Anti-Proliferative Properties and Proapoptotic Function of New CB2 Selective Cannabinoid Receptor Agonist in Jurkat Leukemia Cells

 ,
,  , , and
, , and
Abstract
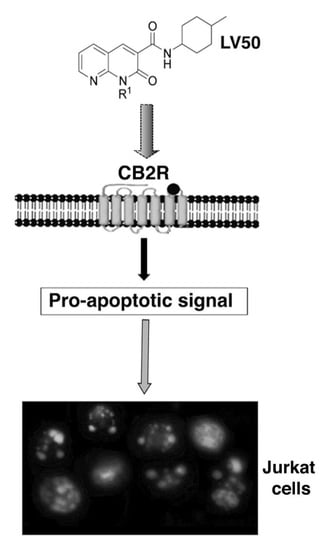
:
1. Introduction
2. Results
2.1. CB1R and CB2R Affinity
2.2. CB2R Expression
2.3. Preliminary Analysis of the Compounds
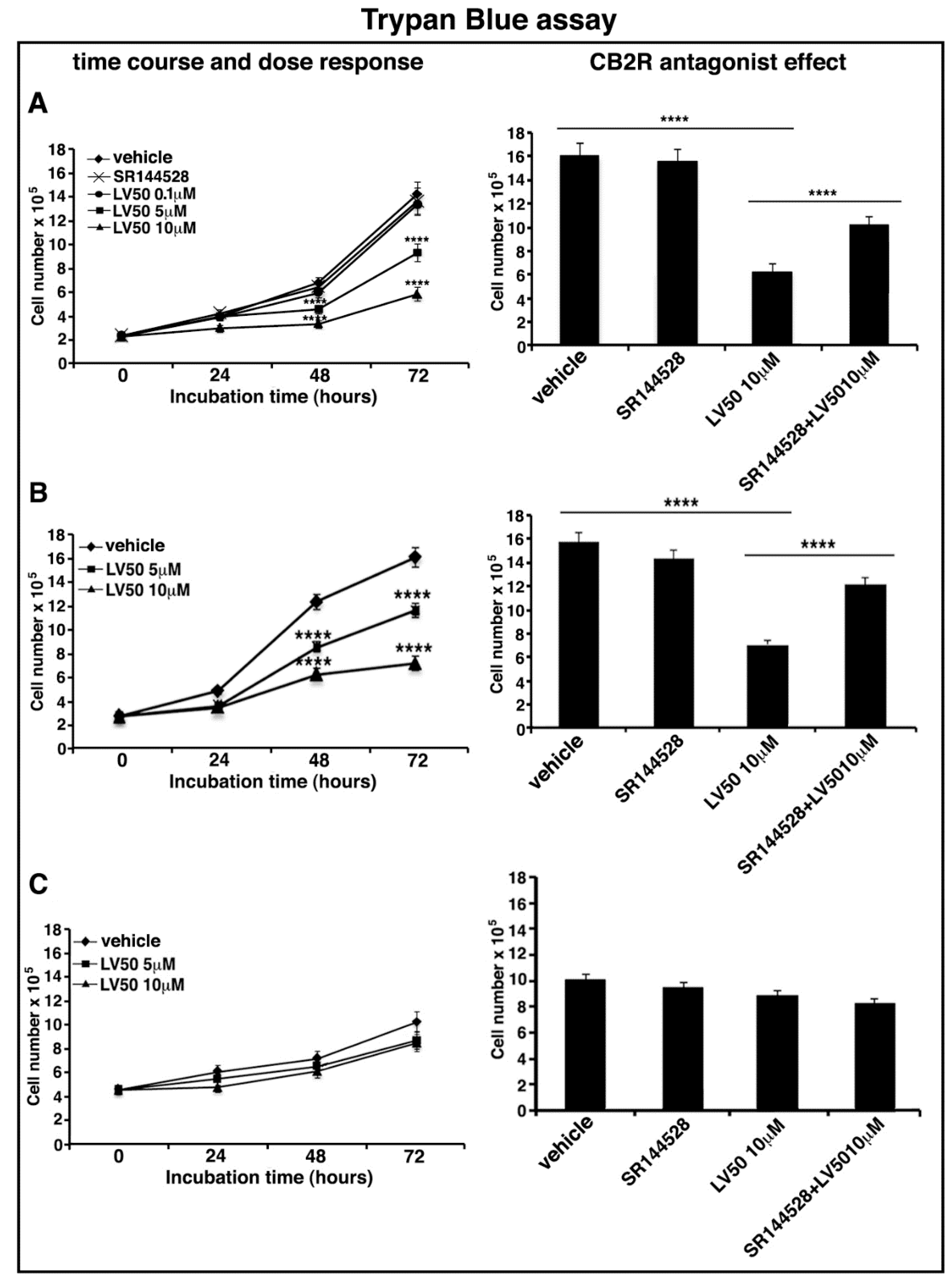
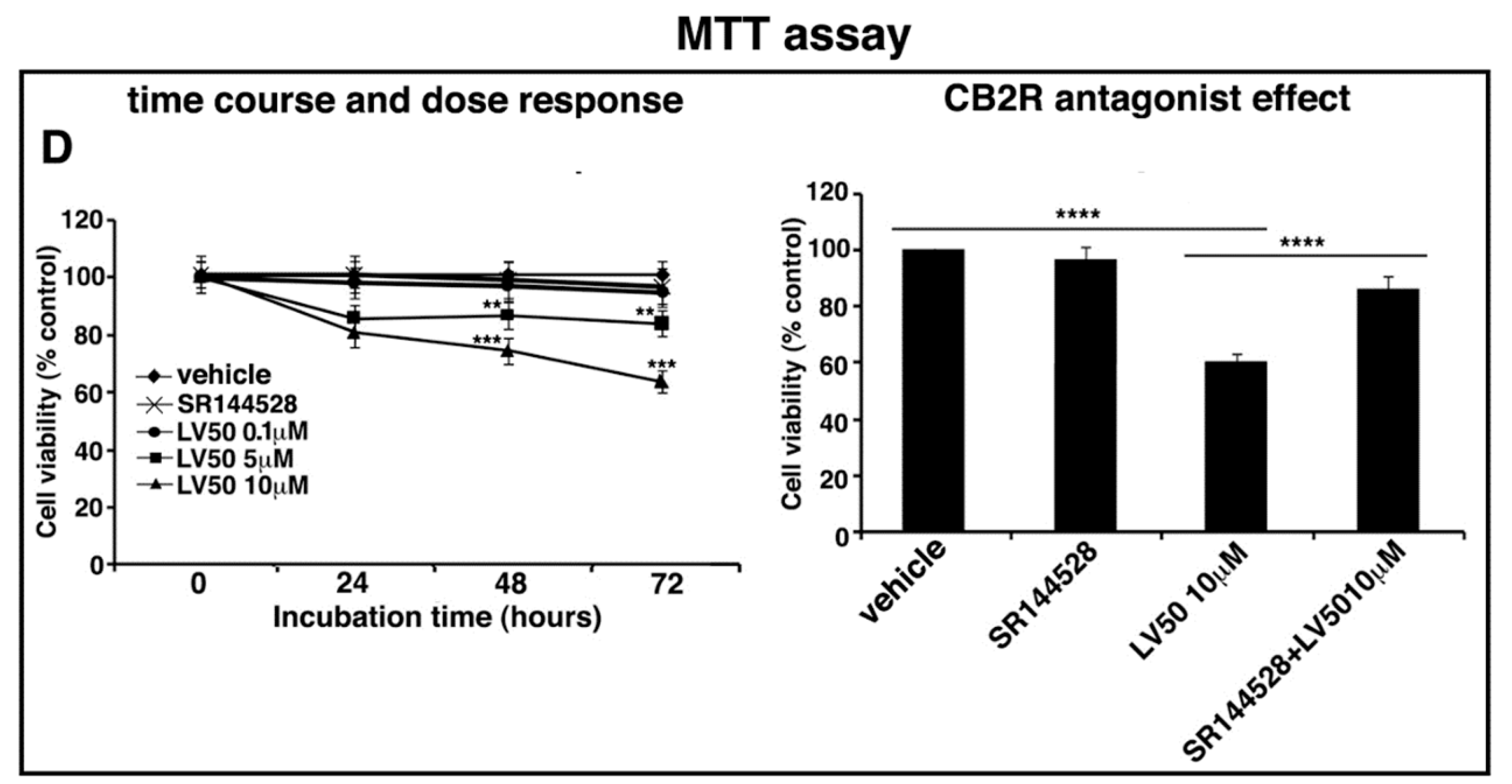
2.4. LV50 Reduces Cell Viability
2.5. Pro-Apoptotic Activity of LV50
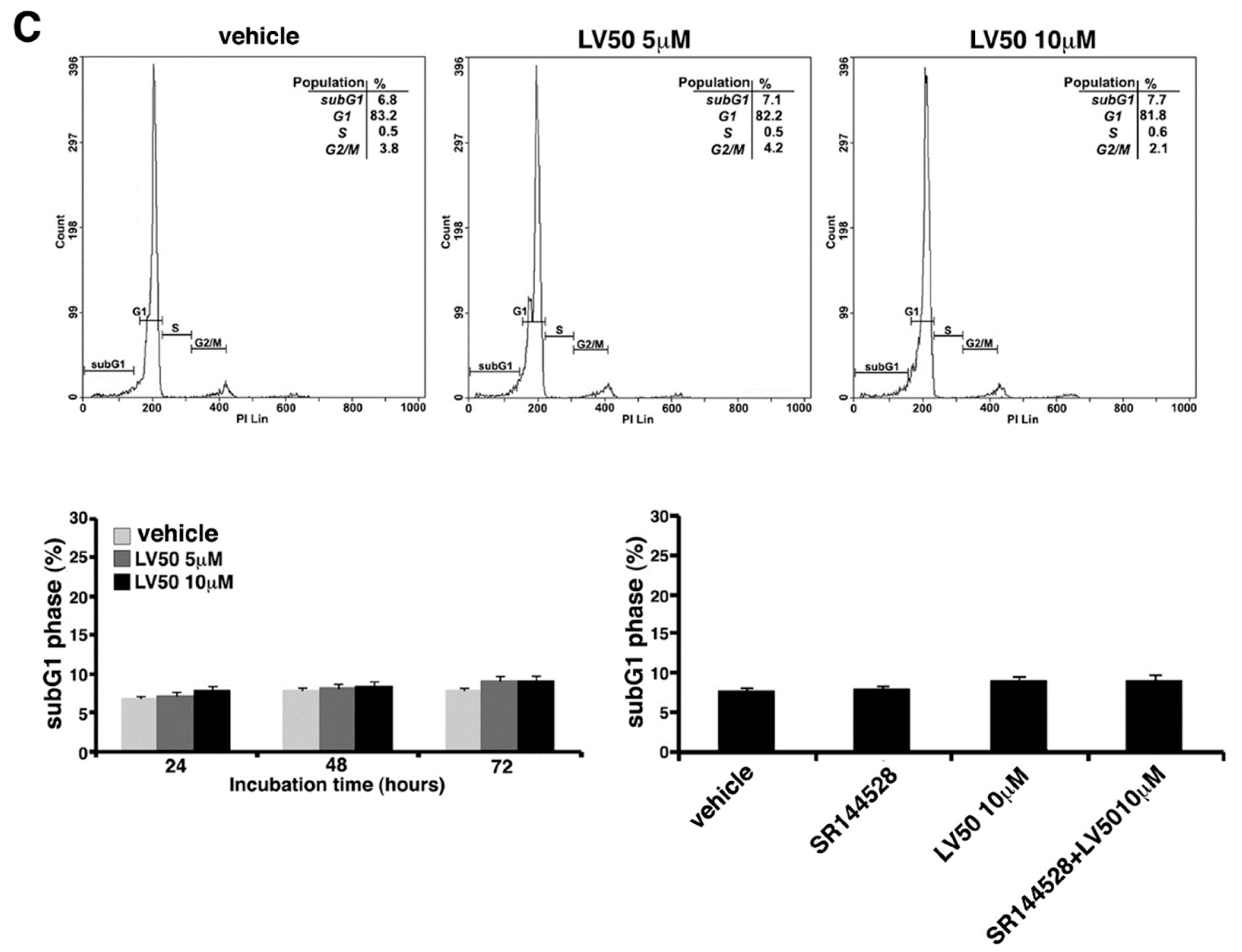
2.5.1. LV50 Increases the Percentage of Cells in Apoptotic Sub-G1 Population and Nuclear Morphological Changes
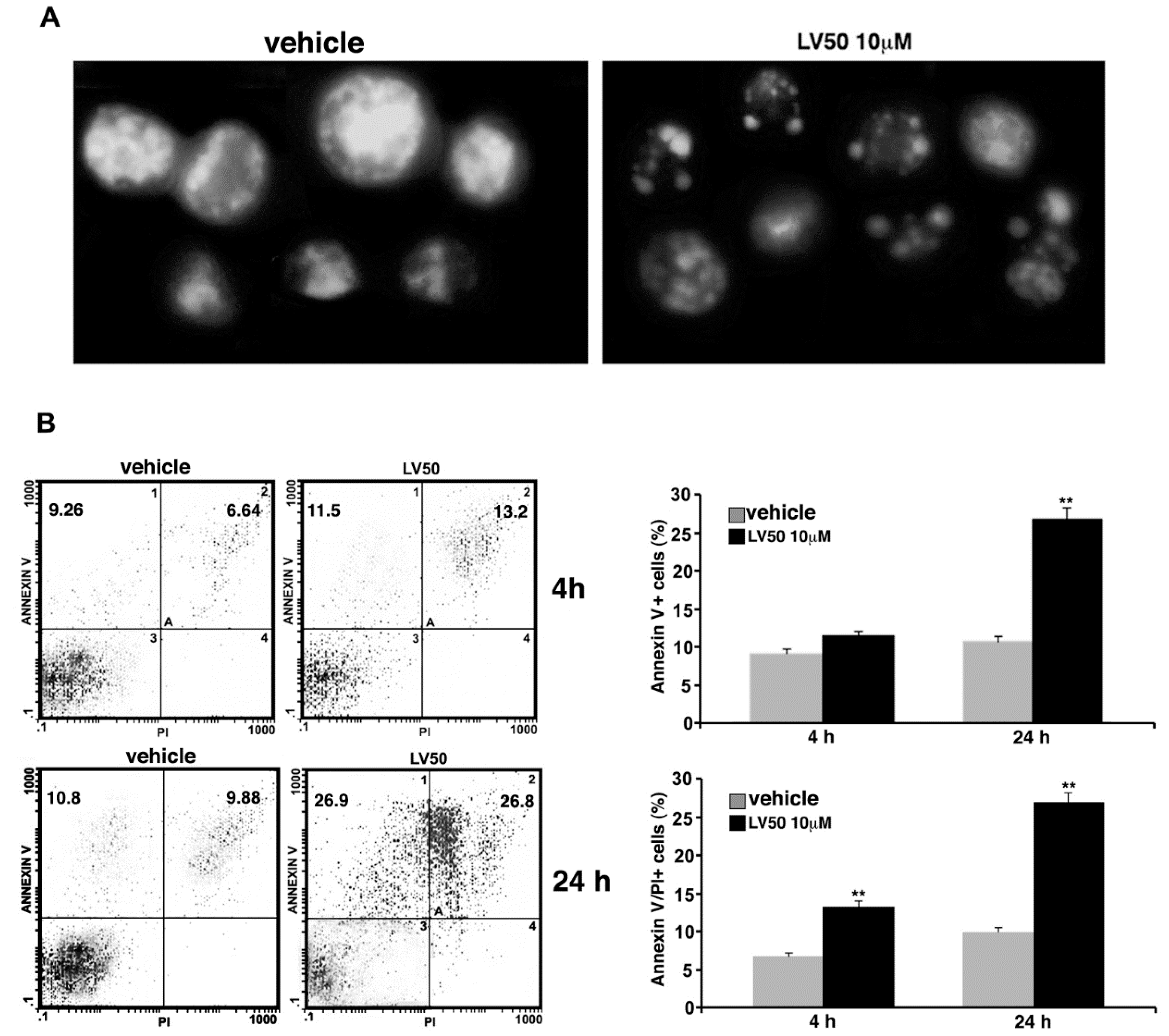
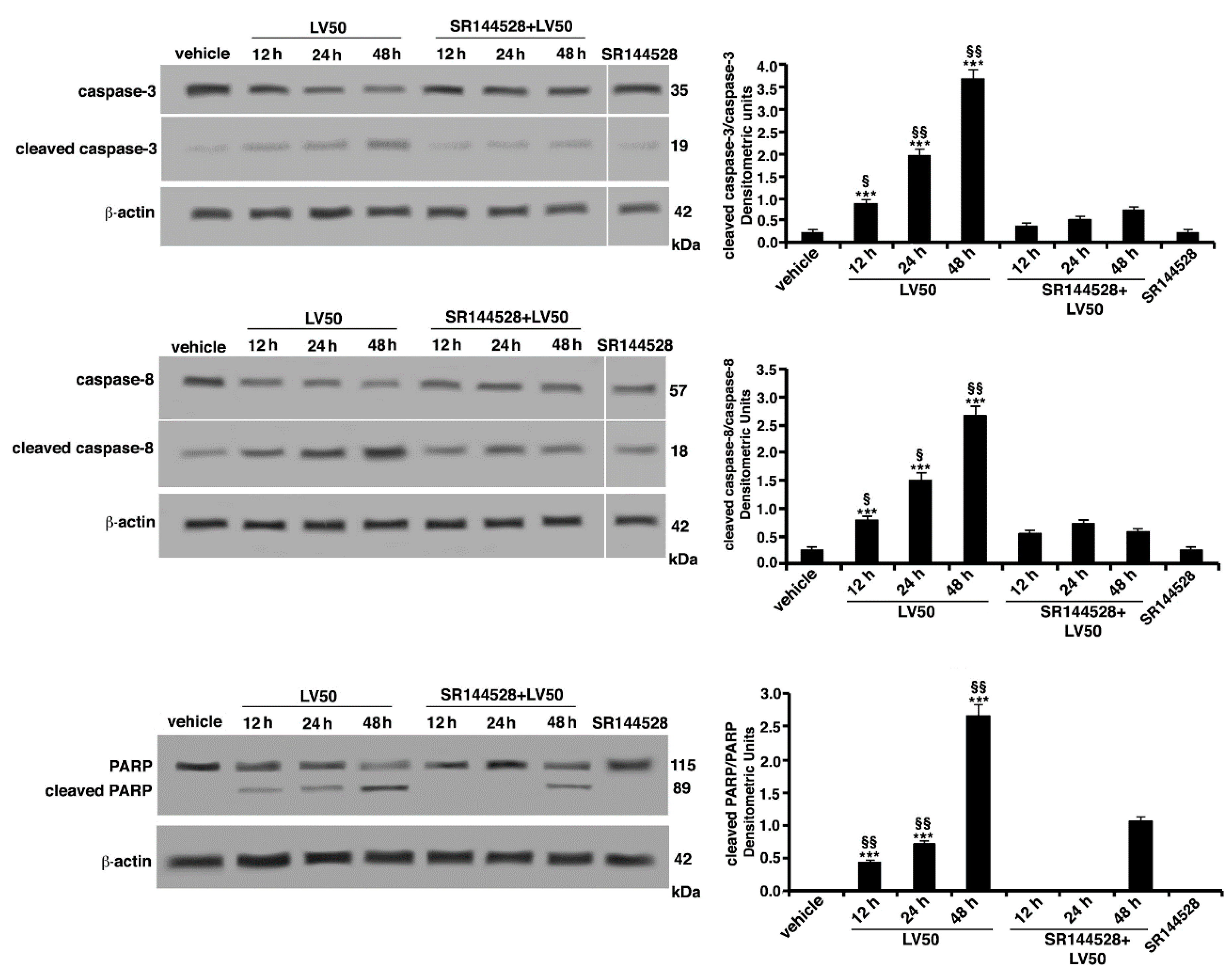
2.5.2. Pro-Apoptotic Activity of LV50 Revealed by Hoechst 33258, Annexin V Staining and Caspases Activation
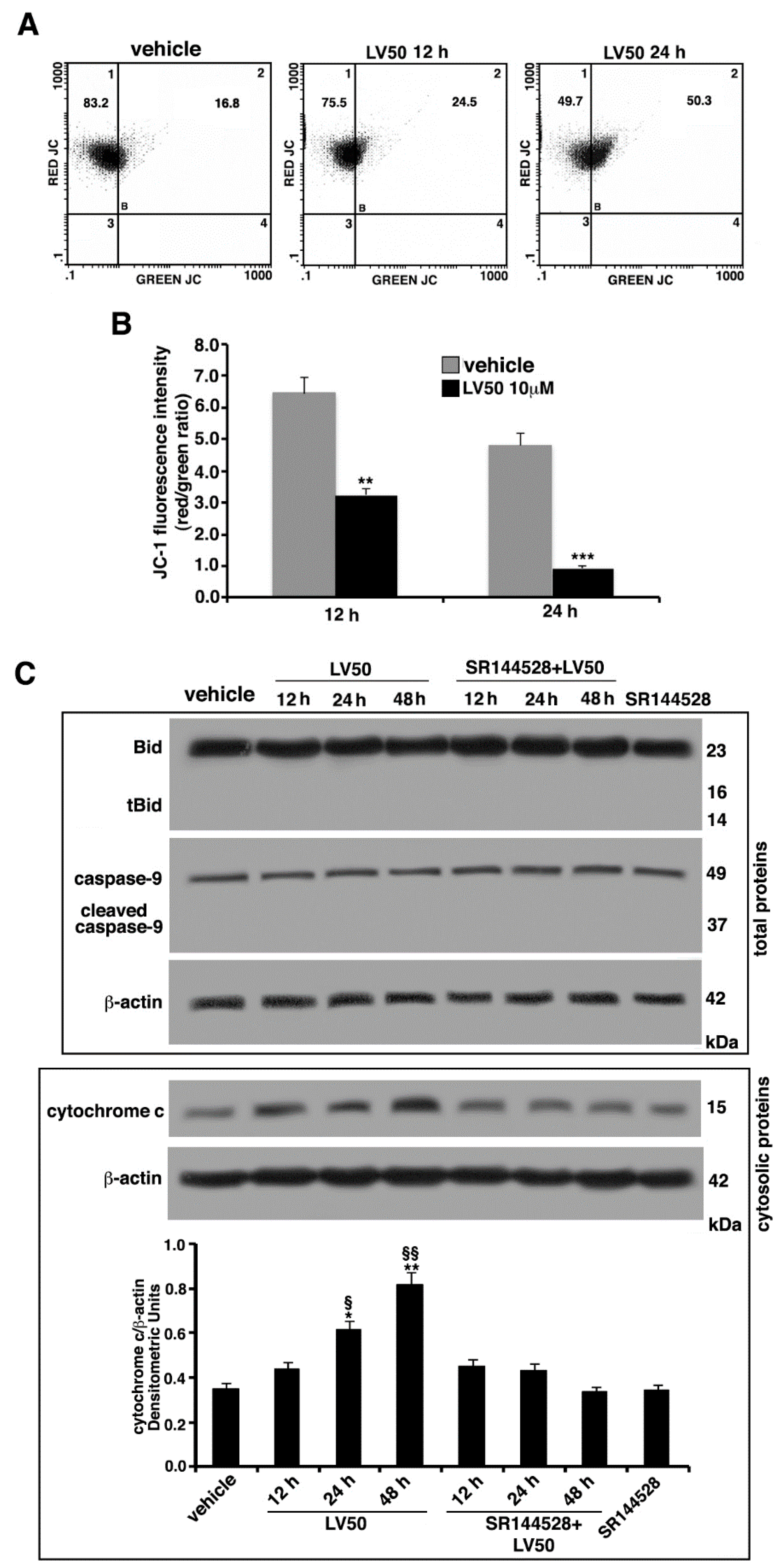
2.6. Molecular Involvement during Apoptotic Execution
3. Discussion
4. Materials and Methods
4.1. Synthesis of 1-(4-Chlorobutyl)-1,2-dihydro-N-(4-methylcyclohexyl)-2-oxo-1,8-naphthyridine-3-carboxamide (LV50)
4.2. CB1 and CB2 Receptor Binding Assays
4.3. Cell Culture and Treatments
4.4. Western Blot Analysis of CB2R Expression
4.5. Proliferation and Cell Viability Assays
4.6. Apoptosis Assays
4.6.1. Propidium Iodide Staining
4.6.2. Hoechst Staining
4.6.3. Annexin V Assay
4.7. Western Blot Analysis of Caspase and PARP Proteins
4.8. Analysis of Mitochondrial Membrane Potential Changes (ΔΨ)
4.9. Analysis of t-Bid Expression and Cytochrome c Release
4.10. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Munro, S.; Thomas, K.L.; Abu-Shaar, M. Molecular characterization of a peripheral receptor for cannabinoids. Nature 1993, 365, 61–65. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Ruiz, J.; Romero, J.; Velasco, G.; Tolón, R.M.; Ramos, J.A.; Guzmán, M. Cannabinoid CB2 receptor: A new target for controlling neural cell survival? Trends Pharmacol. Sci. 2007, 28, 39–45. [Google Scholar] [CrossRef] [PubMed]
- Guzmán, M.; Duarte, M.J.; Blázquez, C.; Ravina, J.; Rosa, M.C.; Galve-Roperh, I.; Sánchez, C.; Velasco, G.; González-Feria, L. A pilot clinical study of Δ9-tetrahydrocannabinol in patients with recurrent glioblastoma multiforme. Br. J. Cancer 2006, 95, 197–203. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sarfaraz, S.; Adhami, V.M.; Syed, D.N.; Afaq, F.; Mukhtar, H. Cannabinoids for cancer treatment: Progress and promise. Cancer Res. 2008, 68, 339–342. [Google Scholar] [CrossRef] [PubMed]
- Velasco, G.; Sánchez, C.; Guzmán, M. Towards the use of cannabinoids as antitumour agents. Nat. Rev. Cancer 2012, 12, 436–444. [Google Scholar] [CrossRef] [PubMed]
- Do, Y.; McKallip, R.J.; Nagarkatti, M.; Nagarkatti, P.S. Activation through cannabinoid receptors 1 and 2 on dendritic cells triggers NF-kappaB-dependent apoptosis: Novel role for endogenous and exogenous cannabinoids in immunoregulation. J. Immunol. 2004, 173, 2373–2382. [Google Scholar] [CrossRef] [PubMed]
- Galve-Roperh, I.; Sanchez, C.; Cortes, M.L.; Gómez del Pulgar, T.; Izquierdo, M.; Guzmán, M. Anti-tumoral action of cannabinoids: Involvement of sustained ceramide accumulation and extracellular signal-regulated kinase activation. Nat. Med. 2000, 6, 313–319. [Google Scholar] [CrossRef] [PubMed]
- Sanchez, C.; de Ceballos, M.L.; del Pulgar, T.G.; Rueda, D.; Corbacho, C.; Velasco, G.; Galve-Roperh, I.; Huffman, J.W.; Ramón y Cajal, S.; Guzmán, M. Inhibition of glioma growth in vivo by selective activation of the CB(2) cannabinoid receptor. Cancer Res. 2001, 61, 5784–5789. [Google Scholar] [PubMed]
- Carracedo, A.; Gironella, M.; Lorente, M.; Garcia, S.; Guzmán, M.; Velasco, G.; Iovanna, J.L. Cannabinoids induce apoptosis of pancreatic tumor cells via endoplasmic reticulum stress-related genes. Cancer Res. 2006, 66, 6748–6755. [Google Scholar] [CrossRef] [PubMed]
- Casanova, M.L.; Blazquez, C.; Martinez-Palacio, J.; Villanueva, C.; Fernández-Aceñero, M.J.; Huffman, J.W.; Jorcano, J.L.; Guzmán, M. Inhibition of skin tumor growth and angiogenesis in vivo by activation of cannabinoid receptors. J. Clin. Investig. 2003, 111, 43–50. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rueda, D.; Navarro, B.; Martinez-Serrano, A.; Guzman, M.; Galve-Roperh, I. The endocannabinoid anandamide inhibits neuronal pro- genitor cell differentiation through attenuation of the Rap1/B-Raf/ERK pathway. J. Biol. Chem. 2002, 277, 46645–46650. [Google Scholar] [CrossRef] [PubMed]
- Rueda, D.; Galve-Roperh, I.; Haro, A.; Guzman, M. The CB1 cannabinoid receptor is coupled to the activation of c-Jun N-terminal kinase. Mol. Pharmacol. 2000, 58, 814–820. [Google Scholar] [CrossRef] [PubMed]
- Greenhough, A.; Patsos, H.A.; Williams, A.C.; Paraskeva, C. The cannabinoid Δ9-tetrahydrocannabinol inhibits RAS-MAPK and PI3K-AKT survival signalling and induces BAD-mediated apoptosis in colorectal cancer cells. Int. J. Cancer 2007, 21, 2172–2180. [Google Scholar] [CrossRef] [PubMed]
- Guzman, M.; Galve-Roperh, I.; Sanchez, C. Ceramide: A new second messenger of cannabinoid action. Trends Pharmacol. Sci. 2001, 22, 19–22. [Google Scholar] [CrossRef]
- Caffarel, M.M.; Moreno-Bueno, G.; Cerutti, C.; Palacios, J.; Guzman, M.; Mechta-Grigoriou, F.; Sanchez, C. JunD is involved in the antiproliferative effect of Delta(9)-tetrahydrocannabinol on human breast cancer cells. Oncogene 2008, 27, 5033–5044. [Google Scholar] [CrossRef] [PubMed]
- Caffarel, M.M.; Sarrio, D.; Palacios, J.; Guzmán, M.; Sánchez, C. Delta9-tetrahydrocannabinol inhibits cell cycle progression in human breast cancer cells through Cdc2 regulation. Cancer Res. 2006, 66, 6615–6621. [Google Scholar] [CrossRef] [PubMed]
- Hohmann, A.G. Spinal and peripheral mechanisms of cannabinoid antinociception: Behavioral, neurophysiological and neuroanatomical perspectives. Chem. Phys. Lipids 2002, 121, 173–190. [Google Scholar] [CrossRef]
- Karsak, M.; Gaffal, E.; Date, R.; Wang-Eckhardt, L.; Rehnelt, J.; Petrosino, S.; Starowicz, K.; Steuder, R.; Schlicker, E.; Cravatt, B.; et al. Attenuation of allergic contact dermatitis through the endocannabinoid system. Science 2007, 316, 1494–1497. [Google Scholar] [CrossRef] [PubMed]
- Guindon, J.; Hohmann, A.G. The endocannabinoid system and cancer: Therapeutic implication. Br. J. Pharmacol. 2011, 163, 1447–1463. [Google Scholar] [CrossRef] [PubMed]
- Oesch, S.; Gertsch, J. Cannabinoid receptor ligands as potential anticancer agents-high hopes for new therapies? J. Pharm. Pharmacol. 2009, 61, 839–853. [Google Scholar] [PubMed]
- Izzo, A.A.; Camilleri, M. Emerging role of cannabinoids in gastrointestinal and liver diseases: Basic and clinical aspects. Gut 2008, 57, 1140–1155. [Google Scholar] [CrossRef] [PubMed]
- Pacher, P.; Bátkai, S.; Kunos, G. The endocannabinoid system as an emerging target of pharmacotherapy. Pharmacol. Rev. 2006, 58, 389–462. [Google Scholar] [CrossRef] [PubMed]
- Benito, C.; Tolón, R.M.; Pazos, M.R.; Núñez, E.; Castillo, A.I.; Romero, J. Cannabinoid CB2 receptors in human brain inflammation. Br. J. Pharmacol. 2008, 153, 277–285. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Malan, T.P.J.; Ibrahim, M.M.; Lai, J.; Vanderah, T.W.; Makriyannis, A.; Porreca, F. CB2 cannabinoid receptor agonists: Pain relief without psychoactive effects? Curr. Opin. Pharmacol. 2003, 3, 62–67. [Google Scholar] [CrossRef]
- Manera, C.; Cascio, M.G.; Benetti, V.; Allarà, M.; Tuccinardi, T.; Martinelli, A.; Saccomanni, G.; Vivoli, E.; Ghelardini, C.; Di Marzo, V.; et al. New 1,8-naphthyridine and quinoline derivatives as CB2 selective agonists. Bioorg. Med. Chem. Lett. 2007, 17, 6505–6510. [Google Scholar] [CrossRef] [PubMed]
- Manera, C.; Saccomanni, G.; Adinolfi, B.; Benetti, V.; Ligresti, A.; Cascio, M.G.; Tuccinardi, T.; Lucchesi, V.; Martinelli, A.; Nieri, P.; et al. Rational design, synthesis, and pharmacological properties of new 1,8-naphthyridin-2(1H)-on-3-carboxamide derivatives as highly selective cannabinoid-2 receptor agonists. J. Med. Chem. 2009, 52, 3644–3651. [Google Scholar] [CrossRef] [PubMed]
- Lucchesi, V.; Hurst, D.P.; Shore, D.M.; Bertini, S.; Ehrmann, B.M.; Allarà, M.; Lawrence, L.; Ligresti, A.; Minutolo, F.; Saccomanni, G. CB2-selective cannabinoid receptor ligands: Synthesis, pharmacological evaluation, and molecular modeling investigation of 1,8-Naphthyridin-2(1H)-one-3-carboxamides. J. Med. Chem. 2014, 57, 8777–8791. [Google Scholar] [CrossRef] [PubMed]
- Malfitano, A.M.; Laezza, C.; D’Alessandro, A.; Procaccini, C.; Saccomanni, G.; Tuccinardi, T.; Manera, C.; Macchia, M.; Matarese, G.; Gazzerro, P.; et al. Effects on immune cells of a new 1,8-naphthyridin-2-one derivative and its analogues as selective CB2 agonists: IMPLICATIONS in multiple sclerosis. PLoS ONE 2013, 8, e62511. [Google Scholar] [CrossRef] [PubMed]
- Malfitano, A.M.; Laezza, C.; Saccomanni, G.; Tuccinardi, T.; Manera, C.; Martinelli, A.; Ciaglia, E.; Pisanti, S.; Vitale, M.; Gazzerro, P.; et al. Immune-modulation and properties of absorption and blood brain barrier permeability of 1,8-naphthyridine derivatives. J. Neuroimmune Pharm. 2013, 8, 1077–1086. [Google Scholar] [CrossRef] [PubMed]
- Manera, C.; Saccomanni, G.; Malfitano, A.M.; Bertini, S.; Castelli, F.; Laezza, C.; Ligresti, A.; Lucchesi, V.; Tuccinardi, T.; Rizzolio, F.; et al. Rational design, synthesis and anti-proliferative properties of new CB2 selective cannabinoid receptor ligands: An investigation of the 1,8-naphthyridin-2(1H)-one scaffold. Eur. J. Med. Chem. 2012, 52, 284–294. [Google Scholar] [CrossRef] [PubMed]
- Ellert-Miklaszewska, A.; Kaminska, B.; Konarska, L. Cannabinoids down-regulate PI3K/Akt and Erk signalling pathways and activate proapoptotic function of Bad protein. Cell Signal. 2005, 17, 25–37. [Google Scholar] [CrossRef] [PubMed]
- McKallip, R.J.; Lombard, C.; Fisher, M.; Martin, B.R.; Ryu, S.; Grant, S.; Nagarkatti, P.S.; Nagarkatti, M. Targeting CB2 cannabinoid receptors as a novel therapy to treat malignant lymphoblastic disease. Blood 2002, 100, 627–634. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kroemer, G.; Galluzzi, L.; Brenner, C. Mitochondrial membrane permeabilization in cell death. Physiol. Rev. 2007, 87, 99–163. [Google Scholar] [CrossRef] [PubMed]
- Velasco, G.; Sánchez, C.; Guzmán, M. Anticancer mechanisms of cannabinoids. Curr. Oncol. 2016, 23, 23–32. [Google Scholar]
- Cianchi, F.; Papucci, L.; Schiavone, N.; Lulli, M.; Magnelli, L.; Vinci, M.C.; Messerini, L.; Manera, C.; Ronconi, E.; Romagnani, P.; et al. Cannabinoid receptor activation induces apoptosis through tumor necrosis factor alpha-mediated ceramide de novo synthesis in colon cancer cells. Clin. Cancer Res. 2008, 14, 7691–7700. [Google Scholar] [CrossRef] [PubMed]
- Capozzi, A.; Mantuano, E.; Matarrese, P.; Saccomanni, G.; Manera, C.; Mattei, V.; Gambardella, L.; Malorni, W.; Sorice, M.; Misasi, R. A new 4-phenyl-1,8-naphthyridine derivative affects carcinoma cell proliferation by impairing cell cycle progression and inducing apoptosis. Anticancer Agents Med. Chem. 2012, 12, 653–662. [Google Scholar] [CrossRef] [PubMed]
- Blazquez, C.; Carracedo, A.; Barrado, L.; Real, P.J.; Fernández-Luna, J.L.; Velasco, G.; Malumbres, M.; Guzmán, M. Cannabinoid receptors as novel targets for the treatment of melanoma. FASEB J. 2006, 20, 2199–2208. [Google Scholar] [CrossRef] [PubMed]
- Blazquez, C.; Gonzalez-Feria, L.; Alvarez, L.; Haro, A.; Casanova, M.L.; Guzmán, M. Cannabinoids inhibit the vascular endothelial growth factor pathway in gliomas. Cancer Res. 2004, 64, 5617–5623. [Google Scholar] [CrossRef] [PubMed]
- Sarfaraz, S.; Afaq, F.; Adhami, V.M.; Malik, A.; Mukhtar, H. Cannabinoid receptor agonist-induced apoptosis of human prostate cancer cells LNCaP proceeds through sustained activation of erk1/2 leading to G1 cell cycle arrest. J. Biol. Chem. 2006, 281, 39480–39491. [Google Scholar] [CrossRef] [PubMed]
- Gustafsson, K.; Christensson, B.; Sander, B.; Flygare, J. Cannabinoid Receptor-Mediated Apoptosis Induced by R(+)-Methanandamide and Win55,212-2 Is Associated with Ceramide Accumulation and p38 Activation in Mantle Cell Lymphoma. Mol. Pharmacol. 2006, 70, 1612–1620. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Munson, A.E.; Harris, L.S.; Friedman, M.A.; Dewey, W.L.; Carchman, R.A. Antineoplastic activity of cannabinoids. J. Natl. Cancer Inst. 1975, 55, 597–602. [Google Scholar] [CrossRef] [PubMed]
- Costa, M.A.; Fonseca, M.B.; Teixeira, N.A.; Correia-da-Silva, G. The endocannabinoid anandamide induces apoptosis in cytotrophoblast cells: Involvement of both mithocondrial and death receptor pathways. Placenta. 2015, 36, 69–76. [Google Scholar] [CrossRef] [PubMed]
- Herrera, B.; Carracedo, A.; Diez-Zaera, M.; Gómez del Pulgar, T.; Guzmán, M.; Velasco, G. The CB2 cannabinoid receptor signals apoptosis via ceramide-dependent activation of the mitochondrial intrinsic pathway. Exp. Cell Res. 2006, 312, 2121–2131. [Google Scholar] [CrossRef] [PubMed]
- Jia, W.; Hegde, V.L.; Singh, N.P.; Sisco, D.; Grant, S.; Nagarkatti, M.; Nagarkatti, P.S. Delta9-Tetrahydrocannabinol-Induced Apoptosis in Jurkat Leukemia T Cells Is Regulated by Translocation of Bad to Mitochondria. Mol. Cancer Res. 2006, 4, 549–562. [Google Scholar] [CrossRef] [PubMed]
- Guzmán, M. Cannabinoids: Potential anticancer agents. Nat. Rev. Cancer 2003, 3, 745–755. [Google Scholar] [CrossRef] [PubMed]
- Gatley, S.J.; Lan, R.; Pyatt, B.; Gifford, A.N.; Volkow, N.D.; Makriyannis, A. Binding of the non-classical cannabinoid CP 55,940, and the diarylpyrazole AM251 to rodent brain cannabinoid receptors. Life Sci. 1997, 61, 191–197. [Google Scholar] [CrossRef]
- Huffman, J.W.; Zengin, G.; Wu, M.J.; Lu, J.; Hynd, G.; Bushell, K.; Thompson, A.L.; Bushell, S.; Tartal, C.; Hurst, D.P.; et al. Structure-activity relationships for 1-alkyl-3-(1-naphthoyl)indoles at the cannabinoid CB(1) and CB(2) receptors: Steric and electronic effects of naphthoyl substituents. New highly selective CB(2) receptor agonists. Bioorg. Med. Chem. Lett. 2005, 13, 89–112. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Y.C.; Prusoff, W.H. Relationship between the inhibition constant (K1) and the concentration of inhibitor which causes 50 per cent inhibition (I50) of an enzymatic reaction. Biochem. Pharmacol. 1973, 22, 3099–3108. [Google Scholar] [PubMed]
- Wilson, A.P. Citotoxicity and viability assays. In Animal Cell Culture: A Practical Approach; Freshney, R.I., Ed.; Oxford University Press: Oxford, UK, 1992; pp. 263–303. [Google Scholar]
- Sorice, M.; Matarrese, P.; Tinari, A.; Giammarioli, A.M.; Garofalo, T.; Manganelli, V.; Ciarlo, L.; Gambardella, L.; Maccari, G.; Botta, M.; et al. Raft component GD3 associates with tubulin following CD95/Fas ligation. FASEB J. 2009, 23, 3298–3308. [Google Scholar] [CrossRef] [PubMed]
- Degli Esposti, M.; Matarrese, P.; Tinari, A.; Longo, A.; Recalchi, S.; Khosravi-Far, R.; Malorni, W.; Misasi, R.; Garofalo, T.; Sorice, M. Changes in membrane lipids drive increased endocytosis following Fas ligation. Apoptosis 2017, 22, 681–695. [Google Scholar] [CrossRef] [PubMed]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}

| Compound | R1 | Ki CB1R (nM) | Ki CB2R (nM) | Ki CB1R/Ki CB2R |
|---|---|---|---|---|
| CB9126 | p–fluorobenzyl | 200 ± 7.7 | 0.90 ± 0.038 | 222 |
| LV6227 | CH2(CH2)4OH | >10,000 | 3.60 ± 0.13 | >187 |
| LV5827 | CH2(CH2)3F | 1011 ± 46.5 | 1.36 ± 0.053 | 743 |
| LV50 | CH2(CH2)3Cl | 224 ± 8.53 | 0.54 ± 0.017 | 415 |
| Compound | Trypan Blue Positive Cells (%) | Viability (%) | subG1 Phase (%) |
|---|---|---|---|
| 24 h | |||
| CB91 | 2.10 ± 0.13 | 83.35 ± 7.34 | 2.35 ± 0.16 |
| LV62 | 7.60 ± 0.55 | 86.75 ± 7.37 | 3.45 ± 0.27 |
| LV58 | 2.85 ± 0.20 | 87.80 ± 7.26 | 2.41 ± 0.18 |
| LV50 | 18.13 ± 1.26 | 77.78 ± 6.22 | 7.85 ± 0.60 |
| 48 h | |||
| CB91 | 2.15 ± 0.16 | 78.80 ± 6.30 | 2.82 ± 0.17 |
| LV62 | 10.5 ± 0.82 | 82.33 ± 6.25 | 8.88 ± 0.61 |
| LV58 | 3.50 ± 0.22 | 85.50 ± 6.85 | 4.50 ± 0.26 |
| LV50 | 23.93 ± 1.91 | 70.90 ± 5.31 | 18.7 ± 1.40 |
| 72 h | |||
| CB91 | 2.30 ± 0.18 | 75.45 ± 4.35 | 3.93 ± 0.18 |
| LV62 | 14.80 ± 0.88 | 70.25 ± 4.21 | 10.60 ± 0.53 |
| LV58 | 3.8 ± 0.25 | 81.25 ± 4.46 | 5.19 ± 0.33 |
| LV50 | 30.00 ± 2.05 | 59.32 ± 3.85 | 24.70 ± 1.95 |
| Compound | Trypan Blue Positive Cells (%) | Viability (%) | subG1 Phase (%) |
|---|---|---|---|
| 24 h | |||
| SR144528 + CB91 | 2.00 ± 0.12 | 82.5 ± 7.30 | 2.31 ± 0.15 |
| SR144528 + LV62 | 7.56 ± 0.54 | 85.73 ± 7.35 | 3.40 ± 0.26 |
| SR144528 + LV58 | 2.8 ± 0.21 | 86.80 ± 7.15 | 2.39 ± 0.17 |
| SR144528 + LV50 | 9.53 ± 0.86 | 87.68 ± 6.12 | 2.55 ± 0.20 |
| 48 h | |||
| SR144528 + CB91 | 2.11 ± 0.16 | 77.50 ± 6.25 | 2.77 ± 0.17 |
| SR144528 + LV62 | 10.10 ± 0.81 | 81.20 ± 6.20 | 8.87 ± 0.61 |
| SR144528 + LV58 | 3.43 ± 0.21 | 84.00 ± 6.75 | 4.48 ± 0.25 |
| SR144528 + LV50 | 12.50 ± 0.95 | 85.60 ± 6.81 | 12.70 ± 1.10 |
| 72 h | |||
| SR144528 + CB91 | 2.31 ± 0.18 | 75.50 ± 4.35 | 3.92 ± 0.18 |
| SR144528 + LV62 | 14.81 ± 0.87 | 70.24 ± 4.22 | 10.61 ± 0.53 |
| SR144528 + LV58 | 3.79 ± 0.26 | 81.20 ± 4.45 | 5.18 ± 0.32 |
| SR144528 + LV50 | 16.58 ± 1.25 | 86.18 ± 4.34 | 19.00 ± 2.05 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Capozzi, A.; Mattei, V.; Martellucci, S.; Manganelli, V.; Saccomanni, G.; Garofalo, T.; Sorice, M.; Manera, C.; Misasi, R. Anti-Proliferative Properties and Proapoptotic Function of New CB2 Selective Cannabinoid Receptor Agonist in Jurkat Leukemia Cells. Int. J. Mol. Sci. 2018, 19, 1958. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms19071958
Capozzi A, Mattei V, Martellucci S, Manganelli V, Saccomanni G, Garofalo T, Sorice M, Manera C, Misasi R. Anti-Proliferative Properties and Proapoptotic Function of New CB2 Selective Cannabinoid Receptor Agonist in Jurkat Leukemia Cells. International Journal of Molecular Sciences. 2018; 19(7):1958. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms19071958
Chicago/Turabian StyleCapozzi, Antonella, Vincenzo Mattei, Stefano Martellucci, Valeria Manganelli, Giuseppe Saccomanni, Tina Garofalo, Maurizio Sorice, Clementina Manera, and Roberta Misasi. 2018. "Anti-Proliferative Properties and Proapoptotic Function of New CB2 Selective Cannabinoid Receptor Agonist in Jurkat Leukemia Cells" International Journal of Molecular Sciences 19, no. 7: 1958. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms19071958