The Role of Na/K-ATPase Signaling in Oxidative Stress Related to Aging: Implications in Obesity and Cardiovascular Disease


Abstract
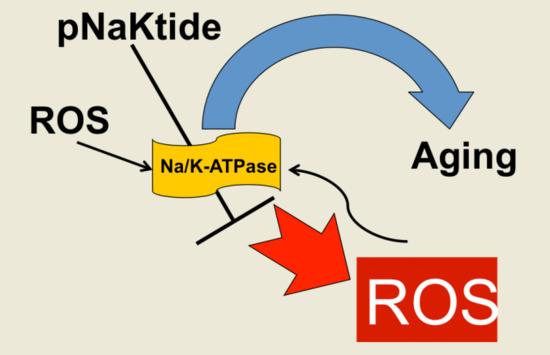
:
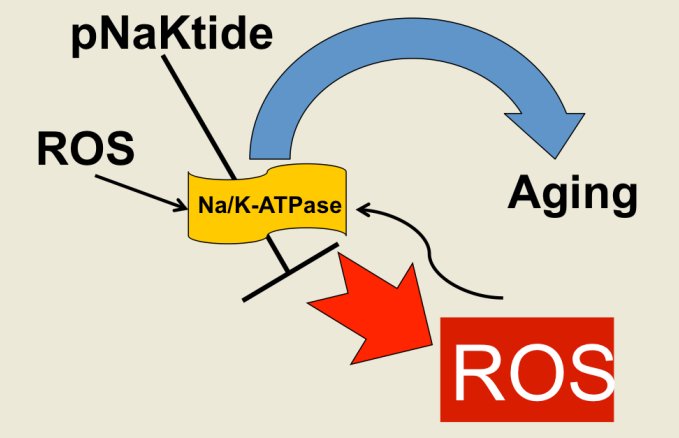{kind=link}
{kind=link}
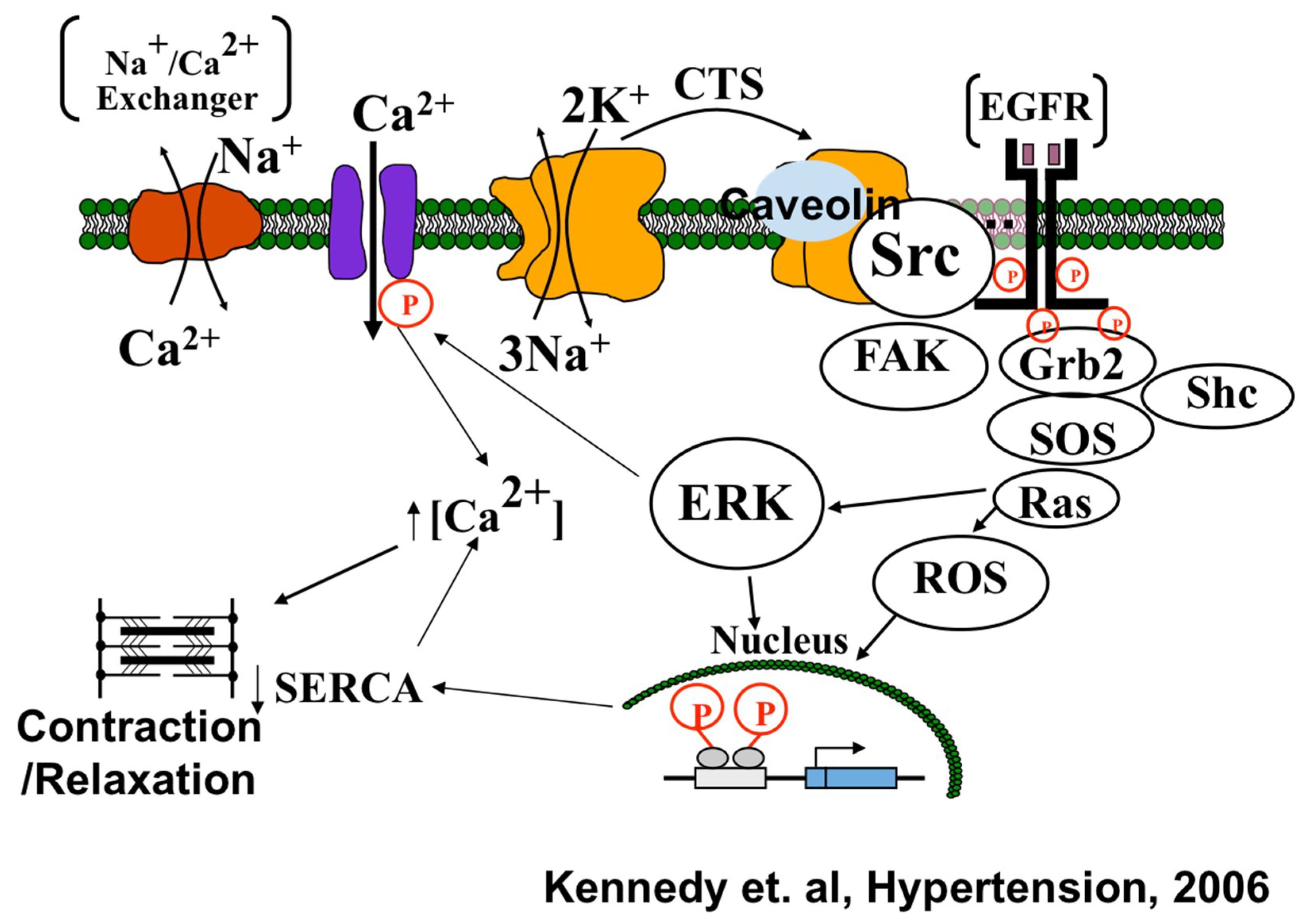{kind=link}
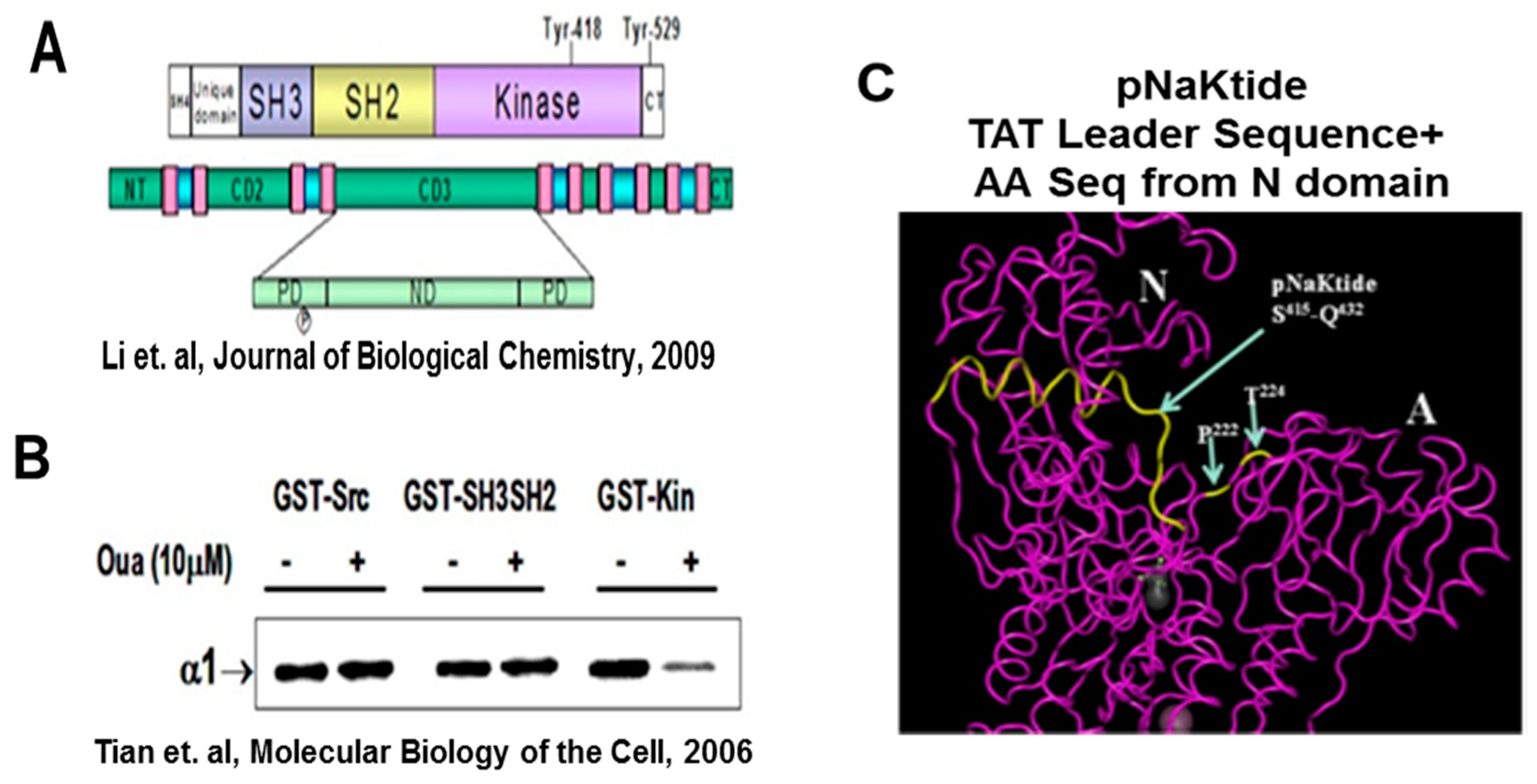{kind=link}
1. Introduction
2. Na/K-ATPase Signaling and Oxidative Stress
3. The Development of pNaKtide
4. The Role of Oxidative Stress and Na/K-ATPase/ROS Signaling in Aging
5. The Role of Oxidative Stress and Na/K-ATPase/ROS Signaling in Aging and Associated Obesity
6. The Role of Oxidative Stress and Na/K-ATPase/ROS Signaling in Aging and Associated Cardiovascular Diseases (CVDs)
7. Pharmacological Interventions for Aging
8. Conclusions
Funding
Conflicts of Interest
References
- Davidovic, M. Genetic stability: The key to longevity? Med. Hypotheses 1999, 53, 329–332. [Google Scholar] [CrossRef] [PubMed]
- Risques, R.A.; Lai, L.A.; Brentnall, T.A.; Li, L.; Feng, Z.; Gallaher, J.; Mandelson, M.T.; Potter, J.D.; Bronner, M.P.; Rabinovitch, P.S. Ulcerative colitis is a disease of accelerated colon aging: Evidence from telomere attrition and DNA damage. Gastroenterology 2008, 135, 410–418. [Google Scholar] [CrossRef] [PubMed]
- Fraga, M.F.; Ballestar, E.; Paz, M.F.; Ropero, S.; Setien, F.; Ballestar, M.L.; Heine-Suner, D.; Cigudosa, J.C.; Urioste, M.; Benitez, J.; et al. Epigenetic differences arise during the lifetime of monozygotic twins. Proc. Natl. Acad. Sci. USA 2005, 102, 10604–10609. [Google Scholar] [CrossRef] [PubMed]
- Baylin, S.B.; Herman, J.G.; Graff, J.R.; Vertino, P.M.; Issa, J.P. Alterations in DNA methylation: A fundamental aspect of neoplasia. Adv. Cancer Res. 1998, 72, 141–196. [Google Scholar] [PubMed]
- Hanahan, D.; Weinberg, R.A. The hallmarks of cancer. Cell 2000, 100, 57–70. [Google Scholar] [CrossRef]
- Abdollahi, M.; Moridani, M.Y.; Aruoma, O.I.; Mostafalou, S. Oxidative stress in aging. Oxidative Med. Cell. Longev. 2014, 2014, 876834. [Google Scholar] [CrossRef] [PubMed]
- North, B.J.; Sinclair, D.A. The intersection between aging and cardiovascular disease. Circ. Res. 2012, 110, 1097–1108. [Google Scholar] [CrossRef] [PubMed]
- Cui, H.; Kong, Y.; Zhang, H. Oxidative stress, mitochondrial dysfunction, and aging. J. Signal Transduct. 2012, 2012, 646354. [Google Scholar] [CrossRef] [PubMed]
- Chandrasekaran, A.; Idelchik, M.; Melendez, J.A. Redox control of senescence and age-related disease. Redox Biol. 2017, 11, 91–102. [Google Scholar] [CrossRef] [PubMed]
- Sallam, N.; Laher, I. Exercise modulates oxidative stress and inflammation in aging and cardiovascular diseases. Oxidative Med. Cell. Longev. 2016, 2016, 7239639. [Google Scholar] [CrossRef] [PubMed]
- Sodhi, K.; Maxwell, K.; Yan, Y.; Liu, J.; Chaudhry, M.A.; Getty, M.; Xie, Z.; Abraham, N.G.; Shapiro, J.I. Pnaktide inhibits Na/K-ATPase reactive oxygen species amplification and attenuates adipogenesis. Sci. Adv. 2015, 1, e1500781. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Tian, J.; Chaudhry, M.; Maxwell, K.; Yan, Y.; Wang, X.; Shah, P.T.; Khawaja, A.A.; Martin, R.; Robinette, T.J.; et al. Attenuation of Na/K-ATPase mediated oxidant amplification with pnaktide ameliorates experimental uremic cardiomyopathy. Sci. Rep. 2016, 6, 34592. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Ye, Q.; Liu, C.; Xie, J.X.; Yan, Y.; Lai, F.; Duan, Q.; Li, X.; Tian, J.; Xie, Z. Involvement of Na/K-ATPase in hydrogen peroxide-induced activation of the Src/ERK pathway in LLC-PK1 cells. Free Radic. Biol. Med. 2014, 71, 415–426. [Google Scholar] [CrossRef] [PubMed]
- Yan, Y.; Shapiro, J.I. The physiological and clinical importance of sodium potassium atpase in cardiovascular diseases. Curr. Opin. Pharmacol. 2016, 27, 43–49. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Zhang, Z.; Xie, J.X.; Li, X.; Tian, J.; Cai, T.; Cui, H.; Ding, H.; Shapiro, J.I.; Xie, Z. Na/K-ATPase mimetic pnaktide peptide inhibits the growth of human cancer cells. J. Biol. Chem. 2011, 286, 32394–32403. [Google Scholar] [CrossRef] [PubMed]
- Sodhi, K.; Nichols, A.; Mallick, A.; Klug, R.L.; Liu, J.; Wang, X.; Srikanthan, K.; Goguet-Rubio, P.; Nawab, A.; Pratt, R.; et al. The Na/K-ATPase oxidant amplification loop regulates aging. Sci. Rep. 2018, 8, 9721. [Google Scholar] [CrossRef] [PubMed]
- Srikanthan, K.; Shapiro, J.I.; Sodhi, K. The role of Na/K-ATPase signaling in oxidative stress related to obesity and cardiovascular disease. Molecules 2016, 21, 1172. [Google Scholar] [CrossRef] [PubMed]
- Sodhi, K.; Srikanthan, K.; Goguet-Rubio, P.; Nichols, A.; Mallick, A.; Nawab, A.; Martin, R.; Shah, P.T.; Chaudhry, M.; Sigdel, S.; et al. PNaKtide attenuates steatohepatitis and atherosclerosis by blocking Na/K-ATPase/ROS amplification in C57Bl6 and ApoE knockout mice fed a western diet. Sci. Rep. 2017, 7, 193. [Google Scholar] [CrossRef] [PubMed]
- Liang, M.; Tian, J.; Liu, L.; Pierre, S.; Liu, J.; Shapiro, J.; Xie, Z.J. Identification of a pool of non-pumping Na/K-ATPase. J. Biol. Chem. 2007, 282, 10585–10593. [Google Scholar] [CrossRef] [PubMed]
- Yan, Y.; Haller, S.; Shapiro, A.; Malhotra, N.; Tian, J.; Xie, Z.; Malhotra, D.; Shapiro, J.I.; Liu, J. Ouabain-stimulated trafficking regulation of the Na/K-ATPase and NHE3 in renal proximal tubule cells. Mol. Cell. Biochem. 2012, 367, 175–183. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, J.; Yan, Y.; Liu, L.; Xie, Z.; Malhotra, D.; Joe, B.; Shapiro, J.I. Impairment of Na/K-ATPase signaling in renal proximal tubule contributes to dahl salt-sensitive hypertension. J. Biol. Chem. 2011, 286, 22806–22813. [Google Scholar] [CrossRef] [PubMed]
- Yan, Y.; Shapiro, A.P.; Haller, S.; Katragadda, V.; Liu, L.; Tian, J.; Basrur, V.; Malhotra, D.; Xie, Z.J.; Abraham, N.G.; et al. Involvement of reactive oxygen species in a feed-forward mechanism of Na/K-ATPase-mediated signaling transduction. J. Biol. Chem. 2013, 288, 34249–34258. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Tian, J.; Haas, M.; Shapiro, J.I.; Askari, A.; Xie, Z. Ouabain interaction with cardiac Na+/K+-ATPase initiates signal cascades independent of changes in intracellular Na+ and Ca2+ concentrations. J. Biol. Chem. 2000, 275, 27838–27844. [Google Scholar] [PubMed]
- Xie, Z.; Kometiani, P.; Liu, J.; Li, J.; Shapiro, J.I.; Askari, A. Intracellular reactive oxygen species mediate the linkage of Na+/K+-ATPase to hypertrophy and its marker genes in cardiac myocytes. J. Biol. Chem. 1999, 274, 19323–19328. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Kennedy, D.J.; Yan, Y.; Shapiro, J.I. Reactive oxygen species modulation of Na/K-ATPase regulates fibrosis and renal proximal tubular sodium handling. Int. J. Nephrol. 2012, 2012, 381320. [Google Scholar] [CrossRef] [PubMed]
- Kennedy, D.J.; Vetteth, S.; Periyasamy, S.M.; Kanj, M.; Fedorova, L.; Khouri, S.; Kahaleh, M.B.; Xie, Z.; Malhotra, D.; Kolodkin, N.I.; et al. Central role for the cardiotonic steroid marinobufagenin in the pathogenesis of experimental uremic cardiomyopathy. Hypertension 2006, 47, 488–495. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Cai, T.; Tian, J.; Xie, J.X.; Zhao, X.; Liu, L.; Shapiro, J.I.; Xie, Z. Naktide, a Na/K-ATPase-derived peptide src inhibitor, antagonizes ouabain-activated signal transduction in cultured cells. J. Biol. Chem. 2009, 284, 21066–21076. [Google Scholar] [CrossRef] [PubMed]
- Tian, J.; Cai, T.; Yuan, Z.; Wang, H.; Liu, L.; Haas, M.; Maksimova, E.; Huang, X.Y.; Xie, Z.J. Binding of SRC to Na+/K+-ATPase forms a functional signaling complex. Mol. Biol. Cell 2006, 17, 317–326. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Otin, C.; Blasco, M.A.; Partridge, L.; Serrano, M.; Kroemer, G. The hallmarks of aging. Cell 2013, 153, 1194–1217. [Google Scholar] [CrossRef] [PubMed]
- Pal, S.; Tyler, J.K. Epigenetics and aging. Sci. Adv. 2016, 2, e1600584. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chakravarti, B.; Chakravarti, D.N. Oxidative modification of proteins: Age-related changes. Gerontology 2007, 53, 128–139. [Google Scholar] [CrossRef] [PubMed]
- Cooke, M.S.; Evans, M.D.; Dizdaroglu, M.; Lunec, J. Oxidative DNA damage: Mechanisms, mutation, and disease. FASEB J. 2003, 17, 1195–1214. [Google Scholar] [CrossRef] [PubMed]
- Evans, M.D.; Dizdaroglu, M.; Cooke, M.S. Oxidative DNA damage and disease: Induction, repair and significance. Mutat. Res. 2004, 567, 1–61. [Google Scholar] [CrossRef] [PubMed]
- Karihtala, P.; Soini, Y. Reactive oxygen species and antioxidant mechanisms in human tissues and their relation to malignancies. APMIS 2007, 115, 81–103. [Google Scholar] [CrossRef] [PubMed]
- Davalli, P.; Mitic, T.; Caporali, A.; Lauriola, A.; D’Arca, D. Ros, cell senescence, and novel molecular mechanisms in aging and age-related diseases. Oxidative Med. Cell. Longev. 2016, 2016, 3565127. [Google Scholar] [CrossRef] [PubMed]
- Van Deursen, J.M. The role of senescent cells in ageing. Nature 2014, 509, 439–446. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Passos, J.F.; Nelson, G.; Wang, C.; Richter, T.; Simillion, C.; Proctor, C.J.; Miwa, S.; Olijslagers, S.; Hallinan, J.; Wipat, A.; et al. Feedback between P21 and reactive oxygen production is necessary for cell senescence. Mol. Syst. Biol. 2010, 6, 347. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Xia, S.; Kalionis, B.; Wan, W.; Sun, T. The role of oxidative stress and inflammation in cardiovascular aging. BioMed Res. Int. 2014, 2014, 615312. [Google Scholar] [CrossRef] [PubMed]
- Harman, D. Aging: A theory based on free radical and radiation chemistry. J. Gerontol. 1956, 11, 298–300. [Google Scholar] [CrossRef] [PubMed]
- Haendeler, J.; Hoffmann, J.; Diehl, J.F.; Vasa, M.; Spyridopoulos, I.; Zeiher, A.M.; Dimmeler, S. Antioxidants inhibit nuclear export of telomerase reverse transcriptase and delay replicative senescence of endothelial cells. Circ. Res. 2004, 94, 768–775. [Google Scholar] [CrossRef] [PubMed]
- Aravinthan, A.; Shannon, N.; Heaney, J.; Hoare, M.; Marshall, A.; Alexander, G.J. The senescent hepatocyte gene signature in chronic liver disease. Exp. Gerontol. 2014, 60, 37–45. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.; Xu, Y. P53, oxidative stress; and aging. Antioxid. Redox Signal. 2011, 15, 1669–1678. [Google Scholar] [CrossRef] [PubMed]
- Gire, V.; Roux, P.; Wynford-Thomas, D.; Brondello, J.M.; Dulic, V. DNA damage checkpoint kinase CHK2 triggers replicative senescence. EMBO J. 2004, 23, 2554–2563. [Google Scholar] [CrossRef] [PubMed]
- Campisi, J. Aging, cellular senescence, and cancer. Annu. Rev. Physiol. 2013, 75, 685–705. [Google Scholar] [CrossRef] [PubMed]
- Chayapong, J.; Madhyastha, H.; Madhyastha, R.; Nurrahmah, Q.I.; Nakajima, Y.; Choijookhuu, N.; Hishikawa, Y.; Maruyama, M. Arsenic trioxide induces ROS activity and DNA damage, leading to G0/G1 extension in skin fibroblasts through the ATM-ATR-associated chk pathway. Environ. Sci. Pollut. Res. Int. 2017, 24, 5316–5325. [Google Scholar] [CrossRef] [PubMed]
- O’Mahony, L.; Holland, J.; Jackson, J.; Feighery, C.; Hennessy, T.P.; Mealy, K. Quantitative intracellular cytokine measurement: Age-related changes in proinflammatory cytokine production. Clin. Exp. Immunol. 1998, 113, 213–219. [Google Scholar] [CrossRef] [PubMed]
- Chiricolo, M.; Bartolini, G.; Orlandi, M.; Peta, G.; Corneli, M.; Spinosa, M.; Tomasi, V.; Franceschi, C. Prostaglandin and thromboxane biosynthesis in resting and activated platelet-free monocytes from aged subjects. Gerontology 1986, 32, 69–73. [Google Scholar] [CrossRef] [PubMed]
- Franceschi, C.; Bonafe, M.; Valensin, S.; Olivieri, F.; De Luca, M.; Ottaviani, E.; De Benedictis, G. Inflamm-aging. An evolutionary perspective on immunosenescence. Ann. N. Y. Acad. Sci. 2000, 908, 244–254. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.; Ames, B.N. Senescence-like growth arrest induced by hydrogen peroxide in human diploid fibroblast F65 cells. Proc. Natl. Acad. Sci. USA. 1994, 91, 4130–4134. [Google Scholar] [CrossRef] [PubMed]
- Hales, C.M.; Carroll, M.D.; Fryar, C.D.; Ogden, C.L. Prevalence of obesity among adults and youth: United states, 2015–2016. NCHS Data Brief 2017, 288, 1–8. [Google Scholar]
- Thorpe, R.J., Jr.; Ferraro, K.F. Aging, obesity, and mortality: Misplaced concern about obese older people? Res. Aging 2004, 26, 108–129. [Google Scholar] [CrossRef] [PubMed]
- Iyer, A.; Fairlie, D.P.; Prins, J.B.; Hammock, B.D.; Brown, L. Inflammatory lipid mediators in adipocyte function and obesity. Nat. Rev. Endocrinol. 2010, 6, 71–82. [Google Scholar] [CrossRef] [PubMed]
- Vidal-Puig, A.; Jimenez-Linan, M.; Lowell, B.B.; Hamann, A.; Hu, E.; Spiegelman, B.; Flier, J.S.; Moller, D.E. Regulation of ppar gamma gene expression by nutrition and obesity in rodents. J. Clin. Investig. 1996, 97, 2553–2561. [Google Scholar] [CrossRef] [PubMed]
- Puri, N.; Sodhi, K.; Haarstad, M.; Kim, D.H.; Bohinc, S.; Foglio, E.; Favero, G.; Abraham, N.G. Heme induced oxidative stress attenuates sirtuin1 and enhances adipogenesis in mesenchymal stem cells and mouse pre-adipocytes. J. Cell. Biochem. 2012, 113, 1926–1935. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takahashi, M.; Kamei, Y.; Ezaki, O. Mest/Peg1 imprinted gene enlarges adipocytes and is a marker of adipocyte size. Am. J. Physiol. Endocrinol. Metab. 2005, 288, E117–E124. [Google Scholar] [CrossRef] [PubMed]
- Monickaraj, F.; Aravind, S.; Nandhini, P.; Prabu, P.; Sathishkumar, C.; Mohan, V.; Balasubramanyam, M. Accelerated fat cell aging links oxidative stress and insulin resistance in adipocytes. J. Biosci. 2013, 38, 113–122. [Google Scholar] [CrossRef] [PubMed]
- Marseglia, L.; Manti, S.; D’Angelo, G.; Nicotera, A.; Parisi, E.; Di Rosa, G.; Gitto, E.; Arrigo, T. Oxidative stress in obesity: A critical component in human diseases. Int. J. Mol. Sci. 2014, 16, 378–400. [Google Scholar] [CrossRef] [PubMed]
- Findeisen, H.M.; Pearson, K.J.; Gizard, F.; Zhao, Y.; Qing, H.; Jones, K.L.; Cohn, D.; Heywood, E.B.; de Cabo, R.; Bruemmer, D. Oxidative stress accumulates in adipose tissue during aging and inhibits adipogenesis. PLoS ONE 2011, 6, e18532. [Google Scholar] [CrossRef] [PubMed]
- Bailey-Downs, L.C.; Tucsek, Z.; Toth, P.; Sosnowska, D.; Gautam, T.; Sonntag, W.E.; Csiszar, A.; Ungvari, Z. Aging exacerbates obesity-induced oxidative stress and inflammation in perivascular adipose tissue in mice: A paracrine mechanism contributing to vascular redox dysregulation and inflammation. J. Gerontol. Ser. A 2013, 68, 780–792. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Kim, D.H.; Tsenovoy, P.L.; Peterson, S.J.; Rezzani, R.; Rodella, L.F.; Aronow, W.S.; Ikehara, S.; Abraham, N.G. Treatment of obese diabetic mice with a heme oxygenase inducer reduces visceral and subcutaneous adiposity, increases adiponectin levels, and improves insulin sensitivity and glucose tolerance. Diabetes 2008, 57, 1526–1535. [Google Scholar] [CrossRef] [PubMed]
- Frasca, D.; Blomberg, B.B.; Paganelli, R. Aging, obesity, and inflammatory age-related diseases. Front. Immunol. 2017, 8, 1745. [Google Scholar] [CrossRef] [PubMed]
- Von Zglinicki, T. Oxidative stress shortens telomeres. Trends Biochem. Sci. 2002, 27, 339–344. [Google Scholar] [CrossRef]
- Lee, M.; Martin, H.; Firpo, M.A.; Demerath, E.W. Inverse association between adiposity and telomere length: The fels longitudinal study. Am. J. Hum. Biol 2011, 23, 100–106. [Google Scholar] [CrossRef] [PubMed]
- Minamino, T.; Orimo, M.; Shimizu, I.; Kunieda, T.; Yokoyama, M.; Ito, T.; Nojima, A.; Nabetani, A.; Oike, Y.; Matsubara, H.; et al. A crucial role for adipose tissue p53 in the regulation of insulin resistance. Nat. Med. 2009, 15, 1082–1087. [Google Scholar] [CrossRef] [PubMed]
- Stout, M.B.; Justice, J.N.; Nicklas, B.J.; Kirkland, J.L. Physiological aging: Links among adipose tissue dysfunction, diabetes, and frailty. Physiology 2017, 32, 9–19. [Google Scholar] [CrossRef] [PubMed]
- Russell, S.J.; Kahn, C.R. Endocrine regulation of ageing. Nat. Rev. Mol. Cell Biol. 2007, 8, 681–691. [Google Scholar] [CrossRef] [PubMed]
- American Heart Association (AHA). Statistical Fact Sheet 2016 Update: Older Americans & Cardiovascular Diseases; AHA: Dallas, TX, USA, 2016. [Google Scholar]
- Mohmand, B.; Malhotra, D.K.; Shapiro, J.I. Uremic cardiomyopathy: Role of circulating digitalis like substances. Front. Biosci. 2005, 10, 2036–2044. [Google Scholar] [CrossRef] [PubMed]
- Bonomini, F.; Tengattini, S.; Fabiano, A.; Bianchi, R.; Rezzani, R. Atherosclerosis and oxidative stress. Histol. Histopathol. 2008, 23, 381–390. [Google Scholar] [PubMed]
- Harrison, D.; Griendling, K.K.; Landmesser, U.; Hornig, B.; Drexler, H. Role of oxidative stress in atherosclerosis. Am. J. Cardiol. 2003, 91, 7A–11A. [Google Scholar] [CrossRef]
- Vogiatzi, G.; Tousoulis, D.; Stefanadis, C. The role of oxidative stress in atherosclerosis. Hell. J. Cardiol. 2009, 50, 402–409. [Google Scholar]
- Boudina, S. Cardiac aging and insulin resistance: Could insulin/insulin-like growth factor (IGF) signaling be used as a therapeutic target? Curr. Pharm. Des. 2013, 19, 5684–5694. [Google Scholar] [CrossRef] [PubMed]
- Lakshmi, S.V.; Padmaja, G.; Kuppusamy, P.; Kutala, V.K. Oxidative stress in cardiovascular disease. Indian J. Biochem. Biophys. 2009, 46, 421–440. [Google Scholar] [PubMed]
- Sudheesh, N.P.; Ajith, T.A.; Ramnath, V.; Janardhanan, K.K. Therapeutic potential of Ganoderma lucidum (Fr.) P. Karst. against the declined antioxidant status in the mitochondria of post-mitotic tissues of aged mice. Clin. Nutr. 2010, 29, 406–412. [Google Scholar] [CrossRef] [PubMed]
- Davalos, A.R.; Coppe, J.P.; Campisi, J.; Desprez, P.Y. Senescent cells as a source of inflammatory factors for tumor progression. Cancer Metastasis Rev. 2010, 29, 273–283. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Freund, A.; Orjalo, A.V.; Desprez, P.Y.; Campisi, J. Inflammatory networks during cellular senescence: Causes and consequences. Trends Mol. Med. 2010, 16, 238–246. [Google Scholar] [CrossRef] [PubMed]
- Laberge, R.M.; Sun, Y.; Orjalo, A.V.; Patil, C.K.; Freund, A.; Zhou, L.; Curran, S.C.; Davalos, A.R.; Wilson-Edell, K.A.; Liu, S.; et al. Mtor regulates the pro-tumorigenic senescence-associated secretory phenotype by promoting il1a translation. Nat. Cell Biol. 2015, 17, 1049–1061. [Google Scholar] [CrossRef] [PubMed]
- Herranz, N.; Gallage, S.; Mellone, M.; Wuestefeld, T.; Klotz, S.; Hanley, C.J.; Raguz, S.; Acosta, J.C.; Innes, A.J.; Banito, A.; et al. Mtor regulates mapkapk2 translation to control the senescence-associated secretory phenotype. Nat. Cell Biol. 2015, 17, 1205–1217. [Google Scholar] [CrossRef] [PubMed]
- Wajapeyee, N.; Serra, R.W.; Zhu, X.; Mahalingam, M.; Green, M.R. Oncogenic braf induces senescence and apoptosis through pathways mediated by the secreted protein IGFBP7. Cell 2008, 132, 363–374. [Google Scholar] [CrossRef] [PubMed]
- Hubackova, S.; Krejcikova, K.; Bartek, J.; Hodny, Z. IL1- and TGFbeta-nox4 signaling, oxidative stress and DNA damage response are shared features of replicative; oncogene-induced; and drug-induced paracrine ‘bystander senescence’. Aging 2012, 4, 932–951. [Google Scholar] [CrossRef] [PubMed]
- McNeal, A.S.; Liu, K.; Nakhate, V.; Natale, C.A.; Duperret, E.K.; Capell, B.C.; Dentchev, T.; Berger, S.L.; Herlyn, M.; Seykora, J.T.; et al. Cdkn2b loss promotes progression from benign melanocytic nevus to melanoma. Cancer Discov. 2015, 5, 1072–1085. [Google Scholar] [CrossRef] [PubMed]
- Soto-Gamez, A.; Demaria, M. Therapeutic interventions for aging: The case of cellular senescence. Drug Discov. Today 2017, 22, 786–795. [Google Scholar] [CrossRef] [PubMed]
- Schlossmacher, G.; Stevens, A.; White, A. Glucocorticoid receptor-mediated apoptosis: Mechanisms of resistance in cancer cells. J. Endocrinol. 2011, 211, 17–25. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Tchkonia, T.; Pirtskhalava, T.; Gower, A.C.; Ding, H.; Giorgadze, N.; Palmer, A.K.; Ikeno, Y.; Hubbard, G.B.; Lenburg, M.; et al. The achilles’ heel of senescent cells: From transcriptome to senolytic drugs. Aging Cell 2015, 14, 644–658. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Tchkonia, T.; Fuhrmann-Stroissnigg, H.; Dai, H.M.; Ling, Y.Y.; Stout, M.B.; Pirtskhalava, T.; Giorgadze, N.; Johnson, K.O.; Giles, C.B.; et al. Identification of a novel senolytic agent, navitoclax, targeting the BCL-2 family of anti-apoptotic factors. Aging Cell 2016, 15, 428–435. [Google Scholar] [CrossRef] [PubMed]



© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bartlett, D.E.; Miller, R.B.; Thiesfeldt, S.; Lakhani, H.V.; Shapiro, J.I.; Sodhi, K. The Role of Na/K-ATPase Signaling in Oxidative Stress Related to Aging: Implications in Obesity and Cardiovascular Disease. Int. J. Mol. Sci. 2018, 19, 2139. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms19072139
Bartlett DE, Miller RB, Thiesfeldt S, Lakhani HV, Shapiro JI, Sodhi K. The Role of Na/K-ATPase Signaling in Oxidative Stress Related to Aging: Implications in Obesity and Cardiovascular Disease. International Journal of Molecular Sciences. 2018; 19(7):2139. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms19072139
Chicago/Turabian StyleBartlett, David E., Richard B. Miller, Scott Thiesfeldt, Hari Vishal Lakhani, Joseph I. Shapiro, and Komal Sodhi. 2018. "The Role of Na/K-ATPase Signaling in Oxidative Stress Related to Aging: Implications in Obesity and Cardiovascular Disease" International Journal of Molecular Sciences 19, no. 7: 2139. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms19072139