Regulation of Energy Expenditure and Brown/Beige Thermogenic Activity by Interleukins: New Roles for Old Actors

Abstract
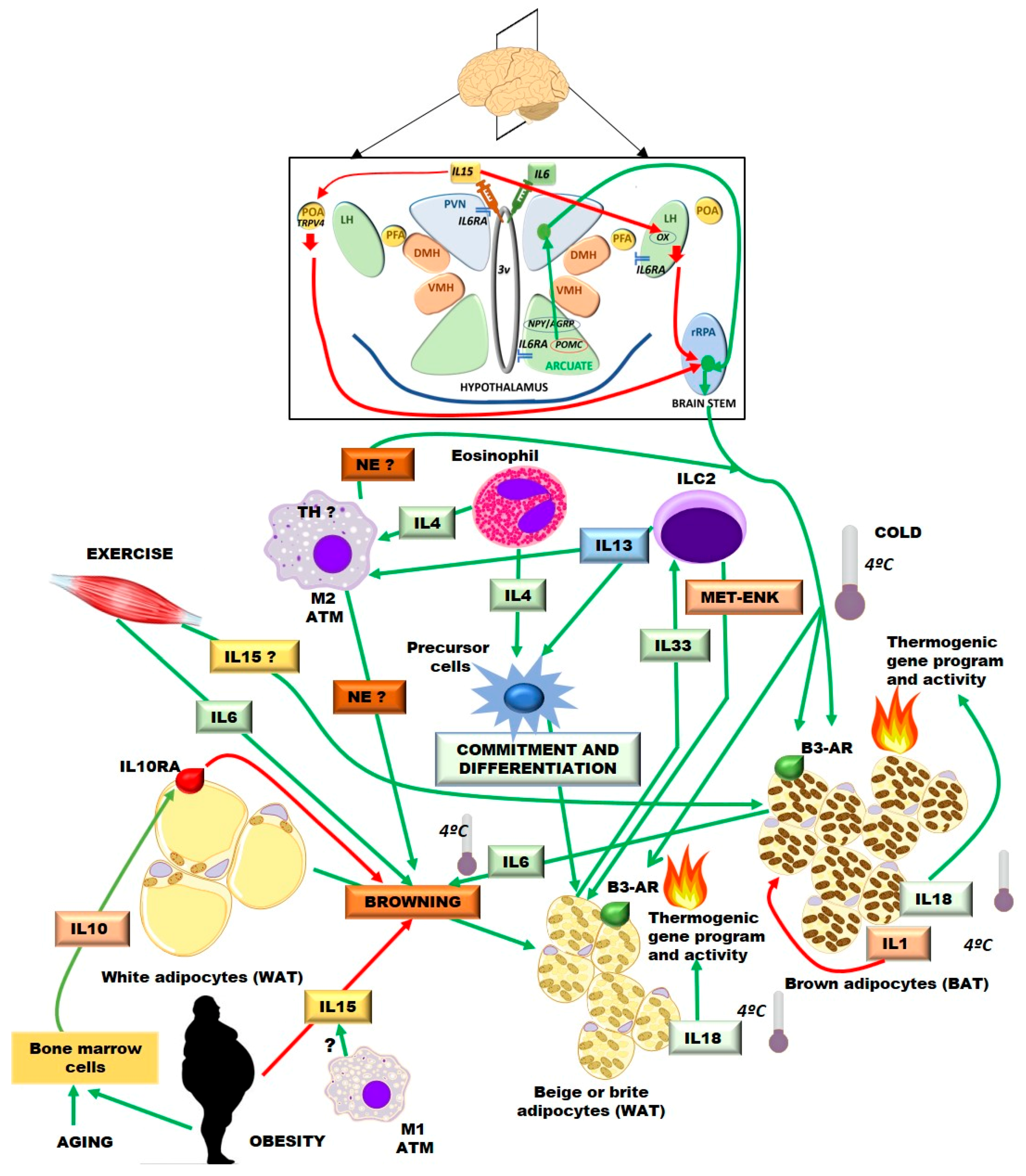
:
1. Introduction
2. The Scenery: Natural History of Thermogenic and Immune Cells in Adipose Tissues
2.1. Commitment and Differentiation of Thermogenic Adipocytes
2.2. Regulation of Thermogenic Activity
2.3. Immune Cells in Brown and Beige Adipose Tissue
3. Cytokines: Immune Actors in BAT Activation and Browning of WAT
3.1. IL4 and IL13, to Be or Not Be: Eosinophils, Macrophages and the Catecolaminergic Controversy
3.1.1. IL4/IL13 Production and Signalling
3.1.2. IL4/IL13 and Energy and Metabolic Homeostasis
3.1.3. IL4/IL13 and the Thermogenic Function of Adipose Tissues
3.2. IL33, an ILC2-Derived Perinatal BAT Licenser and Adult Beige Fat Activator of Thermogenesis
3.2.1. IL33 Production and Signalling
3.2.2. IL33 and Energy and Metabolic Homeostasis
3.2.3. IL33 and Thermogenic Function of Adipose Tissues
3.3. IL6, Central and Peripheral Modulator of AT Thermogenesis
3.3.1. IL6 Production and Signalling
3.3.2. IL6 and Energy and Metabolic Homeostasis
3.3.3. IL6 and Thermogenic Function of Adipose Tissues
3.4. IL15, a Controversial Myokine in Mitochondrial Function and Thermoregulation
3.4.1. IL15 and IL15/IL15RA Complex
3.4.2. IL15 and Energy and Metabolic Homeostasis
3.4.3. IL15 and Thermogenic Function of Adipose Tissues
3.5. IL18/IL18R1, a Complex Immune System in the Control of BAT and Beige Thermogenesis
3.5.1. IL18 Production and Signalling
3.5.2. IL18 and Energy and Metabolic Homeostasis
3.5.3. IL18 and Thermogenic Function of Adipose Tissues
3.6. IL10/IL10RA, an Anti-Inflammatory Brake of Thermogenic Gene Expression and EE
3.6.1. IL10 Production and Signalling
3.6.2. IL10 and Energy and Metabolic Homeostasis
3.6.3. IL10 and Thermogenic Function of Adipose Tissues
4. Conclusions and Future Perspectives
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- GBD 2015 Obesity Collaborators; Afshin, A.; Forouzanfar, M.H.; Reitsma, M.B.; Sur, P.; Estep, K.; Lee, A.; Marczak, L.; Mokdad, A.H.; Moradi-Lakeh, M.; et al. Health Effects of Overweight and Obesity in 195 Countries over 25 Years. N. Engl. J. Med. 2017, 377, 13–27. [Google Scholar] [PubMed] [Green Version]
- Gregg, E.W.; Shaw, J.E. Global Health Effects of Overweight and Obesity. N. Engl. J. Med. 2017, 377, 80–81. [Google Scholar] [CrossRef] [PubMed]
- Minihane, A.M.; Vinoy, S.; Russell, W.R.; Baka, A.; Roche, H.M.; Tuohy, K.M.; Teeling, J.L.; Blaak, E.E.; Fenech, M.; Vauzour, D.; et al. Low-grade inflammation, diet composition and health: Current research evidence and its translation. Br. J. Nutr. 2015, 114, 999–1012. [Google Scholar] [CrossRef] [PubMed]
- Hotamisligil, G.S. Inflammation, metaflammation and immunometabolic disorders. Nature 2017, 542, 177–185. [Google Scholar] [CrossRef] [PubMed]
- Reilly, S.M.; Saltiel, A.R. Adapting to obesity with adipose tissue inflammation. Nat. Rev. Endocrinol. 2017, 13, 633–643. [Google Scholar] [CrossRef] [PubMed]
- Robinson, M.W.; Harmon, C.; O’Farrelly, C. Liver immunology and its role in inflammation and homeostasis. Cell. Mol. Immunol. 2016, 13, 267–276. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lowell, B.B.; Bachman, E.S. β-Adrenergic receptors, diet-induced thermogenesis, and obesity. J. Biol. Chem. 2003, 278, 29385–29388. [Google Scholar] [CrossRef] [PubMed]
- Sakamoto, T.; Nitta, T.; Maruno, K.; Yeh, Y.S.; Kuwata, H.; Tomita, K.; Goto, T.; Takahashi, N.; Kawada, T. Macrophage infiltration into obese adipose tissues suppresses the induction of UCP1 level in mice. Am. J. Physiol. Endocrinol. Metab. 2016, 310, E676–E687. [Google Scholar] [CrossRef] [PubMed]
- Guo, T.; Marmol, P.; Moliner, A.; Bjornholm, M.; Zhang, C.; Shokat, K.M.; Ibanez, C.F. Adipocyte ALK7 links nutrient overload to catecholamine resistance in obesity. eLife 2014, 3, e03245. [Google Scholar] [CrossRef] [PubMed]
- Mowers, J.; Uhm, M.; Reilly, S.M.; Simon, J.; Leto, D.; Chiang, S.H.; Chang, L.; Saltiel, A.R. Inflammation produces catecholamine resistance in obesity via activation of PDE3B by the protein kinases IKKepsilon and TBK1. eLife 2013, 2, e01119. [Google Scholar] [CrossRef] [PubMed]
- Wernstedt Asterholm, I.; Tao, C.; Morley, T.S.; Wang, Q.A.; Delgado-Lopez, F.; Wang, Z.V.; Scherer, P.E. Adipocyte inflammation is essential for healthy adipose tissue expansion and remodeling. Cell Metab. 2014, 20, 103–118. [Google Scholar] [CrossRef] [PubMed]
- Villarroya, F.; Cereijo, R.; Villarroya, J.; Gavalda-Navarro, A.; Giralt, M. Toward an Understanding of How Immune Cells Control Brown and Beige Adipobiology. Cell Metab. 2018, 27, 954–961. [Google Scholar] [CrossRef] [PubMed]
- Ikeda, K.; Maretich, P.; Kajimura, S. The Common and Distinct Features of Brown and Beige Adipocytes. Trends Endocrinol. Metab. 2018, 29, 191–200. [Google Scholar] [CrossRef] [PubMed]
- Giordano, A.; Frontini, A.; Cinti, S. Convertible visceral fat as a therapeutic target to curb obesity. Nat. Rev. Drug Discov. 2016, 15, 405–424. [Google Scholar] [CrossRef] [PubMed]
- Villarroya, F.; Vidal-Puig, A. Beyond the sympathetic tone: The new brown fat activators. Cell Metab. 2013, 17, 638–643. [Google Scholar] [CrossRef] [PubMed]
- Bolus, W.R.; Hasty, A.H. Contributions of Innate Type 2 Inflammation to Adipose Function. J. Lipid Res. 2018. [Google Scholar] [CrossRef] [PubMed]
- Man, K.; Kutyavin, V.I.; Chawla, A. Tissue Immunometabolism: Development, Physiology, and Pathobiology. Cell Metab. 2017, 25, 11–26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van den Berg, S.M.; van Dam, A.D.; Rensen, P.C.; de Winther, M.P.; Lutgens, E. Immune Modulation of Brown(ing) Adipose Tissue in Obesity. Endocr. Rev. 2017, 38, 46–68. [Google Scholar] [CrossRef] [PubMed]
- Saito, M.; Okamatsu-Ogura, Y.; Matsushita, M.; Watanabe, K.; Yoneshiro, T.; Nio-Kobayashi, J.; Iwanaga, T.; Miyagawa, M.; Kameya, T.; Nakada, K.; et al. High incidence of metabolically active brown adipose tissue in healthy adult humans: Effects of cold exposure and adiposity. Diabetes 2009, 58, 1526–1531. [Google Scholar] [CrossRef] [PubMed]
- Cypess, A.M.; Lehman, S.; Williams, G.; Tal, I.; Rodman, D.; Goldfine, A.B.; Kuo, F.C.; Palmer, E.L.; Tseng, Y.H.; Doria, A.; et al. Identification and importance of brown adipose tissue in adult humans. N. Engl. J. Med. 2009, 360, 1509–1517. [Google Scholar] [CrossRef] [PubMed]
- Sidossis, L.; Kajimura, S. Brown and beige fat in humans: Thermogenic adipocytes that control energy and glucose homeostasis. J. Clin. Investig. 2015, 125, 478–486. [Google Scholar] [CrossRef] [PubMed]
- Matsushita, M.; Yoneshiro, T.; Aita, S.; Kameya, T.; Sugie, H.; Saito, M. Impact of brown adipose tissue on body fatness and glucose metabolism in healthy humans. Int. J. Obes. 2014, 38, 812–817. [Google Scholar] [CrossRef] [PubMed]
- Yoneshiro, T.; Aita, S.; Matsushita, M.; Kayahara, T.; Kameya, T.; Kawai, Y.; Iwanaga, T.; Saito, M. Recruited brown adipose tissue as an antiobesity agent in humans. J. Clin. Investig. 2013, 123, 3404–3408. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van der Lans, A.A.; Hoeks, J.; Brans, B.; Vijgen, G.H.; Visser, M.G.; Vosselman, M.J.; Hansen, J.; Jorgensen, J.A.; Wu, J.; Mottaghy, F.M.; et al. Cold acclimation recruits human brown fat and increases nonshivering thermogenesis. J. Clin. Investig. 2013, 123, 3395–3403. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, P.; Smith, S.; Linderman, J.; Courville, A.B.; Brychta, R.J.; Dieckmann, W.; Werner, C.D.; Chen, K.Y.; Celi, F.S. Temperature-acclimated brown adipose tissue modulates insulin sensitivity in humans. Diabetes 2014, 63, 3686–3698. [Google Scholar] [CrossRef] [PubMed]
- Hanssen, M.J.; van der Lans, A.A.; Brans, B.; Hoeks, J.; Jardon, K.M.; Schaart, G.; Mottaghy, F.M.; Schrauwen, P.; van Marken Lichtenbelt, W.D. Short-term Cold Acclimation Recruits Brown Adipose Tissue in Obese Humans. Diabetes 2016, 65, 1179–1189. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Bostrom, P.; Sparks, L.M.; Ye, L.; Choi, J.H.; Giang, A.H.; Khandekar, M.; Virtanen, K.A.; Nuutila, P.; Schaart, G.; et al. Beige adipocytes are a distinct type of thermogenic fat cell in mouse and human. Cell 2012, 150, 366–376. [Google Scholar] [CrossRef] [PubMed]
- Cypess, A.M.; White, A.P.; Vernochet, C.; Schulz, T.J.; Xue, R.; Sass, C.A.; Huang, T.L.; Roberts-Toler, C.; Weiner, L.S.; Sze, C.; et al. Anatomical localization, gene expression profiling and functional characterization of adult human neck brown fat. Nat. Med. 2013, 19, 635–639. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tamucci, K.A.; Namwanje, M.; Fan, L.; Qiang, L. The dark side of browning. Protein Cell 2018, 9, 152–163. [Google Scholar] [CrossRef] [PubMed]
- Petruzzelli, M.; Schweiger, M.; Schreiber, R.; Campos-Olivas, R.; Tsoli, M.; Allen, J.; Swarbrick, M.; Rose-John, S.; Rincon, M.; Robertson, G.; et al. A switch from white to brown fat increases energy expenditure in cancer-associated cachexia. Cell Metab. 2014, 20, 433–447. [Google Scholar] [CrossRef] [PubMed]
- Patsouris, D.; Qi, P.; Abdullahi, A.; Stanojcic, M.; Chen, P.; Parousis, A.; Amini-Nik, S.; Jeschke, M.G. Burn Induces Browning of the Subcutaneous White Adipose Tissue in Mice and Humans. Cell Rep. 2015, 13, 1538–1544. [Google Scholar] [CrossRef] [PubMed]
- Lynes, M.D.; Tseng, Y.H. The Thermogenic Circuit: Regulators of Thermogenic Competency and Differentiation. Genes Dis. 2015, 2, 164–172. [Google Scholar] [CrossRef] [PubMed]
- Lepper, C.; Fan, C.M. Inducible lineage tracing of Pax7-descendant cells reveals embryonic origin of adult satellite cells. Genesis 2010, 48, 424–436. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, W.; Kissig, M.; Rajakumari, S.; Huang, L.; Lim, H.W.; Won, K.J.; Seale, P. Ebf2 is a selective marker of brown and beige adipogenic precursor cells. Proc. Natl. Acad. Sci. USA 2014, 111, 14466–14471. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schoettl, T.; Fischer, I.P.; Ussar, S. Heterogeneity of adipose tissue in development and metabolic function. J. Exp. Biol. 2018, 221. [Google Scholar] [CrossRef] [PubMed]
- Timmons, J.A.; Wennmalm, K.; Larsson, O.; Walden, T.B.; Lassmann, T.; Petrovic, N.; Hamilton, D.L.; Gimeno, R.E.; Wahlestedt, C.; Baar, K.; et al. Myogenic gene expression signature establishes that brown and white adipocytes originate from distinct cell lineages. Proc. Natl. Acad. Sci. USA 2007, 104, 4401–4406. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seale, P.; Bjork, B.; Yang, W.; Kajimura, S.; Chin, S.; Kuang, S.; Scime, A.; Devarakonda, S.; Conroe, H.M.; Erdjument-Bromage, H.; et al. PRDM16 controls a brown fat/skeletal muscle switch. Nature 2008, 454, 961–967. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rajakumari, S.; Wu, J.; Ishibashi, J.; Lim, H.W.; Giang, A.H.; Won, K.J.; Reed, R.R.; Seale, P. EBF2 determines and maintains brown adipocyte identity. Cell Metab. 2013, 17, 562–574. [Google Scholar] [CrossRef] [PubMed]
- Ohno, H.; Shinoda, K.; Ohyama, K.; Sharp, L.Z.; Kajimura, S. EHMT1 controls brown adipose cell fate and thermogenesis through the PRDM16 complex. Nature 2013, 504, 163–167. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harms, M.J.; Ishibashi, J.; Wang, W.; Lim, H.W.; Goyama, S.; Sato, T.; Kurokawa, M.; Won, K.J.; Seale, P. Prdm16 is required for the maintenance of brown adipocyte identity and function in adult mice. Cell Metab. 2014, 19, 593–604. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Gurmaches, J.; Guertin, D.A. Adipocyte lineages: Tracing back the origins of fat. Biochim. Biophys. Acta 2014, 1842, 340–351. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Gurmaches, J.; Hung, C.M.; Sparks, C.A.; Tang, Y.; Li, H.; Guertin, D.A. PTEN loss in the Myf5 lineage redistributes body fat and reveals subsets of white adipocytes that arise from Myf5 precursors. Cell Metab. 2012, 16, 348–362. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Gurmaches, J.; Guertin, D.A. Adipocytes arise from multiple lineages that are heterogeneously and dynamically distributed. Nat. Commun. 2014, 5, 4099. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shinoda, K.; Luijten, I.H.; Hasegawa, Y.; Hong, H.; Sonne, S.B.; Kim, M.; Xue, R.; Chondronikola, M.; Cypess, A.M.; Tseng, Y.H.; et al. Genetic and functional characterization of clonally derived adult human brown adipocytes. Nat. Med. 2015, 21, 389–394. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xue, R.; Lynes, M.D.; Dreyfuss, J.M.; Shamsi, F.; Schulz, T.J.; Zhang, H.; Huang, T.L.; Townsend, K.L.; Li, Y.; Takahashi, H.; et al. Clonal analyses and gene profiling identify genetic biomarkers of the thermogenic potential of human brown and white preadipocytes. Nat. Med. 2015, 21, 760–768. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, Z.; Daquinag, A.C.; Su, F.; Snyder, B.; Kolonin, M.G. PDGFRα/PDGFRβ signaling balance modulates progenitor cell differentiation into white and beige adipocytes. Development 2018, 145, dev155861. [Google Scholar] [CrossRef] [PubMed]
- Inagaki, T.; Sakai, J.; Kajimura, S. Transcriptional and epigenetic control of brown and beige adipose cell fate and function. Nat. Rev. Mol. Cell Biol. 2016, 17, 480–495. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carobbio, S.; Guenantin, A.C.; Samuelson, I.; Bahri, M.; Vidal-Puig, A. Brown and beige fat: From molecules to physiology and pathophysiology. Biochim. Biophys. Acta 2018. [Google Scholar] [CrossRef] [PubMed]
- Nies, V.J.; Sancar, G.; Liu, W.; van Zutphen, T.; Struik, D.; Yu, R.T.; Atkins, A.R.; Evans, R.M.; Jonker, J.W.; Downes, M.R. Fibroblast Growth Factor Signaling in Metabolic Regulation. Front. Endocrinol. 2015, 6, 193. [Google Scholar] [CrossRef] [PubMed]
- Schulz, T.J.; Huang, P.; Huang, T.L.; Xue, R.; McDougall, L.E.; Townsend, K.L.; Cypess, A.M.; Mishina, Y.; Gussoni, E.; Tseng, Y.H. Brown-fat paucity due to impaired BMP signalling induces compensatory browning of white fat. Nature 2013, 495, 379–383. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tseng, Y.H.; Kokkotou, E.; Schulz, T.J.; Huang, T.L.; Winnay, J.N.; Taniguchi, C.M.; Tran, T.T.; Suzuki, R.; Espinoza, D.O.; Yamamoto, Y.; et al. New role of bone morphogenetic protein 7 in brown adipogenesis and energy expenditure. Nature 2008, 454, 1000–1004. [Google Scholar] [CrossRef] [PubMed]
- Okla, M.; Ha, J.H.; Temel, R.E.; Chung, S. BMP7 drives human adipogenic stem cells into metabolically active beige adipocytes. Lipids 2015, 50, 111–120. [Google Scholar] [CrossRef] [PubMed]
- Sharma, A.; Huard, C.; Vernochet, C.; Ziemek, D.; Knowlton, K.M.; Tyminski, E.; Paradis, T.; Zhang, Y.; Jones, J.E.; von Schack, D.; et al. Brown fat determination and development from muscle precursor cells by novel action of bone morphogenetic protein 6. PLoS ONE 2014, 9, e92608. [Google Scholar] [CrossRef] [PubMed]
- Kajimura, S.; Seale, P.; Kubota, K.; Lunsford, E.; Frangioni, J.V.; Gygi, S.P.; Spiegelman, B.M. Initiation of myoblast to brown fat switch by a PRDM16-C/EBP-β transcriptional complex. Nature 2009, 460, 1154–1158. [Google Scholar] [CrossRef] [PubMed]
- Seale, P.; Kajimura, S.; Yang, W.; Chin, S.; Rohas, L.M.; Uldry, M.; Tavernier, G.; Langin, D.; Spiegelman, B.M. Transcriptional control of brown fat determination by PRDM16. Cell Metab. 2007, 6, 38–54. [Google Scholar] [CrossRef] [PubMed]
- Fischer, A.W.; Shabalina, I.G.; Mattsson, C.L.; Abreu-Vieira, G.; Cannon, B.; Nedergaard, J.; Petrovic, N. UCP1 inhibition in Cidea-overexpressing mice is physiologically counteracted by brown adipose tissue hyperrecruitment. Am. J. Physiol. Endocrinol. Metab. 2017, 312, E72–E87. [Google Scholar] [CrossRef] [PubMed]
- Petrovic, N.; Shabalina, I.G.; Timmons, J.A.; Cannon, B.; Nedergaard, J. Thermogenically competent nonadrenergic recruitment in brown preadipocytes by a PPARγ agonist. Am. J. Physiol. Endocrinol. Metab. 2008, 295, E287–E296. [Google Scholar] [CrossRef] [PubMed]
- Puigserver, P.; Wu, Z.; Park, C.W.; Graves, R.; Wright, M.; Spiegelman, B.M. A cold-inducible coactivator of nuclear receptors linked to adaptive thermogenesis. Cell 1998, 92, 829–839. [Google Scholar] [CrossRef]
- Tiraby, C.; Tavernier, G.; Lefort, C.; Larrouy, D.; Bouillaud, F.; Ricquier, D.; Langin, D. Acquirement of brown fat cell features by human white adipocytes. J. Biol. Chem. 2003, 278, 33370–33376. [Google Scholar] [CrossRef] [PubMed]
- Dominy, J.E.; Puigserver, P. Mitochondrial biogenesis through activation of nuclear signaling proteins. Cold Spring Harb. Perspect. Biol. 2013, 5, a015008. [Google Scholar] [CrossRef] [PubMed]
- Mazzucotelli, A.; Viguerie, N.; Tiraby, C.; Annicotte, J.S.; Mairal, A.; Klimcakova, E.; Lepin, E.; Delmar, P.; Dejean, S.; Tavernier, G.; et al. The transcriptional coactivator peroxisome proliferator activated receptor (PPAR)γ coactivator-1 α and the nuclear receptor PPAR α control the expression of glycerol kinase and metabolism genes independently of PPAR γ activation in human white adipocytes. Diabetes 2007, 56, 2467–2475. [Google Scholar] [CrossRef] [PubMed]
- Mottillo, E.P.; Bloch, A.E.; Leff, T.; Granneman, J.G. Lipolytic products activate peroxisome proliferator-activated receptor (PPAR) α and delta in brown adipocytes to match fatty acid oxidation with supply. J. Biol. Chem. 2012, 287, 25038–25048. [Google Scholar] [CrossRef] [PubMed]
- Cao, W.; Daniel, K.W.; Robidoux, J.; Puigserver, P.; Medvedev, A.V.; Bai, X.; Floering, L.M.; Spiegelman, B.M.; Collins, S. p38 mitogen-activated protein kinase is the central regulator of cyclic AMP-dependent transcription of the brown fat uncoupling protein 1 gene. Mol. Cell. Biol. 2004, 24, 3057–3067. [Google Scholar] [CrossRef] [PubMed]
- Defour, M.; Dijk, W.; Ruppert, P.; Nascimento, E.B.M.; Schrauwen, P.; Kersten, S. The Peroxisome Proliferator-Activated Receptor α is dispensable for cold-induced adipose tissue browning in mice. Mol. Metab. 2018, 10, 39–54. [Google Scholar] [CrossRef] [PubMed]
- Kajimura, S.; Spiegelman, B.M.; Seale, P. Brown and Beige Fat: Physiological Roles beyond Heat Generation. Cell Metab. 2015, 22, 546–559. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stine, R.R.; Shapira, S.N.; Lim, H.W.; Ishibashi, J.; Harms, M.; Won, K.J.; Seale, P. EBF2 promotes the recruitment of beige adipocytes in white adipose tissue. Mol. Metab. 2016, 5, 57–65. [Google Scholar] [CrossRef] [PubMed]
- Kajimura, S.; Seale, P.; Tomaru, T.; Erdjument-Bromage, H.; Cooper, M.P.; Ruas, J.L.; Chin, S.; Tempst, P.; Lazar, M.A.; Spiegelman, B.M. Regulation of the brown and white fat gene programs through a PRDM16/CtBP transcriptional complex. Genes Dev. 2008, 22, 1397–1409. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cohen, P.; Levy, J.D.; Zhang, Y.; Frontini, A.; Kolodin, D.P.; Svensson, K.J.; Lo, J.C.; Zeng, X.; Ye, L.; Khandekar, M.J.; et al. Ablation of PRDM16 and beige adipose causes metabolic dysfunction and a subcutaneous to visceral fat switch. Cell 2014, 156, 304–316. [Google Scholar] [CrossRef] [PubMed]
- Nedergaard, J.; Golozoubova, V.; Matthias, A.; Asadi, A.; Jacobsson, A.; Cannon, B. UCP1: The only protein able to mediate adaptive non-shivering thermogenesis and metabolic inefficiency. Biochim. Biophys. Acta 2001, 1504, 82–106. [Google Scholar] [CrossRef]
- Ikeda, K.; Kang, Q.; Yoneshiro, T.; Camporez, J.P.; Maki, H.; Homma, M.; Shinoda, K.; Chen, Y.; Lu, X.; Maretich, P.; et al. UCP1-independent signaling involving SERCA2b-mediated calcium cycling regulates beige fat thermogenesis and systemic glucose homeostasis. Nat. Med. 2017, 23, 1454–1465. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kazak, L.; Chouchani, E.T.; Jedrychowski, M.P.; Erickson, B.K.; Shinoda, K.; Cohen, P.; Vetrivelan, R.; Lu, G.Z.; Laznik-Bogoslavski, D.; Hasenfuss, S.C.; et al. A creatine-driven substrate cycle enhances energy expenditure and thermogenesis in beige fat. Cell 2015, 163, 643–655. [Google Scholar] [CrossRef] [PubMed]
- Long, J.Z.; Svensson, K.J.; Bateman, L.A.; Lin, H.; Kamenecka, T.; Lokurkar, I.A.; Lou, J.; Rao, R.R.; Chang, M.R.; Jedrychowski, M.P.; et al. The Secreted Enzyme PM20D1 Regulates Lipidated Amino Acid Uncouplers of Mitochondria. Cell 2016, 166, 424–435. [Google Scholar] [CrossRef] [PubMed]
- Shabalina, I.G.; Kramarova, T.V.; Nedergaard, J.; Cannon, B. Carboxyatractyloside effects on brown-fat mitochondria imply that the adenine nucleotide translocator isoforms ANT1 and ANT2 may be responsible for basal and fatty-acid-induced uncoupling respectively. Biochem. J. 2006, 399, 405–414. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.I.; Liao, J.; Emont, M.P.; Park, M.J.; Jun, H.; Ramakrishnan, S.K.; Lin, J.D.; Shah, Y.M.; Omary, M.B.; Wu, J. An OLTAM system for analysis of brown/beige fat thermogenic activity. Int. J. Obes. 2018, 42, 939–945. [Google Scholar] [CrossRef] [PubMed]
- Cannon, B.; Nedergaard, J. Brown adipose tissue: Function and physiological significance. Physiol. Rev. 2004, 84, 277–359. [Google Scholar] [CrossRef] [PubMed]
- Flachs, P.; Adamcova, K.; Zouhar, P.; Marques, C.; Janovska, P.; Viegas, I.; Jones, J.G.; Bardova, K.; Svobodova, M.; Hansikova, J.; et al. Induction of lipogenesis in white fat during cold exposure in mice: Link to lean phenotype. Int. J. Obes. 2017, 41, 372–380. [Google Scholar] [CrossRef] [PubMed]
- Bartelt, A.; Bruns, O.T.; Reimer, R.; Hohenberg, H.; Ittrich, H.; Peldschus, K.; Kaul, M.G.; Tromsdorf, U.I.; Weller, H.; Waurisch, C.; et al. Brown adipose tissue activity controls triglyceride clearance. Nat. Med. 2011, 17, 200–205. [Google Scholar] [CrossRef] [PubMed]
- Ortega-Gomez, A.; Varela, L.M.; Lopez, S.; Montserrat de la Paz, S.; Sanchez, R.; Muriana, F.J.G.; Bermudez, B.; Abia, R. Postprandial triglyceride-rich lipoproteins promote lipid accumulation and apolipoprotein B-48 receptor transcriptional activity in human circulating and murine bone marrow neutrophils in a fatty acid-dependent manner. Mol. Nutr. Food Res. 2017, 61. [Google Scholar] [CrossRef] [PubMed]
- Lynes, M.D.; Leiria, L.O.; Lundh, M.; Bartelt, A.; Shamsi, F.; Huang, T.L.; Takahashi, H.; Hirshman, M.F.; Schlein, C.; Lee, A.; et al. The cold-induced lipokine 12,13-diHOME promotes fatty acid transport into brown adipose tissue. Nat. Med. 2017, 23, 631–637. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Warner, A.; Mittag, J. Breaking BAT: Can browning create a better white? J. Endocrinol. 2016, 228, R19–R29. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Seale, P. Control of brown and beige fat development. Nat. Rev. Mol. Cell Biol. 2016, 17, 691–702. [Google Scholar] [CrossRef] [PubMed]
- Contreras, C.; Nogueiras, R.; Dieguez, C.; Rahmouni, K.; Lopez, M. Traveling from the hypothalamus to the adipose tissue: The thermogenic pathway. Redox Biol. 2017, 12, 854–863. [Google Scholar] [CrossRef] [PubMed]
- Luo, X.; Jia, R.; Luo, X.Q.; Wang, G.; Zhang, Q.L.; Qiao, H.; Wang, N.; Yan, J.Q. Cold Exposure Differentially Stimulates Angiogenesis in BAT and WAT of Mice: Implication in Adrenergic Activation. Cell. Physiol. Biochem. 2017, 42, 974–986. [Google Scholar] [CrossRef] [PubMed]
- Park, J.; Kim, M.; Sun, K.; An, Y.A.; Gu, X.; Scherer, P.E. VEGF-A-Expressing Adipose Tissue Shows Rapid Beiging and Enhanced Survival after Transplantation and Confers IL-4-Independent Metabolic Improvements. Diabetes 2017, 66, 1479–1490. [Google Scholar] [CrossRef] [PubMed]
- Sun, K.; Kusminski, C.M.; Luby-Phelps, K.; Spurgin, S.B.; An, Y.A.; Wang, Q.A.; Holland, W.L.; Scherer, P.E. Brown adipose tissue derived VEGF-A modulates cold tolerance and energy expenditure. Mol. Metab. 2014, 3, 474–483. [Google Scholar] [CrossRef] [PubMed]
- Robidoux, J.; Kumar, N.; Daniel, K.W.; Moukdar, F.; Cyr, M.; Medvedev, A.V.; Collins, S. Maximal β3-adrenergic regulation of lipolysis involves Src and epidermal growth factor receptor-dependent ERK1/2 activation. J. Biol. Chem. 2006, 281, 37794–37802. [Google Scholar] [CrossRef] [PubMed]
- Collins, S. β-Adrenoceptor Signaling Networks in Adipocytes for Recruiting Stored Fat and Energy Expenditure. Front. Endocrinol. 2011, 2, 102. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.S.; Wollam, J.; Olefsky, J.M. An Integrated View of Immunometabolism. Cell 2018, 172, 22–40. [Google Scholar] [CrossRef] [PubMed]
- Cautivo, K.M.; Molofsky, A.B. Regulation of metabolic health and adipose tissue function by group 2 innate lymphoid cells. Eur. J. Immunol. 2016, 46, 1315–1325. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murray, P.J. Macrophage Polarization. Annu. Rev. Physiol. 2017, 79, 541–566. [Google Scholar] [CrossRef] [PubMed]
- Stein, M.; Keshav, S.; Harris, N.; Gordon, S. Interleukin 4 potently enhances murine macrophage mannose receptor activity: A marker of alternative immunologic macrophage activation. J. Exp. Med. 1992, 176, 287–292. [Google Scholar] [CrossRef] [PubMed]
- Lumeng, C.N.; DelProposto, J.B.; Westcott, D.J.; Saltiel, A.R. Phenotypic switching of adipose tissue macrophages with obesity is generated by spatiotemporal differences in macrophage subtypes. Diabetes 2008, 57, 3239–3246. [Google Scholar] [CrossRef] [PubMed]
- Kratz, M.; Coats, B.R.; Hisert, K.B.; Hagman, D.; Mutskov, V.; Peris, E.; Schoenfelt, K.Q.; Kuzma, J.N.; Larson, I.; Billing, P.S.; et al. Metabolic dysfunction drives a mechanistically distinct proinflammatory phenotype in adipose tissue macrophages. Cell Metab. 2014, 20, 614–625. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.; Shang, Q.; Pan, Z.; Bai, Y.; Li, Z.; Zhang, H.; Zhang, Q.; Guo, C.; Zhang, L.; Wang, Q. Exosomes from Adipose-Derived Stem Cells Attenuate Adipose Inflammation and Obesity through Polarizing M2 Macrophages and Beiging in White Adipose Tissue. Diabetes 2018, 67, 235–247. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, K.D.; Qiu, Y.; Cui, X.; Goh, Y.P.; Mwangi, J.; David, T.; Mukundan, L.; Brombacher, F.; Locksley, R.M.; Chawla, A. Alternatively activated macrophages produce catecholamines to sustain adaptive thermogenesis. Nature 2011, 480, 104–108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rao, R.R.; Long, J.Z.; White, J.P.; Svensson, K.J.; Lou, J.; Lokurkar, I.; Jedrychowski, M.P.; Ruas, J.L.; Wrann, C.D.; Lo, J.C.; et al. Meteorin-like is a hormone that regulates immune-adipose interactions to increase beige fat thermogenesis. Cell 2014, 157, 1279–1291. [Google Scholar] [CrossRef] [PubMed]
- Qiu, Y.; Nguyen, K.D.; Odegaard, J.I.; Cui, X.; Tian, X.; Locksley, R.M.; Palmiter, R.D.; Chawla, A. Eosinophils and type 2 cytokine signaling in macrophages orchestrate development of functional beige fat. Cell 2014, 157, 1292–1308. [Google Scholar] [CrossRef] [PubMed]
- Fabbiano, S.; Suarez-Zamorano, N.; Rigo, D.; Veyrat-Durebex, C.; Stevanovic Dokic, A.; Colin, D.J.; Trajkovski, M. Caloric Restriction Leads to Browning of White Adipose Tissue through Type 2 Immune Signaling. Cell Metab. 2016, 24, 434–446. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suarez-Zamorano, N.; Fabbiano, S.; Chevalier, C.; Stojanovic, O.; Colin, D.J.; Stevanovic, A.; Veyrat-Durebex, C.; Tarallo, V.; Rigo, D.; Germain, S.; et al. Microbiota depletion promotes browning of white adipose tissue and reduces obesity. Nat. Med. 2015, 21, 1497–1501. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brestoff, J.R.; Kim, B.S.; Saenz, S.A.; Stine, R.R.; Monticelli, L.A.; Sonnenberg, G.F.; Thome, J.J.; Farber, D.L.; Lutfy, K.; Seale, P.; et al. Group 2 innate lymphoid cells promote beiging of white adipose tissue and limit obesity. Nature 2015, 519, 242–246. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.W.; Odegaard, J.I.; Mukundan, L.; Qiu, Y.; Molofsky, A.B.; Nussbaum, J.C.; Yun, K.; Locksley, R.M.; Chawla, A. Activated type 2 innate lymphoid cells regulate beige fat biogenesis. Cell 2015, 160, 74–87. [Google Scholar] [CrossRef] [PubMed]
- Molofsky, A.B.; Nussbaum, J.C.; Liang, H.E.; Van Dyken, S.J.; Cheng, L.E.; Mohapatra, A.; Chawla, A.; Locksley, R.M. Innate lymphoid type 2 cells sustain visceral adipose tissue eosinophils and alternatively activated macrophages. J. Exp. Med. 2013, 210, 535–549. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Medrikova, D.; Sijmonsma, T.P.; Sowodniok, K.; Richards, D.M.; Delacher, M.; Sticht, C.; Gretz, N.; Schafmeier, T.; Feuerer, M.; Herzig, S. Brown adipose tissue harbors a distinct sub-population of regulatory T cells. PLoS ONE 2015, 10, e0118534. [Google Scholar] [CrossRef] [PubMed]
- Lynch, L.; Hogan, A.E.; Duquette, D.; Lester, C.; Banks, A.; LeClair, K.; Cohen, D.E.; Ghosh, A.; Lu, B.; Corrigan, M.; et al. iNKT Cells Induce FGF21 for Thermogenesis and Are Required for Maximal Weight Loss in GLP1 Therapy. Cell Metab. 2016, 24, 510–519. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Dyken, S.J.; Locksley, R.M. Interleukin-4- and interleukin-13-mediated alternatively activated macrophages: Roles in homeostasis and disease. Annu. Rev. Immunol. 2013, 31, 317–343. [Google Scholar] [CrossRef] [PubMed]
- McCormick, S.M.; Heller, N.M. Commentary: IL-4 and IL-13 receptors and signaling. Cytokine 2015, 75, 38–50. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Junttila, I.S.; Creusot, R.J.; Moraga, I.; Bates, D.L.; Wong, M.T.; Alonso, M.N.; Suhoski, M.M.; Lupardus, P.; Meier-Schellersheim, M.; Engleman, E.G.; et al. Redirecting cell-type specific cytokine responses with engineered interleukin-4 superkines. Nat. Chem. Biol. 2012, 8, 990–998. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stanya, K.J.; Jacobi, D.; Liu, S.; Bhargava, P.; Dai, L.; Gangl, M.R.; Inouye, K.; Barlow, J.L.; Ji, Y.; Mizgerd, J.P.; et al. Direct control of hepatic glucose production by interleukin-13 in mice. J. Clin. Investig. 2013, 123, 261–271. [Google Scholar] [CrossRef] [PubMed]
- Khaled, W.T.; Read, E.K.; Nicholson, S.E.; Baxter, F.O.; Brennan, A.J.; Came, P.J.; Sprigg, N.; McKenzie, A.N.; Watson, C.J. The IL-4/IL-13/Stat6 signalling pathway promotes luminal mammary epithelial cell development. Development 2007, 134, 2739–2750. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gadani, S.P.; Cronk, J.C.; Norris, G.T.; Kipnis, J. IL-4 in the brain: A cytokine to remember. J. Immunol. 2012, 189, 4213–4219. [Google Scholar] [CrossRef] [PubMed]
- Ricardo-Gonzalez, R.R.; Red Eagle, A.; Odegaard, J.I.; Jouihan, H.; Morel, C.R.; Heredia, J.E.; Mukundan, L.; Wu, D.; Locksley, R.M.; Chawla, A. IL-4/STAT6 immune axis regulates peripheral nutrient metabolism and insulin sensitivity. Proc. Natl. Acad. Sci. USA 2010, 107, 22617–22622. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oh, I.S.; Thaler, J.P.; Ogimoto, K.; Wisse, B.E.; Morton, G.J.; Schwartz, M.W. Central administration of interleukin-4 exacerbates hypothalamic inflammation and weight gain during high-fat feeding. Am. J. Physiol. Endocrinol. Metab. 2010, 299, E47–E53. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rachmin, I.; O’Meara, C.C.; Ricci-Blair, E.M.; Feng, Y.; Christensen, E.M.; Duffy, J.F.; Zitting, K.M.; Czeisler, C.A.; Pancoast, J.R.; Cannon, C.P.; et al. Soluble interleukin-13rα1: A circulating regulator of glucose. Am. J. Physiol. Endocrinol. Metab. 2017, 313, E663–E671. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.H.; Kim, Y.H.; Son, J.E.; Lee, J.H.; Kim, S.; Choe, M.S.; Moon, J.H.; Zhong, J.; Fu, K.; Lenglin, F.; et al. Intermittent fasting promotes adipose thermogenesis and metabolic homeostasis via VEGF-mediated alternative activation of macrophage. Cell Res. 2017, 27, 1309–1326. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, Y.W.; Liu, P.S.; Adhikari, N.; Hall, J.L.; Wei, L.N. RIP140 contributes to foam cell formation and atherosclerosis by regulating cholesterol homeostasis in macrophages. J. Mol. Cell. Cardiol. 2015, 79, 287–294. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, P.S.; Lin, Y.W.; Burton, F.H.; Wei, L.N. Injecting engineered anti-inflammatory macrophages therapeutically induces white adipose tissue browning and improves diet-induced insulin resistance. Adipocyte 2015, 4, 123–128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shan, B.; Wang, X.; Wu, Y.; Xu, C.; Xia, Z.; Dai, J.; Shao, M.; Zhao, F.; He, S.; Yang, L.; et al. The metabolic ER stress sensor IRE1α suppresses alternative activation of macrophages and impairs energy expenditure in obesity. Nat. Immunol. 2017, 18, 519–529. [Google Scholar] [CrossRef] [PubMed]
- Ruiz de Azua, I.; Mancini, G.; Srivastava, R.K.; Rey, A.A.; Cardinal, P.; Tedesco, L.; Zingaretti, C.M.; Sassmann, A.; Quarta, C.; Schwitter, C.; et al. Adipocyte cannabinoid receptor CB1 regulates energy homeostasis and alternatively activated macrophages. J. Clin. Investig. 2017, 127, 4148–4162. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luo, Y.; Liu, B.; Yang, X.; Ma, X.; Zhang, X.; Bragin, D.E.; Yang, X.O.; Huang, W.; Liu, M. Myeloid adrenergic signaling via CaMKII forms a feedforward loop of catecholamine biosynthesis. J. Mol. Cell. Biol. 2017, 9, 422–434. [Google Scholar] [CrossRef] [PubMed]
- Huang, Z.; Zhong, L.; Lee, J.T.H.; Zhang, J.; Wu, D.; Geng, L.; Wang, Y.; Wong, C.M.; Xu, A. The FGF21-CCL11 Axis Mediates Beiging of White Adipose Tissues by Coupling Sympathetic Nervous System to Type 2 Immunity. Cell Metab. 2017, 26, 493–508. [Google Scholar] [CrossRef] [PubMed]
- Hui, X.; Gu, P.; Zhang, J.; Nie, T.; Pan, Y.; Wu, D.; Feng, T.; Zhong, C.; Wang, Y.; Lam, K.S.; et al. Adiponectin Enhances Cold-Induced Browning of Subcutaneous Adipose Tissue via Promoting M2 Macrophage Proliferation. Cell Metab. 2015, 22, 279–290. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spadaro, O.; Camell, C.D.; Bosurgi, L.; Nguyen, K.Y.; Youm, Y.H.; Rothlin, C.V.; Dixit, V.D. IGF1 Shapes Macrophage Activation in Response to Immunometabolic Challenge. Cell Rep. 2017, 19, 225–234. [Google Scholar] [CrossRef] [PubMed]
- Chang, H.R.; Kim, H.J.; Xu, X.; Ferrante, A.W., Jr. Macrophage and adipocyte IGF1 maintain adipose tissue homeostasis during metabolic stresses. Obesity 2016, 24, 172–183. [Google Scholar] [CrossRef] [PubMed]
- Fischer, K.; Ruiz, H.H.; Jhun, K.; Finan, B.; Oberlin, D.J.; van der Heide, V.; Kalinovich, A.V.; Petrovic, N.; Wolf, Y.; Clemmensen, C.; et al. Alternatively activated macrophages do not synthesize catecholamines or contribute to adipose tissue adaptive thermogenesis. Nat. Med. 2017, 23, 623–630. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jun, H.; Yu, H.; Gong, J.; Jiang, J.; Qiao, X.; Perkey, E.; Kim, D.I.; Emont, M.P.; Zestos, A.G.; Cho, J.S.; et al. An immune-beige adipocyte communication via nicotinic acetylcholine receptor signaling. Nat. Med. 2018, 24, 814–822. [Google Scholar] [CrossRef] [PubMed]
- Pirzgalska, R.M.; Seixas, E.; Seidman, J.S.; Link, V.M.; Sanchez, N.M.; Mahu, I.; Mendes, R.; Gres, V.; Kubasova, N.; Morris, I.; et al. Sympathetic neuron-associated macrophages contribute to obesity by importing and metabolizing norepinephrine. Nat. Med. 2017, 23, 1309–1318. [Google Scholar] [CrossRef] [PubMed]
- Bolus, W.R.; Peterson, K.R.; Hubler, M.J.; Kennedy, A.J.; Gruen, M.L.; Hasty, A.H. Elevating adipose eosinophils in obese mice to physiologically normal levels does not rescue metabolic impairments. Mol. Metab. 2018, 8, 86–95. [Google Scholar] [CrossRef] [PubMed]
- Camell, C.D.; Sander, J.; Spadaro, O.; Lee, A.; Nguyen, K.Y.; Wing, A.; Goldberg, E.L.; Youm, Y.H.; Brown, C.W.; Elsworth, J.; et al. Inflammasome-driven catecholamine catabolism in macrophages blunts lipolysis during ageing. Nature 2017, 550, 119–123. [Google Scholar] [CrossRef] [PubMed]
- Wolf, Y.; Boura-Halfon, S.; Cortese, N.; Haimon, Z.; Sar Shalom, H.; Kuperman, Y.; Kalchenko, V.; Brandis, A.; David, E.; Segal-Hayoun, Y.; et al. Brown-adipose-tissue macrophages control tissue innervation and homeostatic energy expenditure. Nat. Immunol. 2017, 18, 665–674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, Y.H.; Petkova, A.P.; Granneman, J.G. Identification of an adipogenic niche for adipose tissue remodeling and restoration. Cell Metab. 2013, 18, 355–367. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.H.; Kim, S.N.; Kwon, H.J.; Maddipati, K.R.; Granneman, J.G. Adipogenic role of alternatively activated macrophages in β-adrenergic remodeling of white adipose tissue. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2016, 310, R55–R65. [Google Scholar] [CrossRef] [PubMed]
- Chung, K.J.; Chatzigeorgiou, A.; Economopoulou, M.; Garcia-Martin, R.; Alexaki, V.I.; Mitroulis, I.; Nati, M.; Gebler, J.; Ziemssen, T.; Goelz, S.E.; et al. A self-sustained loop of inflammation-driven inhibition of beige adipogenesis in obesity. Nat. Immunol. 2017, 18, 654–664. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cayrol, C.; Girard, J.P. Interleukin-33 (IL-33): A nuclear cytokine from the IL-1 family. Immunol. Rev. 2018, 281, 154–168. [Google Scholar] [CrossRef] [PubMed]
- Molofsky, A.B.; Savage, A.K.; Locksley, R.M. Interleukin-33 in Tissue Homeostasis, Injury, and Inflammation. Immunity 2015, 42, 1005–1019. [Google Scholar] [CrossRef] [PubMed]
- Cayrol, C.; Girard, J.P. IL-33: An alarmin cytokine with crucial roles in innate immunity, inflammation and allergy. Curr. Opin. Immunol. 2014, 31, 31–37. [Google Scholar] [CrossRef] [PubMed]
- Liew, F.Y.; Girard, J.P.; Turnquist, H.R. Interleukin-33 in health and disease. Nat. Rev. Immunol. 2016, 16, 676–689. [Google Scholar] [CrossRef] [PubMed]
- Dohi, E.; Choi, E.Y.; Rose, I.V.L.; Murata, A.S.; Chow, S.; Niwa, M.; Kano, S.I. Behavioral Changes in Mice Lacking Interleukin-33. eNeuro 2017, 4. [Google Scholar] [CrossRef] [PubMed]
- Wood, I.S.; Wang, B.; Trayhurn, P. IL-33, a recently identified interleukin-1 gene family member, is expressed in human adipocytes. Biochem. Biophys. Res. Commun. 2009, 384, 105–109. [Google Scholar] [CrossRef] [PubMed]
- Griesenauer, B.; Paczesny, S. The ST2/IL-33 Axis in Immune Cells during Inflammatory Diseases. Front. Immunol. 2017, 8, 475. [Google Scholar] [CrossRef] [PubMed]
- Miller, A.M.; Asquith, D.L.; Hueber, A.J.; Anderson, L.A.; Holmes, W.M.; McKenzie, A.N.; Xu, D.; Sattar, N.; McInnes, I.B.; Liew, F.Y. Interleukin-33 induces protective effects in adipose tissue inflammation during obesity in mice. Circ. Res. 2010, 107, 650–658. [Google Scholar] [CrossRef] [PubMed]
- Han, J.M.; Wu, D.; Denroche, H.C.; Yao, Y.; Verchere, C.B.; Levings, M.K. IL-33 Reverses an Obesity-Induced Deficit in Visceral Adipose Tissue ST2+ T Regulatory Cells and Ameliorates Adipose Tissue Inflammation and Insulin Resistance. J. Immunol. 2015, 194, 4777–4783. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharma, A.; Rudra, D. Emerging Functions of Regulatory T Cells in Tissue Homeostasis. Front. Immunol. 2018, 9, 883. [Google Scholar] [CrossRef] [PubMed]
- Wagner, N.M.; Brandhorst, G.; Czepluch, F.; Lankeit, M.; Eberle, C.; Herzberg, S.; Faustin, V.; Riggert, J.; Oellerich, M.; Hasenfuss, G.; et al. Circulating regulatory T cells are reduced in obesity and may identify subjects at increased metabolic and cardiovascular risk. Obesity 2013, 21, 461–468. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cipolletta, D.; Feuerer, M.; Li, A.; Kamei, N.; Lee, J.; Shoelson, S.E.; Benoist, C.; Mathis, D. PPAR-γ is a major driver of the accumulation and phenotype of adipose tissue Treg cells. Nature 2012, 486, 549–553. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miller, A.M.; Purves, D.; McConnachie, A.; Asquith, D.L.; Batty, G.D.; Burns, H.; Cavanagh, J.; Ford, I.; McLean, J.S.; Packard, C.J.; et al. Soluble ST2 associates with diabetes but not established cardiovascular risk factors: A new inflammatory pathway of relevance to diabetes? PLoS ONE 2012, 7, e47830. [Google Scholar] [CrossRef] [PubMed]
- Zeyda, M.; Wernly, B.; Demyanets, S.; Kaun, C.; Hammerle, M.; Hantusch, B.; Schranz, M.; Neuhofer, A.; Itariu, B.K.; Keck, M.; et al. Severe obesity increases adipose tissue expression of interleukin-33 and its receptor ST2, both predominantly detectable in endothelial cells of human adipose tissue. Int. J. Obes. 2013, 37, 658–665. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.H.; Zhang, R.C.; Hou, L.B.; Wang, K.J.; Ye, Z.N.; Huang, T.; Zhang, J.; Chen, X.; Kang, J.S. Distribution and clinical association of plasma soluble ST2 during the development of type 2 diabetes. Diabetes Res. Clin. Pract. 2016, 118, 140–145. [Google Scholar] [CrossRef] [PubMed]
- Celic, V.; Majstorovic, A.; Pencic-Popovic, B.; Sljivic, A.; Lopez-Andres, N.; Roy, I.; Escribano, E.; Beunza, M.; Melero, A.; Floridi, F.; et al. Soluble ST2 Levels and Left Ventricular Structure and Function in Patients with Metabolic Syndrome. Ann. Lab. Med. 2016, 36, 542–549. [Google Scholar] [CrossRef] [PubMed]
- Hams, E.; Bermingham, R.; Wurlod, F.A.; Hogan, A.E.; O’Shea, D.; Preston, R.J.; Rodewald, H.R.; McKenzie, A.N.; Fallon, P.G. The helminth T2 RNase omega1 promotes metabolic homeostasis in an IL-33- and group 2 innate lymphoid cell-dependent mechanism. FASEB J. 2016, 30, 824–835. [Google Scholar] [CrossRef] [PubMed]
- Ding, X.; Luo, Y.; Zhang, X.; Zheng, H.; Yang, X.; Yang, X.; Liu, M. IL-33-driven ILC2/eosinophil axis in fat is induced by sympathetic tone and suppressed by obesity. J. Endocrinol. 2016, 231, 35–48. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Odegaard, J.I.; Lee, M.W.; Sogawa, Y.; Bertholet, A.M.; Locksley, R.M.; Weinberg, D.E.; Kirichok, Y.; Deo, R.C.; Chawla, A. Perinatal Licensing of Thermogenesis by IL-33 and ST2. Cell 2016, 166, 841–854. [Google Scholar] [CrossRef] [PubMed]
- Hunter, C.A.; Jones, S.A. IL-6 as a keystone cytokine in health and disease. Nat. Immunol. 2015, 16, 448–457. [Google Scholar] [CrossRef] [PubMed]
- Garbers, C.; Heink, S.; Korn, T.; Rose-John, S. Interleukin-6: Designing specific therapeutics for a complex cytokine. Nat. Rev. Drug Discov. 2018, 17, 395–412. [Google Scholar] [CrossRef] [PubMed]
- Kishimoto, T. IL-6: From its discovery to clinical applications. Int. Immunol. 2010, 22, 347–352. [Google Scholar] [CrossRef] [PubMed]
- Erta, M.; Quintana, A.; Hidalgo, J. Interleukin-6, a major cytokine in the central nervous system. Int. J. Biol. Sci. 2012, 8, 1254–1266. [Google Scholar] [CrossRef] [PubMed]
- Pal, M.; Febbraio, M.A.; Whitham, M. From cytokine to myokine: The emerging role of interleukin-6 in metabolic regulation. Immunol. Cell Biol. 2014, 92, 331–339. [Google Scholar] [CrossRef] [PubMed]
- Schmidt-Arras, D.; Rose-John, S. IL-6 pathway in the liver: From physiopathology to therapy. J. Hepatol. 2016, 64, 1403–1415. [Google Scholar] [CrossRef] [PubMed]
- Scheller, J.; Chalaris, A.; Schmidt-Arras, D.; Rose-John, S. The pro- and anti-inflammatory properties of the cytokine interleukin-6. Biochim. Biophys. Acta 2011, 1813, 878–888. [Google Scholar] [CrossRef] [PubMed]
- Johnson, D.E.; O’Keefe, R.A.; Grandis, J.R. Targeting the IL-6/JAK/STAT3 signalling axis in cancer. Nat. Rev. Clin. Oncol. 2018, 15, 234–248. [Google Scholar] [CrossRef] [PubMed]
- Kishimoto, T.; Tanaka, T. Interleukin 6. In Encyclopedia of Inflammatory Diseases; Parnham, M., Ed.; Springer: Basel, Switzerland, 2015; pp. 1–8. [Google Scholar]
- Toft, A.D.; Falahati, A.; Steensberg, A. Source and kinetics of interleukin-6 in humans during exercise demonstrated by a minimally invasive model. Eur. J. Appl. Physiol. 2011, 111, 1351–1359. [Google Scholar] [CrossRef] [PubMed]
- Pazos, P.; Lima, L.; Casanueva, F.F.; Dieguez, C.; Garcia, M.C. Interleukin 6 deficiency modulates the hypothalamic expression of energy balance regulating peptides during pregnancy in mice. PLoS ONE 2013, 8, e72339. [Google Scholar] [CrossRef] [PubMed]
- Rose-John, S. Interleukin-6 Family Cytokines. Cold Spring Harb. Perspect. Biol. 2018, 10, a028415. [Google Scholar] [CrossRef] [PubMed]
- Rothaug, M.; Becker-Pauly, C.; Rose-John, S. The role of interleukin-6 signaling in nervous tissue. Biochim. Biophys. Acta 2016, 1863, 1218–1227. [Google Scholar] [CrossRef] [PubMed]
- Benrick, A.; Schele, E.; Pinnock, S.B.; Wernstedt-Asterholm, I.; Dickson, S.L.; Karlsson-Lindahl, L.; Jansson, J.O. Interleukin-6 gene knockout influences energy balance regulating peptides in the hypothalamic paraventricular and supraoptic nuclei. J. Neuroendocrinol. 2009, 21, 620–628. [Google Scholar] [CrossRef] [PubMed]
- Anesten, F.; Santos, C.; Gidestrand, E.; Schele, E.; Palsdottir, V.; Swedung-Wettervik, T.; Meister, B.; Patrycja Skibicka, K.; Jansson, J.O. Functional interleukin-6 receptor-α is located in tanycytes at the base of the third ventricle. J. Neuroendocrinol. 2017, 29. [Google Scholar] [CrossRef] [PubMed]
- Bastard, J.P.; Jardel, C.; Bruckert, E.; Blondy, P.; Capeau, J.; Laville, M.; Vidal, H.; Hainque, B. Elevated levels of interleukin 6 are reduced in serum and subcutaneous adipose tissue of obese women after weight loss. J. Clin. Endocrinol. Metab. 2000, 85, 3338–3342. [Google Scholar] [CrossRef] [PubMed]
- Pradhan, A.D.; Manson, J.E.; Rifai, N.; Buring, J.E.; Ridker, P.M. C-reactive protein, interleukin 6, and risk of developing type 2 diabetes mellitus. JAMA 2001, 286, 327–334. [Google Scholar] [CrossRef] [PubMed]
- Carey, A.L.; Bruce, C.R.; Sacchetti, M.; Anderson, M.J.; Olsen, D.B.; Saltin, B.; Hawley, J.A.; Febbraio, M.A. Interleukin-6 and tumor necrosis factor-α are not increased in patients with Type 2 diabetes: Evidence that plasma interleukin-6 is related to fat mass and not insulin responsiveness. Diabetologia 2004, 47, 1029–1037. [Google Scholar] [CrossRef] [PubMed]
- Esposito, K.; Pontillo, A.; Di Palo, C.; Giugliano, G.; Masella, M.; Marfella, R.; Giugliano, D. Effect of weight loss and lifestyle changes on vascular inflammatory markers in obese women: A randomized trial. JAMA 2003, 289, 1799–1804. [Google Scholar] [CrossRef] [PubMed]
- Tournadre, A.; Pereira, B.; Dubost, J.J.; Rincheval, N.; Rat, A.C.; Combe, B.; Soubrier, M. Management of dyslipidaemia in high-risk patients with recent-onset rheumatoid arthritis: Targets still not met despite specific recommendations. Results from the ESPOIR cohort during the first five years of follow-up. Clin. Exp. Rheumatol. 2017, 35, 296–302. [Google Scholar] [PubMed]
- Wallenius, V.; Wallenius, K.; Ahren, B.; Rudling, M.; Carlsten, H.; Dickson, S.L.; Ohlsson, C.; Jansson, J.O. Interleukin-6-deficient mice develop mature-onset obesity. Nat. Med. 2002, 8, 75–79. [Google Scholar] [CrossRef] [PubMed]
- Wernstedt, I.; Edgley, A.; Berndtsson, A.; Faldt, J.; Bergstrom, G.; Wallenius, V.; Jansson, J.O. Reduced stress- and cold-induced increase in energy expenditure in interleukin-6-deficient mice. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2006, 291, R551–R557. [Google Scholar] [CrossRef] [PubMed]
- Faldt, J.; Wernstedt, I.; Fitzgerald, S.M.; Wallenius, K.; Bergstrom, G.; Jansson, J.O. Reduced exercise endurance in interleukin-6-deficient mice. Endocrinology 2004, 145, 2680–2686. [Google Scholar] [CrossRef] [PubMed]
- Ruderman, N.B.; Keller, C.; Richard, A.M.; Saha, A.K.; Luo, Z.; Xiang, X.; Giralt, M.; Ritov, V.B.; Menshikova, E.V.; Kelley, D.E.; et al. Interleukin-6 regulation of AMP-activated protein kinase. Potential role in the systemic response to exercise and prevention of the metabolic syndrome. Diabetes 2006, 55 (Suppl. 2), S48–S54. [Google Scholar] [CrossRef]
- Matthews, V.B.; Allen, T.L.; Risis, S.; Chan, M.H.; Henstridge, D.C.; Watson, N.; Zaffino, L.A.; Babb, J.R.; Boon, J.; Meikle, P.J.; et al. Interleukin-6-deficient mice develop hepatic inflammation and systemic insulin resistance. Diabetologia 2010, 53, 2431–2441. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wunderlich, F.T.; Strohle, P.; Konner, A.C.; Gruber, S.; Tovar, S.; Bronneke, H.S.; Juntti-Berggren, L.; Li, L.S.; van Rooijen, N.; Libert, C.; et al. Interleukin-6 signaling in liver-parenchymal cells suppresses hepatic inflammation and improves systemic insulin action. Cell Metab. 2010, 12, 237–249. [Google Scholar] [CrossRef] [PubMed]
- Mauer, J.; Chaurasia, B.; Goldau, J.; Vogt, M.C.; Ruud, J.; Nguyen, K.D.; Theurich, S.; Hausen, A.C.; Schmitz, J.; Bronneke, H.S.; et al. Signaling by IL-6 promotes alternative activation of macrophages to limit endotoxemia and obesity-associated resistance to insulin. Nat. Immunol. 2014, 15, 423–430. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Braune, J.; Weyer, U.; Hobusch, C.; Mauer, J.; Bruning, J.C.; Bechmann, I.; Gericke, M. IL-6 Regulates M2 Polarization and Local Proliferation of Adipose Tissue Macrophages in Obesity. J. Immunol. 2017, 198, 2927–2934. [Google Scholar] [CrossRef] [PubMed]
- Ellingsgaard, H.; Hauselmann, I.; Schuler, B.; Habib, A.M.; Baggio, L.L.; Meier, D.T.; Eppler, E.; Bouzakri, K.; Wueest, S.; Muller, Y.D.; et al. Interleukin-6 enhances insulin secretion by increasing glucagon-like peptide-1 secretion from L cells and α cells. Nat. Med. 2011, 17, 1481–1489. [Google Scholar] [CrossRef] [PubMed]
- Wueest, S.; Laesser, C.I.; Boni-Schnetzler, M.; Item, F.; Lucchini, F.C.; Borsigova, M.; Muller, W.; Donath, M.Y.; Konrad, D. IL-6-Type Cytokine Signaling in Adipocytes Induces Intestinal GLP-1 Secretion. Diabetes 2018, 67, 36–45. [Google Scholar] [CrossRef] [PubMed]
- Juttler, E.; Tarabin, V.; Schwaninger, M. Interleukin-6 (IL-6): A possible neuromodulator induced by neuronal activity. Neuroscientist 2002, 8, 268–275. [Google Scholar] [CrossRef] [PubMed]
- Rohde, T.; MacLean, D.A.; Richter, E.A.; Kiens, B.; Pedersen, B.K. Prolonged submaximal eccentric exercise is associated with increased levels of plasma IL-6. Am. J. Physiol. 1997, 273, E85–E91. [Google Scholar] [CrossRef] [PubMed]
- Ropelle, E.R.; Flores, M.B.; Cintra, D.E.; Rocha, G.Z.; Pauli, J.R.; Morari, J.; de Souza, C.T.; Moraes, J.C.; Prada, P.O.; Guadagnini, D.; et al. IL-6 and IL-10 anti-inflammatory activity links exercise to hypothalamic insulin and leptin sensitivity through IKKβ and ER stress inhibition. PLoS Biol. 2010, 8, e1000465. [Google Scholar] [CrossRef] [PubMed]
- Wallenius, K.; Wallenius, V.; Sunter, D.; Dickson, S.L.; Jansson, J.O. Intracerebroventricular interleukin-6 treatment decreases body fat in rats. Biochem. Biophys. Res. Commun. 2002, 293, 560–565. [Google Scholar] [CrossRef]
- Li, G.; Klein, R.L.; Matheny, M.; King, M.A.; Meyer, E.M.; Scarpace, P.J. Induction of uncoupling protein 1 by central interleukin-6 gene delivery is dependent on sympathetic innervation of brown adipose tissue and underlies one mechanism of body weight reduction in rats. Neuroscience 2002, 115, 879–889. [Google Scholar] [CrossRef]
- Quintana, A.; Erta, M.; Ferrer, B.; Comes, G.; Giralt, M.; Hidalgo, J. Astrocyte-specific deficiency of interleukin-6 and its receptor reveal specific roles in survival, body weight and behavior. Brain Behav. Immun. 2013, 27, 162–173. [Google Scholar] [CrossRef] [PubMed]
- Senaris, R.M.; Trujillo, M.L.; Navia, B.; Comes, G.; Ferrer, B.; Giralt, M.; Hidalgo, J. Interleukin-6 regulates the expression of hypothalamic neuropeptides involved in body weight in a gender-dependent way. J. Neuroendocrinol. 2011, 23, 675–686. [Google Scholar] [CrossRef] [PubMed]
- Timper, K.; Denson, J.L.; Steculorum, S.M.; Heilinger, C.; Engstrom-Ruud, L.; Wunderlich, C.M.; Rose-John, S.; Wunderlich, F.T.; Bruning, J.C. IL-6 Improves Energy and Glucose Homeostasis in Obesity via Enhanced Central IL-6 trans-Signaling. Cell Rep. 2017, 19, 267–280. [Google Scholar] [CrossRef] [PubMed]
- Shirazi, R.; Palsdottir, V.; Collander, J.; Anesten, F.; Vogel, H.; Langlet, F.; Jaschke, A.; Schurmann, A.; Prevot, V.; Shao, R.; et al. Glucagon-like peptide 1 receptor induced suppression of food intake, and body weight is mediated by central IL-1 and IL-6. Proc. Natl. Acad. Sci. USA 2013, 110, 16199–16204. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Conti, B.; Tabarean, I.; Andrei, C.; Bartfai, T. Cytokines and fever. Front. Biosci. 2004, 9, 1433–1449. [Google Scholar] [CrossRef] [PubMed]
- Chai, Z.; Gatti, S.; Toniatti, C.; Poli, V.; Bartfai, T. Interleukin (IL)-6 gene expression in the central nervous system is necessary for fever response to lipopolysaccharide or IL-1 β: A study on IL-6-deficient mice. J. Exp. Med. 1996, 183, 311–316. [Google Scholar] [CrossRef] [PubMed]
- Kozak, W.; Kluger, M.J.; Soszynski, D.; Conn, C.A.; Rudolph, K.; Leon, L.R.; Zheng, H. IL-6 and IL-1 β in fever. Studies using cytokine-deficient (knockout) mice. Ann. N. Y. Acad. Sci. 1998, 856, 33–47. [Google Scholar] [CrossRef] [PubMed]
- Eskilsson, A.; Mirrasekhian, E.; Dufour, S.; Schwaninger, M.; Engblom, D.; Blomqvist, A. Immune-induced fever is mediated by IL-6 receptors on brain endothelial cells coupled to STAT3-dependent induction of brain endothelial prostaglandin synthesis. J. Neurosci. 2014, 34, 15957–15961. [Google Scholar] [CrossRef] [PubMed]
- Ferrer, B.; Navia, B.; Giralt, M.; Comes, G.; Carrasco, J.; Molinero, A.; Quintana, A.; Senaris, R.M.; Hidalgo, J. Muscle-specific interleukin-6 deletion influences body weight and body fat in a sex-dependent manner. Brain Behav. Immun. 2014, 40, 121–130. [Google Scholar] [CrossRef] [PubMed]
- Egecioglu, E.; Anesten, F.; Schéle, E.; Palsdottir, V. Interleukin-6 is important for regulation of core body temperature during long-term cold exposure in mice. Biomed. Rep. 2018, 9, 206–212. [Google Scholar] [CrossRef]
- Knudsen, J.G.; Murholm, M.; Carey, A.L.; Bienso, R.S.; Basse, A.L.; Allen, T.L.; Hidalgo, J.; Kingwell, B.A.; Febbraio, M.A.; Hansen, J.B.; et al. Role of IL-6 in exercise training- and cold-induced UCP1 expression in subcutaneous white adipose tissue. PLoS ONE 2014, 9, e84910. [Google Scholar] [CrossRef] [PubMed]
- Sidossis, L.S.; Porter, C.; Saraf, M.K.; Borsheim, E.; Radhakrishnan, R.S.; Chao, T.; Ali, A.; Chondronikola, M.; Mlcak, R.; Finnerty, C.C.; et al. Browning of Subcutaneous White Adipose Tissue in Humans after Severe Adrenergic Stress. Cell Metab. 2015, 22, 219–227. [Google Scholar] [CrossRef] [PubMed]
- Abdullahi, A.; Chen, P.; Stanojcic, M.; Sadri, A.R.; Coburn, N.; Jeschke, M.G. IL-6 Signal from the Bone Marrow is Required for the Browning of White Adipose Tissue Post Burn Injury. Shock 2017, 47, 33–39. [Google Scholar] [CrossRef] [PubMed]
- Grabstein, K.H.; Eisenman, J.; Shanebeck, K.; Rauch, C.; Srinivasan, S.; Fung, V.; Beers, C.; Richardson, J.; Schoenborn, M.A.; Ahdieh, M.; et al. Cloning of a T cell growth factor that interacts with the β chain of the interleukin-2 receptor. Science 1994, 264, 965–968. [Google Scholar] [CrossRef] [PubMed]
- Castillo, E.F.; Schluns, K.S. Regulating the immune system via IL-15 transpresentation. Cytokine 2012, 59, 479–490. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mathieu, C.; Beltra, J.C.; Charpentier, T.; Bourbonnais, S.; Di Santo, J.P.; Lamarre, A.; Decaluwe, H. IL-2 and IL-15 regulate CD8+ memory T-cell differentiation but are dispensable for protective recall responses. Eur. J. Immunol. 2015, 45, 3324–3338. [Google Scholar] [CrossRef] [PubMed]
- Desbois, M.; Le Vu, P.; Coutzac, C.; Marcheteau, E.; Beal, C.; Terme, M.; Gey, A.; Morisseau, S.; Teppaz, G.; Boselli, L.; et al. IL-15 Trans-Signaling with the Superagonist RLI Promotes Effector/Memory CD8+ T Cell Responses and Enhances Antitumor Activity of PD-1 Antagonists. J. Immunol. 2016, 197, 168–178. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vicari, A.P.; Schoepfer, A.M.; Meresse, B.; Goffin, L.; Leger, O.; Josserand, S.; Guegan, N.; Yousefi, S.; Straumann, A.; Cerf-Bensussan, N.; et al. Discovery and characterization of a novel humanized anti-IL-15 antibody and its relevance for the treatment of refractory celiac disease and eosinophilic esophagitis. MAbs 2017, 9, 927–944. [Google Scholar] [CrossRef] [PubMed]
- Schluns, K.S.; Stoklasek, T.; Lefrancois, L. The roles of interleukin-15 receptor α: Trans-presentation, receptor component, or both? Int. J. Biochem. Cell Biol. 2005, 37, 1567–1571. [Google Scholar] [CrossRef] [PubMed]
- Bergamaschi, C.; Rosati, M.; Jalah, R.; Valentin, A.; Kulkarni, V.; Alicea, C.; Zhang, G.M.; Patel, V.; Felber, B.K.; Pavlakis, G.N. Intracellular interaction of interleukin-15 with its receptor α during production leads to mutual stabilization and increased bioactivity. J. Biol. Chem. 2008, 283, 4189–4199. [Google Scholar] [CrossRef] [PubMed]
- Tamzalit, F.; Barbieux, I.; Plet, A.; Heim, J.; Nedellec, S.; Morisseau, S.; Jacques, Y.; Mortier, E. IL-15.IL-15Rα complex shedding following trans-presentation is essential for the survival of IL-15 responding NK and T cells. Proc. Natl. Acad. Sci. USA 2014, 111, 8565–8570. [Google Scholar] [CrossRef] [PubMed]
- Bergamaschi, C.; Bear, J.; Rosati, M.; Beach, R.K.; Alicea, C.; Sowder, R.; Chertova, E.; Rosenberg, S.A.; Felber, B.K.; Pavlakis, G.N. Circulating IL-15 exists as heterodimeric complex with soluble IL-15Rα in human and mouse serum. Blood 2012, 120, e1–e8. [Google Scholar] [CrossRef] [PubMed]
- Pistilli, E.E.; Bogdanovich, S.; Garton, F.; Yang, N.; Gulbin, J.P.; Conner, J.D.; Anderson, B.G.; Quinn, L.S.; North, K.; Ahima, R.S.; et al. Loss of IL-15 receptor α alters the endurance, fatigability, and metabolic characteristics of mouse fast skeletal muscles. J. Clin. Investig. 2011, 121, 3120–3132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anderson, B.G.; Quinn, L.S. Free IL-15 Is More Abundant than IL-15 Complexed with Soluble IL-15 Receptor-α in Murine Serum: Implications for the Mechanism of IL-15 Secretion. Endocrinology 2016, 157, 1315–1320. [Google Scholar] [CrossRef] [PubMed]
- Argiles, J.M.; Lopez-Soriano, F.J.; Busquets, S. Therapeutic potential of interleukin-15: A myokine involved in muscle wasting and adiposity. Drug Discov. Today 2009, 14, 208–213. [Google Scholar] [CrossRef] [PubMed]
- Quinn, L.S.; Anderson, B.G. Interleukin-15, IL-15 Receptor-A, and Obesity: Concordance of Laboratory Animal and Human Genetic Studies. J. Obes. 2011, 2011, 456347. [Google Scholar] [CrossRef] [PubMed]
- Duan, Y.; Li, F.; Wang, W.; Guo, Q.; Wen, C.; Li, Y.; Yin, Y. Interleukin-15 in obesity and metabolic dysfunction: Current understanding and future perspectives. Obes. Rev. 2017, 18, 1147–1158. [Google Scholar] [CrossRef] [PubMed]
- Quinn, L.S.; Haugk, K.L.; Grabstein, K.H. Interleukin-15: A novel anabolic cytokine for skeletal muscle. Endocrinology 1995, 136, 3669–3672. [Google Scholar] [CrossRef] [PubMed]
- Pistilli, E.E.; Quinn, L.S. From anabolic to oxidative: Reconsidering the roles of IL-15 and IL-15Rα in skeletal muscle. Exerc. Sport Sci. Rev. 2013, 41, 100–106. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, A.R.; Mounier, R.; Plomgaard, P.; Mortensen, O.H.; Penkowa, M.; Speerschneider, T.; Pilegaard, H.; Pedersen, B.K. Expression of interleukin-15 in human skeletal muscle effect of exercise and muscle fibre type composition. J. Physiol. 2007, 584, 305–312. [Google Scholar] [CrossRef] [PubMed]
- Dieli-Conwright, C.M.; Spektor, T.M.; Rice, J.C.; Sattler, F.R.; Schroeder, E.T. Hormone therapy attenuates exercise-induced skeletal muscle damage in postmenopausal women. J. Appl. Physiol. 2009, 107, 853–858. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Molanouri Shamsi, M.; Hassan, Z.H.; Gharakhanlou, R.; Quinn, L.S.; Azadmanesh, K.; Baghersad, L.; Isanejad, A.; Mahdavi, M. Expression of interleukin-15 and inflammatory cytokines in skeletal muscles of STZ-induced diabetic rats: Effect of resistance exercise training. Endocrine 2014, 46, 60–69. [Google Scholar] [CrossRef] [PubMed]
- Molanouri Shamsi, M.; Hassan, Z.M.; Quinn, L.S.; Gharakhanlou, R.; Baghersad, L.; Mahdavi, M. Time course of IL-15 expression after acute resistance exercise in trained rats: Effect of diabetes and skeletal muscle phenotype. Endocrine 2015, 49, 396–403. [Google Scholar] [CrossRef] [PubMed]
- Riechman, S.E.; Balasekaran, G.; Roth, S.M.; Ferrell, R.E. Association of interleukin-15 protein and interleukin-15 receptor genetic variation with resistance exercise training responses. J. Appl. Physiol. 2004, 97, 2214–2219. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tamura, Y.; Watanabe, K.; Kantani, T.; Hayashi, J.; Ishida, N.; Kaneki, M. Upregulation of circulating IL-15 by treadmill running in healthy individuals: Is IL-15 an endocrine mediator of the beneficial effects of endurance exercise? Endocr. J. 2011, 58, 211–215. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quinn, L.S.; Anderson, B.G.; Conner, J.D.; Wolden-Hanson, T.; Marcell, T.J. IL-15 is required for postexercise induction of the pro-oxidative mediators PPARdelta and SIRT1 in male mice. Endocrinology 2014, 155, 143–155. [Google Scholar] [CrossRef] [PubMed]
- Rinnov, A.; Yfanti, C.; Nielsen, S.; Akerstrom, T.C.; Peijs, L.; Zankari, A.; Fischer, C.P.; Pedersen, B.K. Endurance training enhances skeletal muscle interleukin-15 in human male subjects. Endocrine 2014, 45, 271–278. [Google Scholar] [CrossRef] [PubMed]
- Catoire, M.; Mensink, M.; Kalkhoven, E.; Schrauwen, P.; Kersten, S. Identification of human exercise-induced myokines using secretome analysis. Physiol. Genom. 2014, 46, 256–267. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pierce, J.R.; Maples, J.M.; Hickner, R.C. IL-15 concentrations in skeletal muscle and subcutaneous adipose tissue in lean and obese humans: Local effects of IL-15 on adipose tissue lipolysis. Am. J. Physiol. Endocrinol. Metab. 2015, 308, E1131–E1139. [Google Scholar] [CrossRef] [PubMed]
- Ye, J. Beneficial metabolic activities of inflammatory cytokine interleukin 15 in obesity and type 2 diabetes. Front. Med. 2015, 9, 139–145. [Google Scholar] [CrossRef] [PubMed]
- Barra, N.G.; Reid, S.; MacKenzie, R.; Werstuck, G.; Trigatti, B.L.; Richards, C.; Holloway, A.C.; Ashkar, A.A. Interleukin-15 contributes to the regulation of murine adipose tissue and human adipocytes. Obesity 2010, 18, 1601–1607. [Google Scholar] [CrossRef] [PubMed]
- Barra, N.G.; Palanivel, R.; Denou, E.; Chew, M.V.; Gillgrass, A.; Walker, T.D.; Kong, J.; Richards, C.D.; Jordana, M.; Collins, S.M.; et al. Interleukin-15 modulates adipose tissue by altering mitochondrial mass and activity. PLoS ONE 2014, 9, e114799. [Google Scholar] [CrossRef] [PubMed]
- Quinn, L.S.; Anderson, B.G.; Strait-Bodey, L.; Stroud, A.M.; Argiles, J.M. Oversecretion of interleukin-15 from skeletal muscle reduces adiposity. Am. J. Physiol. Endocrinol. Metab. 2009, 296, E191–E202. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quinn, L.S.; Anderson, B.G.; Conner, J.D.; Wolden-Hanson, T. IL-15 overexpression promotes endurance, oxidative energy metabolism, and muscle PPARdelta, SIRT1, PGC-1α, and PGC-1β expression in male mice. Endocrinology 2013, 154, 232–245. [Google Scholar] [CrossRef] [PubMed]
- Quinn, L.S.; Anderson, B.G.; Conner, J.D.; Pistilli, E.E.; Wolden-Hanson, T. Overexpression of interleukin-15 in mice promotes resistance to diet-induced obesity, increased insulin sensitivity, and markers of oxidative skeletal muscle metabolism. Int. J. Interferon Cytokine Mediat. Res. 2011, 3, 29–42. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, A.R.; Hojman, P.; Erikstrup, C.; Fischer, C.P.; Plomgaard, P.; Mounier, R.; Mortensen, O.H.; Broholm, C.; Taudorf, S.; Krogh-Madsen, R.; et al. Association between interleukin-15 and obesity: Interleukin-15 as a potential regulator of fat mass. J. Clin. Endocrinol. Metab. 2008, 93, 4486–4493. [Google Scholar] [CrossRef] [PubMed]
- Quinn, L.S.; Strait-Bodey, L.; Anderson, B.G.; Argiles, J.M.; Havel, P.J. Interleukin-15 stimulates adiponectin secretion by 3T3-L1 adipocytes: Evidence for a skeletal muscle-to-fat signaling pathway. Cell Biol. Int. 2005, 29, 449–457. [Google Scholar] [CrossRef] [PubMed]
- Al-Attar, A.; Presnell, S.R.; Clasey, J.L.; Long, D.E.; Walton, R.G.; Sexton, M.; Starr, M.E.; Kern, P.A.; Peterson, C.A.; Lutz, C.T. Human Body Composition and Immunity: Visceral Adipose Tissue Produces IL-15 and Muscle Strength Inversely Correlates with NK Cell Function in Elderly Humans. Front. Immunol. 2018, 9, 440. [Google Scholar] [CrossRef] [PubMed]
- Fehniger, T.A.; Suzuki, K.; Ponnappan, A.; VanDeusen, J.B.; Cooper, M.A.; Florea, S.M.; Freud, A.G.; Robinson, M.L.; Durbin, J.; Caligiuri, M.A. Fatal leukemia in interleukin 15 transgenic mice follows early expansions in natural killer and memory phenotype CD8+ T cells. J. Exp. Med. 2001, 193, 219–231. [Google Scholar] [CrossRef] [PubMed]
- He, Y.; Wu, X.; Khan, R.S.; Kastin, A.J.; Cornelissen-Guillaume, G.G.; Hsuchou, H.; Robert, B.; Halberg, F.; Pan, W. IL-15 receptor deletion results in circadian changes of locomotor and metabolic activity. J. Mol. Neurosci. 2010, 41, 315–321. [Google Scholar] [CrossRef] [PubMed]
- Loro, E.; Seifert, E.L.; Moffat, C.; Romero, F.; Mishra, M.K.; Sun, Z.; Krajacic, P.; Anokye-Danso, F.; Summer, R.S.; Ahima, R.S.; et al. IL-15Rα is a determinant of muscle fuel utilization, and its loss protects against obesity. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2015, 309, R835–R844. [Google Scholar] [CrossRef] [PubMed]
- Lacraz, G.; Rakotoarivelo, V.; Labbe, S.M.; Vernier, M.; Noll, C.; Mayhue, M.; Stankova, J.; Schwertani, A.; Grenier, G.; Carpentier, A.; et al. Deficiency of Interleukin-15 Confers Resistance to Obesity by Diminishing Inflammation and Enhancing the Thermogenic Function of Adipose Tissues. PLoS ONE 2016, 11, e0162995. [Google Scholar]
- Barra, N.G.; Chew, M.V.; Reid, S.; Ashkar, A.A. Interleukin-15 treatment induces weight loss independent of lymphocytes. PLoS ONE 2012, 7, e39553. [Google Scholar] [CrossRef] [PubMed]
- Almendro, V.; Fuster, G.; Busquets, S.; Ametller, E.; Figueras, M.; Argiles, J.M.; Lopez-Soriano, F.J. Effects of IL-15 on rat brown adipose tissue: Uncoupling proteins and PPARs. Obesity 2008, 16, 285–289. [Google Scholar] [CrossRef] [PubMed]
- Sun, H.; Ma, Y.; Gao, M.; Liu, D. IL-15/sIL-15Rα gene transfer induces weight loss and improves glucose homeostasis in obese mice. Gene Ther. 2016, 23, 349–356. [Google Scholar] [CrossRef] [PubMed]
- Novick, D.; Kim, S.; Kaplanski, G.; Dinarello, C.A. Interleukin-18, more than a Th1 cytokine. Semin. Immunol. 2013, 25, 439–448. [Google Scholar] [CrossRef] [PubMed]
- Afonina, I.S.; Muller, C.; Martin, S.J.; Beyaert, R. Proteolytic Processing of Interleukin-1 Family Cytokines: Variations on a Common Theme. Immunity 2015, 42, 991–1004. [Google Scholar] [CrossRef] [PubMed]
- Latz, E.; Xiao, T.S.; Stutz, A. Activation and regulation of the inflammasomes. Nat. Rev. Immunol. 2013, 13, 397–411. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murphy, A.J.; Kraakman, M.J.; Kammoun, H.L.; Dragoljevic, D.; Lee, M.K.; Lawlor, K.E.; Wentworth, J.M.; Vasanthakumar, A.; Gerlic, M.; Whitehead, L.W.; et al. IL-18 Production from the NLRP1 Inflammasome Prevents Obesity and Metabolic Syndrome. Cell Metab. 2016, 23, 155–164. [Google Scholar] [CrossRef] [PubMed]
- Pazos, P.; Lima, L.; Tovar, S.; Gonzalez-Touceda, D.; Dieguez, C.; Garcia, M.C. Divergent responses to thermogenic stimuli in BAT and subcutaneous adipose tissue from interleukin 18 and interleukin 18 receptor 1-deficient mice. Sci. Rep. 2015, 5, 17977. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.K.; Kim, S.H.; Lewis, E.C.; Azam, T.; Reznikov, L.L.; Dinarello, C.A. Differences in signaling pathways by IL-1β and IL-18. Proc. Natl. Acad. Sci. USA 2004, 101, 8815–8820. [Google Scholar] [CrossRef] [PubMed]
- Lindegaard, B.; Matthews, V.B.; Brandt, C.; Hojman, P.; Allen, T.L.; Estevez, E.; Watt, M.J.; Bruce, C.R.; Mortensen, O.H.; Syberg, S.; et al. Interleukin-18 activates skeletal muscle AMPK and reduces weight gain and insulin resistance in mice. Diabetes 2013, 62, 3064–3074. [Google Scholar] [CrossRef] [PubMed]
- Alboni, S.; Montanari, C.; Benatti, C.; Blom, J.M.; Simone, M.L.; Brunello, N.; Caggia, F.; Guidotti, G.; Marcondes, M.C.; Sanchez-Alavez, M.; et al. Constitutive and LPS-regulated expression of interleukin-18 receptor β variants in the mouse brain. Brain Behav. Immun. 2011, 25, 483–493. [Google Scholar] [CrossRef] [PubMed]
- Esposito, K.; Pontillo, A.; Ciotola, M.; Di Palo, C.; Grella, E.; Nicoletti, G.; Giugliano, D. Weight loss reduces interleukin-18 levels in obese women. J. Clin. Endocrinol. Metab. 2002, 87, 3864–3866. [Google Scholar] [CrossRef] [PubMed]
- Hung, J.; McQuillan, B.M.; Chapman, C.M.; Thompson, P.L.; Beilby, J.P. Elevated interleukin-18 levels are associated with the metabolic syndrome independent of obesity and insulin resistance. Arterioscler. Thromb. Vasc. Biol. 2005, 25, 1268–1273. [Google Scholar] [CrossRef] [PubMed]
- Netea, M.G.; Joosten, L.A.; Lewis, E.; Jensen, D.R.; Voshol, P.J.; Kullberg, B.J.; Tack, C.J.; van Krieken, H.; Kim, S.H.; Stalenhoef, A.F.; et al. Deficiency of interleukin-18 in mice leads to hyperphagia, obesity and insulin resistance. Nat. Med. 2006, 12, 650–656. [Google Scholar] [CrossRef] [PubMed]
- Zorrilla, E.P.; Conti, B. Interleukin-18 null mutation increases weight and food intake and reduces energy expenditure and lipid substrate utilization in high-fat diet fed mice. Brain Behav. Immun. 2014, 37, 45–53. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zorrilla, E.P.; Sanchez-Alavez, M.; Sugama, S.; Brennan, M.; Fernandez, R.; Bartfai, T.; Conti, B. Interleukin-18 controls energy homeostasis by suppressing appetite and feed efficiency. Proc. Natl. Acad. Sci. USA 2007, 104, 11097–11102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zilverschoon, G.R.; Tack, C.J.; Joosten, L.A.; Kullberg, B.J.; van der Meer, J.W.; Netea, M.G. Interleukin-18 resistance in patients with obesity and type 2 diabetes mellitus. Int. J. Obes. 2008, 32, 1407–1414. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Esposito, K.; Nappo, F.; Giugliano, F.; Di Palo, C.; Ciotola, M.; Barbieri, M.; Paolisso, G.; Giugliano, D. Meal modulation of circulating interleukin 18 and adiponectin concentrations in healthy subjects and in patients with type 2 diabetes mellitus. Am. J. Clin. Nutr. 2003, 78, 1135–1140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rajbhandari, P.; Thomas, B.J.; Feng, A.C.; Hong, C.; Wang, J.; Vergnes, L.; Sallam, T.; Wang, B.; Sandhu, J.; Seldin, M.M.; et al. IL-10 Signaling Remodels Adipose Chromatin Architecture to Limit Thermogenesis and Energy Expenditure. Cell 2018, 172, 218–233. [Google Scholar] [CrossRef] [PubMed]
- Couper, K.N.; Blount, D.G.; Riley, E.M. IL-10: The master regulator of immunity to infection. J. Immunol. 2008, 180, 5771–5777. [Google Scholar] [CrossRef] [PubMed]
- Saraiva, M.; O’Garra, A. The regulation of IL-10 production by immune cells. Nat. Rev. Immunol. 2010, 10, 170–181. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Commins, S.; Steinke, J.W.; Borish, L. The extended IL-10 superfamily: IL-10, IL-19, IL-20, IL-22, IL-24, IL-26, IL-28, and IL-29. J. Allergy Clin. Immunol. 2008, 121, 1108–1111. [Google Scholar] [CrossRef] [PubMed]
- Moore, K.W.; de Waal Malefyt, R.; Coffman, R.L.; O’Garra, A. Interleukin-10 and the interleukin-10 receptor. Annu. Rev. Immunol. 2001, 19, 683–765. [Google Scholar] [CrossRef] [PubMed]
- Verma, R.; Balakrishnan, L.; Sharma, K.; Khan, A.A.; Advani, J.; Gowda, H.; Tripathy, S.P.; Suar, M.; Pandey, A.; Gandotra, S.; et al. A network map of Interleukin-10 signaling pathway. J. Cell Commun. Signal. 2016, 10, 61–67. [Google Scholar] [CrossRef] [PubMed]
- Lang, R.; Patel, D.; Morris, J.J.; Rutschman, R.L.; Murray, P.J. Shaping gene expression in activated and resting primary macrophages by IL-10. J. Immunol. 2002, 169, 2253–2263. [Google Scholar] [CrossRef] [PubMed]
- Bluher, M.; Fasshauer, M.; Tonjes, A.; Kratzsch, J.; Schon, M.R.; Paschke, R. Association of interleukin-6, C-reactive protein, interleukin-10 and adiponectin plasma concentrations with measures of obesity, insulin sensitivity and glucose metabolism. Exp. Clin. Endocrinol. Diabetes 2005, 113, 534–537. [Google Scholar] [CrossRef] [PubMed]
- Manigrasso, M.R.; Ferroni, P.; Santilli, F.; Taraborelli, T.; Guagnano, M.T.; Michetti, N.; Davi, G. Association between circulating adiponectin and interleukin-10 levels in android obesity: Effects of weight loss. J. Clin. Endocrinol. Metab. 2005, 90, 5876–5879. [Google Scholar] [CrossRef] [PubMed]
- Esposito, K.; Pontillo, A.; Giugliano, F.; Giugliano, G.; Marfella, R.; Nicoletti, G.; Giugliano, D. Association of low interleukin-10 levels with the metabolic syndrome in obese women. J. Clin. Endocrinol. Metab. 2003, 88, 1055–1058. [Google Scholar] [CrossRef] [PubMed]
- Charles, B.A.; Doumatey, A.; Huang, H.; Zhou, J.; Chen, G.; Shriner, D.; Adeyemo, A.; Rotimi, C.N. The roles of IL-6, IL-10, and IL-1RA in obesity and insulin resistance in African-Americans. J. Clin. Endocrinol. Metab. 2011, 96, E2018–E2022. [Google Scholar] [CrossRef] [PubMed]
- Juge-Aubry, C.E.; Somm, E.; Pernin, A.; Alizadeh, N.; Giusti, V.; Dayer, J.M.; Meier, C.A. Adipose tissue is a regulated source of interleukin-10. Cytokine 2005, 29, 270–274. [Google Scholar] [PubMed]
- Verthelyi, D.; Klinman, D.M. Sex hormone levels correlate with the activity of cytokine-secreting cells in vivo. Immunology 2000, 100, 384–390. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stowe, R.P.; Peek, M.K.; Cutchin, M.P.; Goodwin, J.S. Plasma cytokine levels in a population-based study: Relation to age and ethnicity. J. Gerontol. A Biol. Sci. Med. Sci. 2010, 65, 429–433. [Google Scholar] [CrossRef] [PubMed]
- Van Exel, E.; Gussekloo, J.; de Craen, A.J.; Frolich, M.; Bootsma-Van Der Wiel, A.; Westendorp, R.G. Low production capacity of interleukin-10 associates with the metabolic syndrome and type 2 diabetes: The Leiden 85-Plus Study. Diabetes 2002, 51, 1088–1092. [Google Scholar] [CrossRef] [PubMed]
- Petersen, A.M.; Pedersen, B.K. The anti-inflammatory effect of exercise. J. Appl. Physiol. 2005, 98, 1154–1162. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clementi, A.H.; Gaudy, A.M.; van Rooijen, N.; Pierce, R.H.; Mooney, R.A. Loss of Kupffer cells in diet-induced obesity is associated with increased hepatic steatosis, STAT3 signaling, and further decreases in insulin signaling. Biochim. Biophys. Acta 2009, 1792, 1062–1072. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Den Boer, M.A.; Voshol, P.J.; Schroder-van der Elst, J.P.; Korsheninnikova, E.; Ouwens, D.M.; Kuipers, F.; Havekes, L.M.; Romijn, J.A. Endogenous interleukin-10 protects against hepatic steatosis but does not improve insulin sensitivity during high-fat feeding in mice. Endocrinology 2006, 147, 4553–4558. [Google Scholar] [CrossRef] [PubMed]
- Miller, A.M.; Wang, H.; Bertola, A.; Park, O.; Horiguchi, N.; Ki, S.H.; Yin, S.; Lafdil, F.; Gao, B. Inflammation-associated interleukin-6/signal transducer and activator of transcription 3 activation ameliorates alcoholic and nonalcoholic fatty liver diseases in interleukin-10-deficient mice. Hepatology 2011, 54, 846–856. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Faulkner, J.L.; Gomolak, J.R.; Didion, S.P. Interleukin-10 deficiency limits the development of obesity and insulin resistance produced by a high fat diet. FASEB J. 2013, 27, 1183. [Google Scholar]
- Cintra, D.E.; Pauli, J.R.; Araujo, E.P.; Moraes, J.C.; de Souza, C.T.; Milanski, M.; Morari, J.; Gambero, A.; Saad, M.J.; Velloso, L.A. Interleukin-10 is a protective factor against diet-induced insulin resistance in liver. J. Hepatol. 2008, 48, 628–637. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.J.; Higashimori, T.; Park, S.Y.; Choi, H.; Dong, J.; Kim, Y.J.; Noh, H.L.; Cho, Y.R.; Cline, G.; Kim, Y.B.; et al. Differential effects of interleukin-6 and -10 on skeletal muscle and liver insulin action in vivo. Diabetes 2004, 53, 1060–1067. [Google Scholar] [CrossRef] [PubMed]
- Hong, E.G.; Ko, H.J.; Cho, Y.R.; Kim, H.J.; Ma, Z.; Yu, T.Y.; Friedline, R.H.; Kurt-Jones, E.; Finberg, R.; Fischer, M.A.; et al. Interleukin-10 prevents diet-induced insulin resistance by attenuating macrophage and cytokine response in skeletal muscle. Diabetes 2009, 58, 2525–2535. [Google Scholar] [CrossRef] [PubMed]
- Dagdeviren, S.; Jung, D.Y.; Lee, E.; Friedline, R.H.; Noh, H.L.; Kim, J.H.; Patel, P.R.; Tsitsilianos, N.; Tsitsilianos, A.V.; Tran, D.A.; et al. Altered Interleukin-10 Signaling in Skeletal Muscle Regulates Obesity-Mediated Inflammation and Insulin Resistance. Mol. Cell. Biol. 2016, 36, 2956–2966. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, M.; Zhang, C.; Ma, Y.; Bu, L.; Yan, L.; Liu, D. Hydrodynamic delivery of mIL10 gene protects mice from high-fat diet-induced obesity and glucose intolerance. Mol. Ther. 2013, 21, 1852–1861. [Google Scholar] [CrossRef] [PubMed]
- Goto, T.; Naknukool, S.; Yoshitake, R.; Hanafusa, Y.; Tokiwa, S.; Li, Y.; Sakamoto, T.; Nitta, T.; Kim, M.; Takahashi, N.; et al. Proinflammatory cytokine interleukin-1β suppresses cold-induced thermogenesis in adipocytes. Cytokine 2016, 77, 107–114. [Google Scholar] [CrossRef] [PubMed]
- Nisoli, E.; Briscini, L.; Giordano, A.; Tonello, C.; Wiesbrock, S.M.; Uysal, K.T.; Cinti, S.; Carruba, M.O.; Hotamisligil, G.S. Tumor necrosis factor α mediates apoptosis of brown adipocytes and defective brown adipocyte function in obesity. Proc. Natl. Acad. Sci. USA 2000, 97, 8033–8038. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
| Reference | Interventional Approach | Cell/Cytokine/Intracellular Mediator | Rodent Model (Genetic Background) | Age (week) | External Cue (T °C, Diet, Treatment) | Gender | Effects on EE, Thermogenesis and Metabolic Homeostasis |
|---|---|---|---|---|---|---|---|
| Nguyen, 2011 | Global knockout | IL4/IL13 STAT6 | BALB/cJ BALB/cJ or C5BL6J | 8–12 | 4 °C, 6 h | male | Decreased weight loss Cold-induced hypothermia Decreased BAT thermogenic gene expression Exhausted lipid stores in BAT Decreased serum FFA Blunted M2-like markers in BAT and WAT |
| Conditional knockout, myeloid-specific | IL4RA | BALB/cJ IL4RαL/LLysMCre | |||||
| Global knockout | IL4/IL13 | BALB/cJ | 4 °C, 6 h Acute β3-agonist treatment | Normalized weight loss Increased EE Increased core body temperature Increased thermogenic gene expression Increased lipid storage in BAT | |||
| Global deletion, clodronate liposomes treatment | Macrophages | 4 °C, 6 h | Cold-induced hypothermia Decreased BAT thermogenic gene expression Blunted M2-like markers in BAT and WAT | ||||
| Qiu, 2014 | Global knockout | IL4/IL13 STAT6 IL4RA | BALB/cJ | 12 | 4 °C, 48 h | male | Decreased cold induced EE (VO2) (STAT6 and IL4RA KO) Cold-induced hypothermia Impaired browning Reduced sc WAT thermogenic gene expression Decreased scWAT oxygen consumption (IL4/IL13 KO) |
| Eosinophil deficient 4get/ΔdblGata mice | Eosinophils | Decreased cold induced EE (VO2) Impaired browning Reduced sc WAT thermogenic gene expression | |||||
| Global knockout | CCR2 | Decreased cold-induced ATM recruitment Decreased cold induced EE (VO2) Impaired browning Reduced sc WAT thermogenic gene expression | |||||
| IL4 i.p. treatment (IL4 complexed) | - | DIO C57BL6/J | HFD 10 weeks 30 °C IL4 treatment 14 days | Decreased body weight Decreased fat mass Improved insulin sensitivity Increased browning | |||
| Brestoff, 2015 | Global knockout | IL33 | C57BL6/J | 7 | LFD 12 weeks | male | Increased body weight Increased fat mass Insulin resistance Decreased beige adipocytes in scWAT Decreased ILC2s content in scWAT |
| IL33 i.p. treatment | - | 8 | LFD 12 weeks IL33 treatment 7 days | Decreased fat mass Increased EE Increased browning in scWAT | |||
| HFD and IL33 treatment 4 weeks | Counteracts DIO Abrogates glucose intolerance Increases ILC2s and Treg content in WAT | ||||||
| adoptively transferred congenic ILC2 | ILC2-deficient Rag 2 mice | IL33 treatment 7 days | Increased UCP1 protein in iWAT Increased iWAT browning dependent on ILC2s | ||||
| Lee, 2015 | IL33 i.p. treatment IL13 i.p. treatment IL4 i.p. treatment | C57BL6/J or IL5Red5/+, R5 BALB/cJ | 8–12 | Cytokine treatment 8 days 30 °C | male | Increased browning of scWAT Elicited beige progenitors (IL33, R5 mice) Increased scWAT UCP1 protein levels Increase cold-induced EE (IL33 treatment) | |
| Global knockout | IL5 (eosinophil growth factor) Normal IL13 secretion | IL5Red5/Red5 BALB/cJ | IL33 treatment 8 days 30 °C | Elicited proliferation of beige progenitors | |||
| IL4/IL13 IL4RA | BALB/cJ | Failed to increase proliferation of beige progenitors | |||||
| IL4 i.p. treatment (IL4 complexed) | C57BL6/J | 30 °C IL4 treatment, 24–48 h | Elicited proliferation of beige progenitors | ||||
| Conditional knockout, Progenitor cells specific | IL4RA | IL4RAf/fPdgfraCre | Failed to increase proliferation of beige progenitors | ||||
| Odegaard, 2016 | Global knockout | IL33 IL1R1 (ST2) | Adult: 8–12 Perinatal: 3–4 | 5 °C 48 h | male and female | Impaired cold-induced iWAT UCP1 expression Impaired browning Decreased survival in cold | |
| Fisher, 2017 | IL4 i.p. treatment Global knockout | None IL4RA | C57BL6/J | 12 | Daily treatment 14 days Declining T-30–5 °C | male | Unchanged body weight and EE No activation of thermogenic gene program in iWAT |
| Ding, 2016 | IL33 i.p. treatment | - | C57BL6/J | 6 | HFD 11 weeks IL33 treatment 7 days | male | Restoration of ILC2s and eosinophils content in scWAT Increased UCP1 protein level in scWAT |
| ST2 antibody treatment | 7 | ST2 antibody treatment 4 °C 48 h | Blunted ILC2s and eosinophils recruitment Decreased UCP1 protein levels in WAT | ||||
| Wallenius, 2002 | IL6 icv administration | - | Sprague-Dawley rats | Acute IL6 treatment | male | Increased EE (VO2) Lowers body weight and fat mass Unchanged food intake and activity | |
| Wernstedt, 2003 Wallenius, 2002 | Global knockout | IL6 | C57BL6/J | 8 | Cold challenge (6 h 4 °C) Stress challenge (1 h) | male | Spontaneous mature onset obesity Decreased EE in cold and stress Lower body core temperature Decreased NE serum levels |
| Li, 2002 | Adenoviral IL6 gene delivery icv | IL6 | Sprague-Dawley rats | - | 5 weeks | male | Supressed weight gain and adiposity Increased BAT UCP1 protein levels Blunted by denervation of BAT |
| Knudsen, 2014 | Global knockout | IL6 | C57BL6/J | 8 | Treadmill running 5 weeks or 4 °C, 3 days | male | Reduced sc WAT browning and UCP1 levels Partially reversed by IL6 treatment |
| IL6 i.p. treatment | - | C57BL6/J | 7 days | male | Increased sc WAT UCP1 levels | ||
| Petruzzelli,2014 | Transgenic mice with epithelial cell specific overexpression (cancer cachexia) | SOS-F | K5-SOS (skin tumours) C57BL6/J | 5 | Anti-IL6 Ab BAT denervation | Loss of body weight Fat and muscle wasting Increased UCP1 in sc WAT Increased EE Effects blunted by blocking IL6 or denervation | |
| Patsouris, 2015 | Global knockout Burn mice | IL6 | C57BL6/J | Burn back by 98 °C for 10 s Evaluation 2 days post-burn | Increase scWAT browning in WT Increased scWAT UCP1 levels Effects blunted in IL6KO mice Effects blunted after propranolol treatment | ||
| Almendro, 2008 | IL15 i.p. treatment | - | Wistar rats | - | Daily administration for 7 days | male | Decreased WAT and BAT mass Increased BAT UCP1 gene expression Increased expression of FA oxidation genes |
| Sun, 2016 | Hydrodynamic gene delivery Untargeted overexpression | IL15:IL15A | DIO C57BL6/J | 6 | 10 weeks Administration every 10 days | male | Reduced body weight Reduced adiposity Increased thermogenic markers in BAT and iWAT Improved insulin sensitivity |
| Lacraz, 2016 | Global knockout | IL15 | C57BL6/J | 4 | 16 weeks on HFD or 10 °C, 20 h or β3-agonist treatment | male | Resistance to DIO and IR Higher EE than controls Increased expression of genes associated with thermogenesis Elevated basal core temperature Increased BAT activation and iWAT browning in response to cold |
| Pazos, 2015 | Global knockout | IL18 | C57BL6/J | 8 | 10 weeks of HFD 4 °C 6 h 4 °C 5 days | male | HFD obesity prone Decreased UCP1 expression in BAT and iWAT Hypothermic after short cold challenge Null browning of scWAT in response to cold |
| IL18R1 | C57BL6/J | 10 weeks of HFD Acute HFD challenge 4 °C 6 h 4 °C 5 days | DIO resistant Increased UCP1 expression in sc WAT Increased EE in response to HFD challenge Maintenance of body temperature in cold Increased browning of scWAT and activation of thermogenic program |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
García, M.D.C.; Pazos, P.; Lima, L.; Diéguez, C. Regulation of Energy Expenditure and Brown/Beige Thermogenic Activity by Interleukins: New Roles for Old Actors. Int. J. Mol. Sci. 2018, 19, 2569. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms19092569
García MDC, Pazos P, Lima L, Diéguez C. Regulation of Energy Expenditure and Brown/Beige Thermogenic Activity by Interleukins: New Roles for Old Actors. International Journal of Molecular Sciences. 2018; 19(9):2569. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms19092569
Chicago/Turabian StyleGarcía, María Del Carmen, Patricia Pazos, Luis Lima, and Carlos Diéguez. 2018. "Regulation of Energy Expenditure and Brown/Beige Thermogenic Activity by Interleukins: New Roles for Old Actors" International Journal of Molecular Sciences 19, no. 9: 2569. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms19092569