, G.; Schuit, F.C. Evidence for co-evolution between human microRNAs and Alu-repeats. PLoS ONE 2009, 4, e4456. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Ruocco, F.; Basso, V.; Rivoire, M.; Mehlen, P.; Ambati, J.; de Falco, S.; Tarallo, V. Alu RNA accumulation induces epithelial-to-mesenchymal transition by modulating miR-566 and is associated with cancer progression. Oncogene 2018, 37, 627–637. [Google Scholar] [CrossRef] [PubMed]
- Pandey, R.; Bhattacharya, A.; Bhardwaj, V.; Jha, V.; Mandal, A.K.; Mukerji, M. Alu-miRNA interactions modulate transcript isoform diversity in stress response and reveal signatures of positive selection. Sci. Rep. 2016, 6, 32348. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoffman, Y.; Bublik, D.R.; Pilpel, Y.; Oren, M. miR-661 downregulates both Mdm2 and Mdm4 to activate p53. Cell Death Differ. 2014, 21, 302–309. [Google Scholar] [CrossRef] [PubMed]
- Hernández-Presa, M.; Bustos, C.; Ortego, M.; Tuñon, J.; Renedo, G.; Ruiz-Ortega, M.; Egido, J. Angiotensin-converting enzyme inhibition prevents arterial nuclear factor-kappa B activation, monocyte chemoattractant protein-1 expression, and macrophage infiltration in a rabbit model of early accelerated atherosclerosis. Circulation 1997, 95, 1532–1541. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Xing, S.S.; Sun, X.L.; Xing, Q.C. Overexpression of activated nuclear factor-kappa B in aorta of patients with coronary atherosclerosis. Clin. Cardiol. 2009, 32, E42–E47. [Google Scholar] [CrossRef] [PubMed]
- Hueso, M.; Torras, J.; Carrera, M.; Vidal, A.; Navarro, E.; Grinyó, J. Chronic Kidney Disease is associated with an increase of Intimal Dendritic cells in a comparative autopsy study. J. Inflamm. 2015, 12, 26. [Google Scholar] [CrossRef] [PubMed]
- Vallabhapurapu, S.; Karin, M. Regulation and function of NF-κB transcription factors in the immune system. Annu. Rev. Immunol. 2009, 27, 693–733. [Google Scholar] [CrossRef] [PubMed]
- Farnham, P.J. Insights from genomic profiling of transcription factors. Nat. Rev. Genet. 2009, 10, 605–616. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Antonaki, A.; Demetriades, C.; Polyzos, A.; Banos, A.; Vatsellas, G.; Lavigne, M.D.; Apostolou, E.; Mantouvalou, E.; Papadopoulou, D.; Mosialos, G.; et al. Genomic analysis reveals a novel nuclear factor-κB (NF-κB)-binding site in Alu-repetitive elements. J. Biol. Chem. 2011, 286, 38768–38782. [Google Scholar] [CrossRef] [PubMed]
- Kasowski, M.; Grubert, F.; Heffelfinger, C.; Hariharan, M.; Asabere, A.; Waszak, S.M.; Habegger, L.; Rozowsky, J.; Shi, M.; Urban, A.E.; et al. Variation in transcription factor binding among humans. Science 2010, 328, 232–235. [Google Scholar] [CrossRef] [PubMed]
- Ruiz-Ortega, M.; Lorenzo, O.; Rupérez, M.; Esteban, V.; Suzuki, Y.; Mezzano, S.; Plaza, J.J.; Egido, J. Role of the renin-angiotensin system in vascular diseases: Expanding the field. Hypertension 2001, 38, 1382–1387. [Google Scholar] [CrossRef] [PubMed]
- Sahar, S.; Dwarakanath, R.; Reddy, M.; Lanting, L.; Todorov, I.; Natarajan, R. Angiotensin II enhances interleukin-18 mediated inflammatory gene expression in vascular smooth Muscle cells-A novel Cross-Talk in the Pathogenesis of Atherosclerosis. Circ. Res. 2005, 96, 1064–1071. [Google Scholar] [CrossRef] [PubMed]
- Pfeffer, M.A.; Braunwald, E.; Moyé, L.A.; Basta, L.; Brown, E.J.; Cuddy, T.E.; Davis, B.R.; Geltman, E.M.; Goldman, S.; Flaker, G.C. Effect of captopril on mortality and morbidity in patients with left ventricular dysfunction after myocardial infarction. Results of the survival and ventricular enlargement trial. The SAVE Investigators. N. Engl. J. Med. 1992, 327, 669–677. [Google Scholar] [CrossRef] [PubMed]
- Hueso, M.; Alía, P.; Moreso, F.; Beltrán-Sastre, V.; Riera, L.; González, C.; Navarro, M.A.; Grinyó, J.M.; Navarro, E.; Serón, D. Angiotensin converting enzyme genotype and chronic allograft nephropathy in protocol biopsies. J. Am. Soc. Nephrol. 2004, 15, 2229–2236. [Google Scholar] [CrossRef] [PubMed]
- Cambien, F.; Poirier, O.; Lecerf, L.; Evans, A.; Cambou, J.P.; Arveiler, D.; Luc, G.; Bard, J.M.; Bara, L.; Ricard, S. Deletion polymorphism in the gene for angiotensin-converting enzyme is a potent risk factor for myocardial infarction. Nature 1992, 359, 641–644. [Google Scholar] [CrossRef] [PubMed]
- Rigat, B.; Hubert, C.; Alhenc-Gelas, F.; Cambien, F.; Corvol, P.; Soubrier, F. An insertion/deletion polymorphism in the angiotensin I-converting enzyme gene accounting for half the variance of serum enzyme levels. J. Clin. Investig. 1990, 86, 1343–1346. [Google Scholar] [CrossRef] [PubMed]
- Woods, D.R.; Brull, D.; Montgomery, H.E. Endurance and the ACE I/D polymorphism. Sci. Prog. 2000, 83 Pt 4, 317–336. [Google Scholar] [PubMed]
- Wu, S.J.; Hsieh, T.J.; Kuo, M.C.; Tsai, M.L.; Tsai, K.L.; Chen, C.H.; Yang, Y.H. Functional regulation of Alu element of human angiotensin-converting enzyme gene in neuron cells. Neurobiol. Aging 2013, 34, 1921.e1–1921.e7. [Google Scholar] [CrossRef] [PubMed]
- Sayed-Tabatabaei, F.A.; Oostra, B.A.; Isaacs, A.; van Duijn, C.M.; Witteman, J.C. ACE polymorphisms. Circ. Res. 2006, 98, 1123–1133. [Google Scholar] [CrossRef] [PubMed]
- Luo, R.; Li, X.; Wang, Y.; Li, Y.; Deng, Y.; Wan, Y.; Jiang, Z.; Hua, W.; Wu, X. The influence of Angiotensin converting enzyme and angiotensinogen gene polymorphisms on hypertrophic cardiomyopathy. PLoS ONE 2013, 8, e77030. [Google Scholar] [CrossRef] [PubMed]
- Wei, L.K.; Au, A.; Menon, S.; Griffiths, L.R.; Kooi, C.W.; Irene, L.; Zhao, J.; Lee, C.; Alekseevna, A.M.; Hassan, M.R.A.; et al. Polymorphisms of MTHFR, eNOS, ACE, AGT, ApoE, PON1, PDE4D, and Ischemic Stroke: Meta-Analysis. J. Stroke Cerebrovasc. Dis. 2017, 26, 2482–2493. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.; Yang, H.Y.; Wu, C.C.; Lee, H.S.; Lin, Y.F.; Lu, K.C.; Chu, C.M.; Lin, F.H.; Kao, S.Y.; Su, S.L. Angiotensin-converting enzyme insertion/deletion polymorphism contributes high risk for chronic kidney disease in Asian male with hypertension--a meta-regression analysis of 98 observational studies. PLoS ONE 2014, 9, e87604. [Google Scholar] [CrossRef] [PubMed]
- Song, G.G.; Kim, J.H.; Lee, Y.H. Associations between the insertion/deletion polymorphism of the angiotensin-converting enzyme and susceptibility to aortic aneurysms: A meta-analysis. J. Renin Angiotensin Aldosterone Syst. 2015, 16, 211–218. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Qin, X.; Li, S.; Zeng, Z. Association between the ACE I/D polymorphism and risk of ischemic stroke: An updated meta-analysis of 47026 subjects from 105 case-control studies. J. Neurol. Sci. 2014, 345, 37–47. [Google Scholar] [CrossRef] [PubMed]
- Yan, Q. From pharmacogenomics and systems biology to personalized care: A framework of systems and dynamical medicine. Methods Mol. Biol. 2014, 1175, 3–17. [Google Scholar] [CrossRef] [PubMed]
- Derry, P.S.; Derry, G.N. Menstruation, perimenopause, and chaos theory. Perspect. Biol. Med. 2012, 55, 26–42. [Google Scholar] [CrossRef] [PubMed]
- Badimon, L.; Vilahur, G. LDL-cholesterol versus HDL-cholesterol in the atherosclerotic plaque: Inflammatory resolution versus thrombotic chaos. Ann. N. Y. Acad. Sci. 2012, 1254, 18–32. [Google Scholar] [CrossRef] [PubMed]
- Mangin, L.; Lesèche, G.; Duprey, A.; Clerici, C. Ventilatory chaos is impaired in carotid atherosclerosis. PLoS ONE 2011, 6, e16297. [Google Scholar] [CrossRef] [PubMed]
- Martín-Araguz, A.; Ruiz-Aláez, A.; García de la Rocha, M.L.; Fernández-Armayor, V.; Delgado-Reyes, S.; Moreno-Martínez, J.M. [Kinematic deterministic chaos of fluids and fractal geometry in the carotid system]. Rev. Neurol. 1997, 25, 2021–2031. [Google Scholar] [PubMed]


© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hueso, M.; Cruzado, J.M.; Torras, J.; Navarro, E. ALUminating the Path of Atherosclerosis Progression: Chaos Theory Suggests a Role for Alu Repeats in the Development of Atherosclerotic Vascular Disease. Int. J. Mol. Sci. 2018, 19, 1734. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms19061734
Hueso M, Cruzado JM, Torras J, Navarro E. ALUminating the Path of Atherosclerosis Progression: Chaos Theory Suggests a Role for Alu Repeats in the Development of Atherosclerotic Vascular Disease. International Journal of Molecular Sciences. 2018; 19(6):1734. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms19061734
Chicago/Turabian StyleHueso, Miguel, Josep M. Cruzado, Joan Torras, and Estanislao Navarro. 2018. "ALUminating the Path of Atherosclerosis Progression: Chaos Theory Suggests a Role for Alu Repeats in the Development of Atherosclerotic Vascular Disease" International Journal of Molecular Sciences 19, no. 6: 1734. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms19061734