An Apoptotic and Endosymbiotic Explanation of the Warburg and the Inverse Warburg Hypotheses


Abstract
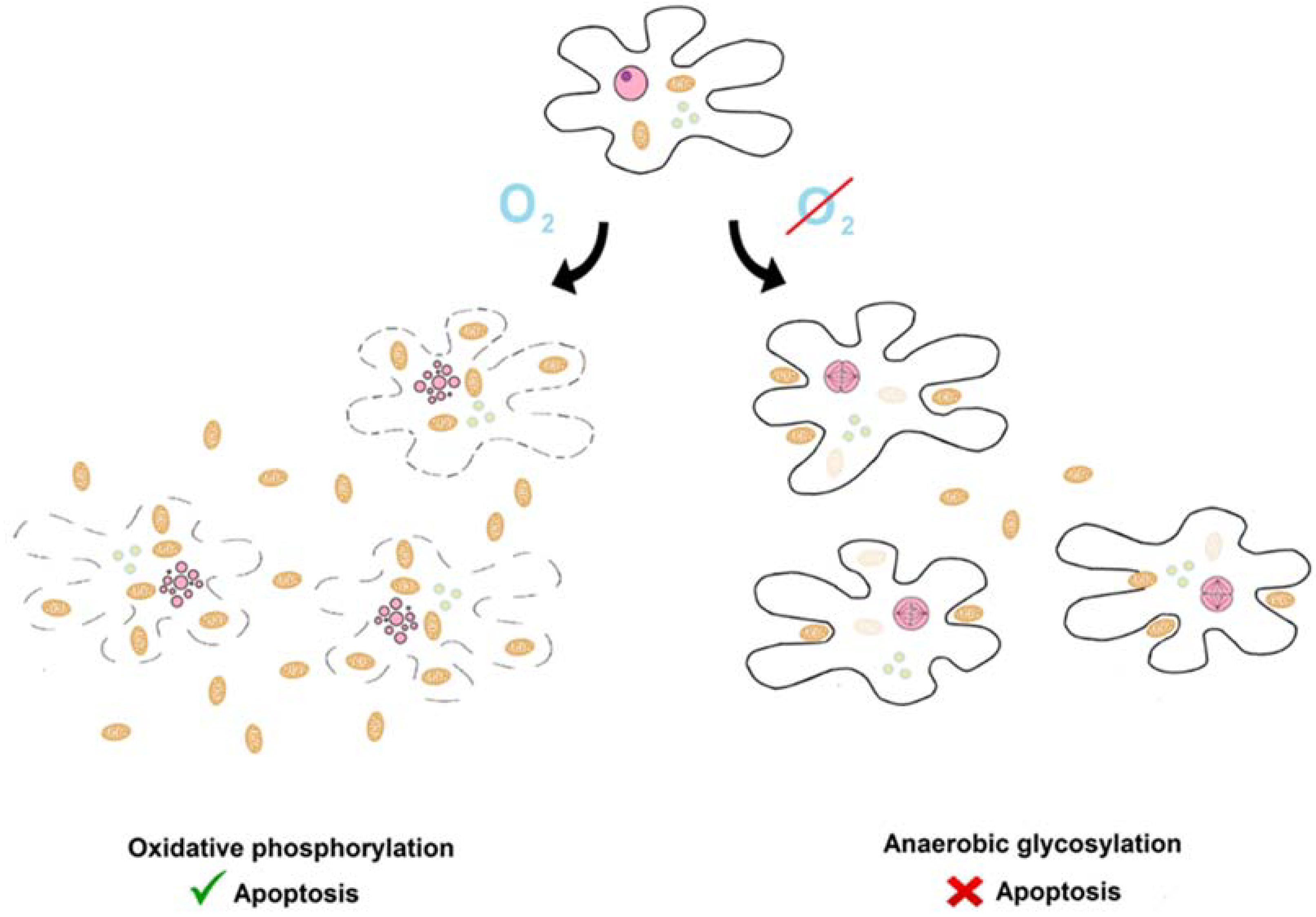
:1. Introduction
2. Aging
3. Animal Apoptosis—A Form of Programmed Cell Death?
4. Cancer and Apoptosis
5. Neurodegenerative Diseases and Apoptosis
6. Inverse Comorbidity of Cancer and Apoptosis
7. Warburg Hypothesis
8. The Inverse Warburg Hypothesis
9. Mitochondria Are Key Players in Oxidative Respiration and Apoptosis
10. The Endosymbiotic Theory of Origin of Apoptosis and Oxidative Respiration
11. Apoptotic-Like Cell Death (Apoptosis?) of Non-Animal Organisms
12. Ancestral State Reconstruction of Apoptosis Machinery
13. An Apoptotic Explanation of the Warburg and the Inverse Warburg Hypotheses
14. The Warburg and the Inverse Warburg Hypotheses in Yeast
15. Medical Implications of Our Observations
16. Conclusions
- -
- The induction of apoptosis causes a metabolic shift towards aerobic respiration;
- -
- The stimulation of aerobic respiration induces apoptosis;
- -
- The suppression of apoptosis causes a metabolic shift towards anaerobic respiration;
- -
- The suppression of aerobic respiration inhibits apoptosis.
Author Contributions
Funding
Conflicts of Interest
References
- Alzheimer’s Association. 2016 Alzheimer’s disease facts and figures. Alzheimers Dement. 2016, 12, 459–509. [Google Scholar] [CrossRef]
- Alzheimer’s Association. 2015 Alzheimer’s disease facts and figures. Alzheimers Dement. 2015, 11, 332–384. [Google Scholar] [CrossRef]
- Heron, M.P. Deaths: Leading causes for 2016. Natl. Vital. Stat. Rep. 2018, 67, 1–77. [Google Scholar] [PubMed]
- Kerr, J.F.; Wyllie, A.H.; Currie, A.R. Apoptosis: A basic biological phenomenon with wide-ranging implications in tissue kinetics. Br. J. Cancer 1972, 26, 239–257. [Google Scholar] [CrossRef] [PubMed]
- Hengartner, M.O.; Horvitz, H.R. C. elegans cell survival gene ced-9 encodes a functional homolog of the mammalian proto-oncogene bcl-2. Cell 1994, 76, 665–676. [Google Scholar] [CrossRef]
- Yuan, J.; Shaham, S.; Ledoux, S.; Ellis, H.M.; Horvitz, H.R. The, C. elegans cell death gene ced-3 encodes a protein similar to mammalian interleukin-1 beta-converting enzyme. Cell 1993, 75, 641–652. [Google Scholar] [CrossRef]
- Qi, S.; Pang, Y.; Hu, Q.; Liu, Q.; Li, H.; Zhou, Y.; He, T.; Liang, Q.; Liu, Y.; Yuan, X.; et al. Crystal structure of the Caenorhabditis elegans apoptosome reveals an octameric assembly of CED-4. Cell 2010, 141, 446–457. [Google Scholar] [CrossRef] [PubMed]
- Yan, N.; Chai, J.; Lee, E.S.; Gu, L.; Liu, Q.; He, J.; Wu, J.W.; Kokel, D.; Li, H.; Hao, Q.; et al. Structure of the CED-4-CED-9 complex provides insights into programmed cell death in Caenorhabditis elegans. Nature 2005, 437, 831–837. [Google Scholar] [CrossRef] [PubMed]
- Sakamaki, K.; Imai, K.; Tomii, K.; Miller, D.J. Evolutionary analyses of caspase-8 and its paralogs: Deep origins of the apoptotic signaling pathways. Bioessays 2015, 37, 767–776. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Green, D.R.; Fitzgerald, P. Just So Stories about the Evolution of Apoptosis. Curr. Biol. 2016, 26, R620–R627. [Google Scholar] [CrossRef] [PubMed]
- Klim, J.; Gładki, A.; Kucharczyk, R.; Zielenkiewicz, U.; Kaczanowski, S. Ancestral State Reconstruction of the Apoptosis Machinery in the Common Ancestor of Eukaryotes. G3 (Bethesda) 2018, 8, 2121–2134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Evan, G.I.; Vousden, K.H. Proliferation, cell cycle and apoptosis in cancer. Nature 2001, 411, 342–348. [Google Scholar] [CrossRef] [PubMed]
- Lowe, S.W.; Cepero, E.; Evan, G. Intrinsic tumour suppression. Nature 2004, 432, 307–315. [Google Scholar] [CrossRef] [PubMed]
- LaFerla, F.M.; Tinkle, B.T.; Bieberich, C.J.; Haudenschild, C.C.; Jay, G. The Alzheimer’s A beta peptide induces neurodegeneration and apoptotic cell death in transgenic mice. Nat. Genet. 1995, 9, 21–30. [Google Scholar] [CrossRef] [PubMed]
- Mochizuki, H.; Goto, K.; Mori, H.; Mizuno, Y. Histochemical detection of apoptosis in Parkinson’s disease. J. Neurol. Sci. 1996, 137, 120–123. [Google Scholar] [CrossRef]
- Warburg, O. On the origin of cancer cells. Science 1956, 123, 309–314. [Google Scholar] [CrossRef] [PubMed]
- Tabarés-Seisdedos, R.; Dumont, N.; Baudot, A.; Valderas, J.M.; Climent, J.; Valencia, A.; Crespo-Facorro, B.; Vieta, E.; Gómez-Beneyto, M.; Martínez, S.; et al. No paradox, no progress: Inverse cancer comorbidity in people with other complex diseases. Lancet Oncol. 2011, 12, 604–608. [Google Scholar] [CrossRef]
- Driver, J.A.; Beiser, A.; Au, R.; Kreger, B.E.; Splansky, G.L.; Kurth, T.; Kiel, D.P.; Lu, K.P.; Seshadri, S.; Wolf, P.A. Inverse association between cancer and Alzheimer’s disease: Results from the Framingham Heart Study. BMJ 2012, 344, e1442. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jansson, B.; Jankovic, J. Low cancer rates among patients with Parkinson’s disease. Ann. Neurol. 1985, 17, 505–509. [Google Scholar] [CrossRef] [PubMed]
- West, A.B.; Dawson, V.L.; Dawson, T.M. To die or grow: Parkinson’s disease and cancer. Trends Neurosci. 2005, 28, 348–352. [Google Scholar] [CrossRef] [PubMed]
- Musicco, M.; Adorni, F.; Di Santo, S.; Prinelli, F.; Pettenati, C.; Caltagirone, C.; Palmer, K.; Russo, A. Inverse occurrence of cancer and Alzheimer disease: A population-based incidence study. Neurology 2013, 81, 322–328. [Google Scholar] [CrossRef] [PubMed]
- Lodato, M.A.; Rodin, R.E.; Bohrson, C.L.; Coulter, M.E.; Barton, A.R.; Kwon, M.; Sherman, M.A.; Vitzthum, C.M.; Luquette, L.J.; Yandava, C.N.; et al. Aging and neurodegeneration are associated with increased mutations in single human neurons. Science 2018, 359, 555–559. [Google Scholar] [CrossRef] [PubMed]
- Luu, J.; Palczewski, K. Human aging and disease: Lessons from age-related macular degeneration. Proc. Natl. Acad. Sci. USA 2018, 115, 2866–2872. [Google Scholar] [CrossRef] [PubMed]
- David, D.C.; Ollikainen, N.; Trinidad, J.C.; Cary, M.P.; Burlingame, A.L.; Kenyon, C. Widespread protein aggregation as an inherent part of aging in C. elegans. PLoS Biol. 2010, 8, e1000450. [Google Scholar] [CrossRef] [PubMed]
- Chiti, F.; Dobson, C.M. Protein Misfolding, Amyloid Formation, and Human Disease: A Summary of Progress Over the Last Decade. Ann. Rev. Biochem. 2017, 86, 27–68. [Google Scholar] [CrossRef] [PubMed]
- Reed, J.C. Dysregulation of apoptosis in cancer. J. Clin. Oncol. 1999, 17, 2941–2953. [Google Scholar] [CrossRef] [PubMed]
- Pistritto, G.; Trisciuoglio, D.; Ceci, C.; Garufi, A.; D’Orazi, G. Apoptosis as anticancer mechanism: Function and dysfunction of its modulators and targeted therapeutic strategies. Aging 2016, 8, 603–619. [Google Scholar] [CrossRef] [PubMed]
- Sarosiek, K.A.; Fraser, C.; Muthalagu, N.; Bhola, P.D.; Chang, W.; McBrayer, S.K.; Cantlon, A.; Fisch, S.; Golomb-Mello, G.; Ryan, J.A.; et al. Developmental Regulation of Mitochondrial Apoptosis by c-Myc Governs Age- and Tissue-Specific Sensitivity to Cancer Therapeutics. Cancer Cell 2017, 31, 142–156. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaczanowski, S. Apoptosis: Its origin, history, maintenance and the medical implications for cancer and aging. Phys. Biol. 2016, 13, 031001. [Google Scholar] [CrossRef] [PubMed]
- Sulston, J.E.; Horvitz, H.R. Post-embryonic cell lineages of the nematode, Caenorhabditis elegans. Dev. Biol. 1977, 56, 110–156. [Google Scholar] [CrossRef]
- Sulston, J.E.; Schierenberg, E.; White, J.G.; Thomson, J.N. The embryonic cell lineage of the nematode Caenorhabditis elegans. Dev. Biol. 1983, 100, 64–119. [Google Scholar] [CrossRef]
- Robertson, A.M.; Thomson, J. Morphology of programmed cell death in the ventral nerve cord of Caenorhabditis elegans larvae. Development 1982, 67, 89–100. [Google Scholar]
- Visconti, R.; Della Monica, R.; Grieco, D. Cell cycle checkpoint in cancer: A therapeutically targetable double-edged sword. J. Exp. Clin. Cancer Res. 2016, 35, 153. [Google Scholar] [CrossRef] [PubMed]
- Bao, S.; Wu, Q.; McLendon, R.E.; Hao, Y.; Shi, Q.; Hjelmeland, A.B.; Dewhirst, M.W.; Bigner, D.D.; Rich, J.N. Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nature 2006, 444, 756–760. [Google Scholar] [CrossRef] [PubMed]
- Puig, P.E.; Guilly, M.N.; Bouchot, A.; Droin, N.; Cathelin, D.; Bouyer, F.; Favier, L.; Ghiringhelli, F.; Kroemer, G.; Solary, E.; et al. Tumor cells can escape DNA-damaging cisplatin through DNA endoreduplication and reversible polyploidy. Cell Biol. Int. 2008, 32, 1031–1043. [Google Scholar] [CrossRef] [PubMed]
- Bacus, S.S.; Gudkov, A.V.; Lowe, M.; Lyass, L.; Yung, Y.; Komarov, A.P.; Keyomarsi, K.; Yarden, Y.; Seger, R. Taxol-induced apoptosis depends on MAP kinase pathways (ERK and p38) and is independent of p53. Oncogene 2001, 20, 147–155. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, F.; Ambrosini, G.; Chu, E.Y.; Plescia, J.; Tognin, S.; Marchisio, P.C.; Altieri, D.C. Control of apoptosis and mitotic spindle checkpoint by survivin. Nature 1998, 396, 580–584. [Google Scholar] [CrossRef] [PubMed]
- Zhou, M.; Zhao, Y.; Ding, Y.; Liu, H.; Liu, Z.; Fodstad, O.; Riker, A.I.; Kamarajugadda, S.; Lu, J.; Owen, L.B.; et al. Warburg effect in chemosensitivity: Targeting lactate dehydrogenase-A re-sensitizes taxol-resistant cancer cells to taxol. Mol. Cancer 2010, 9, 33. [Google Scholar] [CrossRef] [PubMed]
- Dobson, C.M. The structural basis of protein folding and its links with human disease. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2001, 356, 133–145. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Büttner, S.; Habernig, L.; Broeskamp, F.; Ruli, D.; Vögtle, F.N.; Vlachos, M.; Macchi, F.; Küttner, V.; Carmona-Gutierrez, D.; Eisenberg, T.; et al. Endonuclease G mediates α-synuclein cytotoxicity during Parkinson’s disease. EMBO J. 2013, 32, 3041–3054. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Flagmeier, P.; Meisl, G.; Vendruscolo, M.; Knowles, T.P.; Dobson, C.M.; Buell, A.K.; Galvagnion, C. Mutations associated with familial Parkinson’s disease alter the initiation and amplification steps of α-synuclein aggregation. Proc. Natl. Acad. Sci. USA 2016, 113, 10328–10333. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spillantini, M.G.; Crowther, R.A.; Jakes, R.; Hasegawa, M.; Goedert, M. Alpha-Synuclein in filamentous inclusions of Lewy bodies from Parkinson’s disease and dementia with lewy bodies. Proc. Natl. Acad. Sci. USA 1998, 95, 6469–6473. [Google Scholar] [CrossRef] [PubMed]
- Selkoe, D.J.; Hardy, J. The amyloid hypothesis of Alzheimer’s disease at 25 years. EMBO Mol. Med. 2016, 8, 595–608. [Google Scholar] [CrossRef] [PubMed]
- Glenner, G.G.; Wong, C.W. Alzheimer’s disease: Initial report of the purification and characterization of a novel cerebrovascular amyloid protein. Biochem. Biophys. Res. Commun. 1984, 120, 885–890. [Google Scholar] [CrossRef]
- Vassar, R.; Bennett, B.D.; Babu-Khan, S.; Kahn, S.; Mendiaz, E.A.; Denis, P.; Teplow, D.B.; Ross, S.; Amarante, P.; Loeloff, R.; et al. Beta-secretase cleavage of Alzheimer’s amyloid precursor protein by the transmembrane aspartic protease BACE. Science 1999, 286, 735–741. [Google Scholar] [CrossRef] [PubMed]
- Ong, E.L.; Goldacre, R.; Goldacre, M. Differential risks of cancer types in people with Parkinson’s disease: A national record-linkage study. Eur. J. Cancer 2014, 50, 2456–2462. [Google Scholar] [CrossRef] [PubMed]
- Altenberg, B.; Greulich, K.O. Genes of glycolysis are ubiquitously overexpressed in 24 cancer classes. Genomics 2004, 84, 1014–1020. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez-Enríquez, S.; Carreño-Fuentes, L.; Gallardo-Pérez, J.C.; Saavedra, E.; Quezada, H.; Vega, A.; Marín-Hernández, A.; Olín-Sandoval, V.; Torres-Márquez, M.E.; Moreno-Sánchez, R. Oxidative phosphorylation is impaired by prolonged hypoxia in breast and possibly in cervix carcinoma. Int. J. Biochem. Cell Biol. 2010, 42, 1744–1751. [Google Scholar] [CrossRef] [PubMed]
- Vander Heiden, M.G.; Cantley, L.C.; Thompson, C.B. Understanding the Warburg effect: The metabolic requirements of cell proliferation. Science 2009, 324, 1029–1033. [Google Scholar] [CrossRef] [PubMed]
- Christofk, H.R.; Vander Heiden, M.G.; Harris, M.H.; Ramanathan, A.; Gerszten, R.E.; Wei, R.; Fleming, M.D.; Schreiber, S.L.; Cantley, L.C. The M2 splice isoform of pyruvate kinase is important for cancer metabolism and tumour growth. Nature 2008, 452, 230–233. [Google Scholar] [CrossRef] [PubMed]
- Faubert, B.; Boily, G.; Izreig, S.; Griss, T.; Samborska, B.; Dong, Z.; Dupuy, F.; Chambers, C.; Fuerth, B.J.; Viollet, B.; et al. AMPK is a negative regulator of the Warburg effect and suppresses tumor growth in vivo. Cell Metab. 2013, 17, 113–124. [Google Scholar] [CrossRef] [PubMed]
- Zivieri, R.; Pacini, N.; Finocchio, G.; Carpentieri, M. Rate of entropy model for irreversible processes in living systems. Sci. Rep. 2017, 7, 134. [Google Scholar] [CrossRef] [PubMed]
- Zheng, J. Energy metabolism of cancer: Glycolysis versus oxidative phosphorylation (Review). Oncol. Lett. 2012, 4, 1151–1157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liberti, M.V.; Locasale, J.W. The Warburg Effect: How Does it Benefit Cancer Cells? Trends Biochem. Sci. 2016, 41, 211–218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, X.; Qian, Y.; Wu, S. The Warburg effect: Evolving interpretations of an established concept. Free Radic. Biol. Med. 2015, 79, 253–263. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Demetrius, L.A.; Coy, J.F.; Tuszynski, J.A. Cancer proliferation and therapy: The Warburg effect and quantum metabolism. Theor. Biol. Med. Model 2010, 7, 2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shestov, A.A.; Liu, X.; Ser, Z.; Cluntun, A.A.; Hung, Y.P.; Huang, L.; Kim, D.; Le, A.; Yellen, G.; Albeck, J.G.; et al. Quantitative determinants of aerobic glycolysis identify flux through the enzyme GAPDH as a limiting step. Elife 2014, 3, e03342. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Estrella, V.; Chen, T.; Lloyd, M.; Wojtkowiak, J.; Cornnell, H.H.; Ibrahim-Hashim, A.; Bailey, K.; Balagurunathan, Y.; Rothberg, J.M.; Sloane, B.F.; et al. Acidity generated by the tumor microenvironment drives local invasion. Cancer Res. 2013, 73, 1524–1535. [Google Scholar] [CrossRef] [PubMed]
- San-Millán, I.; Brooks, G.A. Reexamining cancer metabolism: Lactate production for carcinogenesis could be the purpose and explanation of the Warburg Effect. Carcinogenesis 2017, 38, 119–133. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.H.; Qiu, J.; O’Sullivan, D.; Buck, M.D.; Noguchi, T.; Curtis, J.D.; Chen, Q.; Gindin, M.; Gubin, M.M.; van der Windt, G.J.; et al. Metabolic Competition in the Tumor Microenvironment Is a Driver of Cancer Progression. Cell 2015, 162, 1229–1241. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Levine, A.J.; Puzio-Kuter, A.M. The control of the metabolic switch in cancers by oncogenes and tumor suppressor genes. Science 2010, 330, 1340–1344. [Google Scholar] [CrossRef] [PubMed]
- Attardi, L.D. The role of p53-mediated apoptosis as a crucial anti-tumor response to genomic instability: Lessons from mouse models. Mutat. Res. 2005, 569, 145–157. [Google Scholar] [CrossRef] [PubMed]
- Weng, L.; Brown, J.; Eng, C. PTEN induces apoptosis and cell cycle arrest through phosphoinositol-3-kinase/Akt-dependent and -independent pathways. Hum. Mol. Genet. 2001, 10, 237–242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choi, Y.; Zhang, J.; Murga, C.; Yu, H.; Koller, E.; Monia, B.P.; Gutkind, J.S.; Li, W. PTEN, but not SHIP and SHIP2, suppresses the PI3K/Akt pathway and induces growth inhibition and apoptosis of myeloma cells. Oncogene 2002, 21, 5289–5300. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cordero-Espinoza, L.; Hagen, T. Increased concentrations of fructose 2,6-bisphosphate contribute to the Warburg effect in phosphatase and tensin homolog (PTEN)-deficient cells. J. Biol. Chem. 2013, 288, 36020–36028. [Google Scholar] [CrossRef] [PubMed]
- Itahana, Y.; Itahana, K. Emerging Roles of p53 Family Members in Glucose Metabolism. Int. J. Mol. Sci. 2018, 19, 776. [Google Scholar] [CrossRef] [PubMed]
- Eriksson, M.; Ambroise, G.; Ouchida, A.T.; Lima Queiroz, A.; Smith, D.; Gimenez-Cassina, A.; Iwanicki, M.P.; Muller, P.A.; Norberg, E.; Vakifahmetoglu-Norberg, H. Effect of Mutant p53 Proteins on Glycolysis and Mitochondrial Metabolism. Mol. Cell. Biol. 2017, 37. [Google Scholar] [CrossRef] [PubMed]
- Sasabe, E.; Tatemoto, Y.; Li, D.; Yamamoto, T.; Osaki, T. Mechanism of HIF-1alpha-dependent suppression of hypoxia-induced apoptosis in squamous cell carcinoma cells. Cancer Sci. 2005, 96, 394–402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mimeault, M.; Batra, S.K. Hypoxia-inducing factors as master regulators of stemness properties and altered metabolism of cancer- and metastasis-initiating cells. J. Cell. Mol. Med. 2013, 17, 30–54. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Delbridge, A.R.; Strasser, A. The BCL-2 protein family, BH3-mimetics and cancer therapy. Cell Death Differ. 2015, 22, 1071–1080. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sinicrope, F.A.; Ruan, S.B.; Cleary, K.R.; Stephens, L.C.; Lee, J.J.; Levin, B. Bcl-2 and p53 oncoprotein expression during colorectal tumorigenesis. Cancer Res. 1995, 55, 237–241. [Google Scholar] [PubMed]
- Hagenbuchner, J.; Kiechl-Kohlendorfer, U.; Obexer, P.; Ausserlechner, M.J. BIRC5/Survivin as a target for glycolysis inhibition in high-stage neuroblastoma. Oncogene 2016, 35, 2052–2061. [Google Scholar] [CrossRef] [PubMed]
- Hagenbuchner, J.; Kuznetsov, A.V.; Obexer, P.; Ausserlechner, M.J. BIRC5/Survivin enhances aerobic glycolysis and drug resistance by altered regulation of the mitochondrial fusion/fission machinery. Oncogene 2013, 32, 4748–4757. [Google Scholar] [CrossRef] [PubMed]
- Mahmood, B.; Wilson, J.; Ahmad, M.; Olino, P.; King, J.; Dejean, L. Bcl-2 Overexpression Stimulates Glycolysis and Lactic Fermentation in a Bax-Dependent Fashion. Biophys. J. 2015, 108, 613a. [Google Scholar] [CrossRef]
- Semenza, G.L. HIF-1 mediates the Warburg effect in clear cell renal carcinoma. J. Bioenerg. Biomembr. 2007, 39, 231–234. [Google Scholar] [CrossRef] [PubMed]
- Vaughn, A.E.; Deshmukh, M. Glucose metabolism inhibits apoptosis in neurons and cancer cells by redox inactivation of cytochrome c. Nat. Cell Biol. 2008, 10, 1477–1483. [Google Scholar] [CrossRef] [PubMed]
- Bonnet, S.; Archer, S.L.; Allalunis-Turner, J.; Haromy, A.; Beaulieu, C.; Thompson, R.; Lee, C.T.; Lopaschuk, G.D.; Puttagunta, L.; Harry, G.; et al. A mitochondria-K+ channel axis is suppressed in cancer and its normalization promotes apoptosis and inhibits cancer growth. Cancer Cell 2007, 11, 37–51. [Google Scholar] [CrossRef] [PubMed]
- Jia, Y.; Ma, Z.; Liu, X.; Zhou, W.; He, S.; Xu, X.; Ren, G.; Xu, G.; Tian, K. Metformin prevents DMH-induced colorectal cancer in diabetic rats by reversing the warburg effect. Cancer Med. 2015, 4, 1730–1741. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harada, K.; Ferdous, T.; Harada, T.; Ueyama, Y. Metformin in combination with 5-fluorouracil suppresses tumor growth by inhibiting the Warburg effect in human oral squamous cell carcinoma. Int. J. Oncol. 2016, 49, 276–284. [Google Scholar] [CrossRef] [PubMed]
- Flaveny, C.A.; Griffett, K.; El-Gendy, B.D.; Kazantzis, M.; Sengupta, M.; Amelio, A.L.; Chatterjee, A.; Walker, J.; Solt, L.A.; Kamenecka, T.M.; et al. Broad Anti-tumor Activity of a Small Molecule that Selectively Targets the Warburg Effect and Lipogenesis. Cancer Cell 2015, 28, 42–56. [Google Scholar] [CrossRef] [PubMed]
- Takai, T.; Yoshikawa, Y.; Inamoto, T.; Minami, K.; Taniguchi, K.; Sugito, N.; Kuranaga, Y.; Shinohara, H.; Kumazaki, M.; Tsujino, T.; et al. A Novel Combination RNAi toward Warburg Effect by Replacement with miR-145 and Silencing of PTBP1 Induces Apoptotic Cell Death in Bladder Cancer Cells. Int. J. Mol. Sci. 2017, 18, 179. [Google Scholar] [CrossRef] [PubMed]
- Bianchi, G.; Martella, R.; Ravera, S.; Marini, C.; Capitanio, S.; Orengo, A.; Emionite, L.; Lavarello, C.; Amaro, A.; Petretto, A.; et al. Fasting induces anti-Warburg effect that increases respiration but reduces ATP-synthesis to promote apoptosis in colon cancer models. Oncotarget 2015, 6, 11806–11819. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Demetrius, L.A.; Magistretti, P.J.; Pellerin, L. Alzheimer’s disease: The amyloid hypothesis and the Inverse Warburg effect. Front Physiol. 2014, 5, 522. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Demetrius, L.A.; Simon, D.K. The inverse association of cancer and Alzheimer’s: A bioenergetic mechanism. J. R. Soc. Interface 2013, 10, 20130006. [Google Scholar] [CrossRef] [PubMed]
- Zhu, X.; Perry, G.; Moreira, P.I.; Aliev, G.; Cash, A.D.; Hirai, K.; Smith, M.A. Mitochondrial abnormalities and oxidative imbalance in Alzheimer disease. J. Alzheimers Dis. 2006, 9, 147–153. [Google Scholar] [CrossRef] [PubMed]
- Ibáñez, K.; Boullosa, C.; Tabarés-Seisdedos, R.; Baudot, A.; Valencia, A. Molecular evidence for the inverse comorbidity between central nervous system disorders and cancers detected by transcriptomic meta-analyses. PLoS Genet. 2014, 10, e1004173. [Google Scholar] [CrossRef] [PubMed]
- Marchetti, P.; Castedo, M.; Susin, S.A.; Zamzami, N.; Hirsch, T.; Macho, A.; Haeffner, A.; Hirsch, F.; Geuskens, M.; Kroemer, G. Mitochondrial permeability transition is a central coordinating event of apoptosis. J. Exp. Med. 1996, 184, 1155–1160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kroemer, G.; Reed, J. Mitochondrial control of cell death. Nat. Med. 2000, 6, 513–519. [Google Scholar] [CrossRef] [PubMed]
- Petit, P.; Susin, S.; Zamzami, N.; Mignotte, B.; Kroemer, G. Mitochondria and programmed cell death: Back to the future. FEBS Lett. 1996, 396, 7–13. [Google Scholar] [CrossRef]
- Kluck, R.; Bossy-Wetzel, E.; Green, D.; Newmeyer, D. The release of cytochrome c from mitochondria: A primary site for Bcl-2 regulation of apoptosis. Science 1997, 275, 1132–1136. [Google Scholar] [CrossRef] [PubMed]
- Margulis, L. Symbiosis in Cell Evolution; Freeman: New York, NY, USA, 1993. [Google Scholar]
- Kroemer, G. Mitochondrial implication in apoptosis. Towards an endosymbiont hypothesis of apoptosis evolution. Cell Death Differ. 1997, 4, 443–456. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koonin, E.V.; Aravind, L. Origin and evolution of eukaryotic apoptosis: The bacterial connection. Cell Death Differ. 2002, 9, 394–404. [Google Scholar] [CrossRef] [PubMed]
- Aravind, L.; Dixit, V.M.; Koonin, E.V. Apoptotic molecular machinery: Vastly increased complexity in vertebrates revealed by genome comparisons. Science 2001, 291, 1279–1284. [Google Scholar] [CrossRef] [PubMed]
- Carmona-Gutierrez, D.; Bauer, M.A.; Zimmermann, A.; Aguilera, A.; Austriaco, N.; Ayscough, K.; Balzan, R.; Bar-Nun, S.; Barrientos, A.; Belenky, P.; et al. Guidelines and recommendations on yeast cell death nomenclature. Microbiol. Cell 2018, 5, 4–31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaczanowski, S.; Sajid, M.; Reece, S.E. Evolution of apoptosis-like programmed cell death in unicellular protozoan parasites. Parasit. Vectors 2011, 4, 44. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Proto, W.R.; Coombs, G.H.; Mottram, J.C. Cell death in parasitic protozoa: Regulated or incidental? Nat. Rev. Microbiol. 2013, 11, 58–66. [Google Scholar] [CrossRef] [PubMed]
- Taylor-Brown, E.; Hurd, H. The first suicides: A legacy inherited by parasitic protozoans from prokaryote ancestors. Parasit. Vectors 2013, 6, 108. [Google Scholar] [CrossRef] [PubMed]
- Duszenko, M.; Figarella, K.; Macleod, E.; Welburn, S. Death of a trypanosome: A selfish altruism. Trends Parasitol. 2006, 22, 536–542. [Google Scholar] [CrossRef] [PubMed]
- Durand, P.M.; Choudhury, R.; Rashidi, A.; Michod, R.E. Programmed death in a unicellular organism has species-specific fitness effects. Biol. Lett. 2014, 10, 20131088. [Google Scholar] [CrossRef] [PubMed]
- Amigoni, L.; Frigerio, G.; Martegani, E.; Colombo, S. Involvement of Aif1 in apoptosis triggered by lack of Hxk2 in the yeast Saccharomyces cerevisiae. FEMS Yeast Res. 2016, 16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amigoni, L.; Martegani, E.; Colombo, S. Lack of HXK2 induces localization of active Ras in mitochondria and triggers apoptosis in the yeast Saccharomyces cerevisiae. Oxid. Med. Cell. Longev. 2013, 2013, 678473. [Google Scholar] [CrossRef] [PubMed]
- Büttner, S.; Bitto, A.; Ring, J.; Augsten, M.; Zabrocki, P.; Eisenberg, T.; Jungwirth, H.; Hutter, S.; Carmona-Gutierrez, D.; Kroemer, G.; et al. Functional mitochondria are required for alpha-synuclein toxicity in aging yeast. J. Biol. Chem. 2008, 283, 7554–7560. [Google Scholar] [CrossRef] [PubMed]
- Treusch, S.; Hamamichi, S.; Goodman, J.L.; Matlack, K.E.; Chung, C.Y.; Baru, V.; Shulman, J.M.; Parrado, A.; Bevis, B.J.; Valastyan, J.S.; et al. Functional links between Aβ toxicity, endocytic trafficking, and Alzheimer’s disease risk factors in yeast. Science 2011, 334, 1241–1245. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Bisschops, M.M.M.; Agarwal, N.R.; Ji, B.; Shanmugavel, K.P.; Petranovic, D. Interplay of Energetics and ER Stress Exacerbates Alzheimer’s Amyloid-β (Aβ) Toxicity in Yeast. Front. Mol. Neurosci. 2017, 10, 232. [Google Scholar] [CrossRef] [PubMed]
- Hubbard, B.P.; Sinclair, D.A. Small molecule SIRT1 activators for the treatment of aging and age-related diseases. Trends Pharmacol. Sci. 2014, 35, 146–154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fontana, L.; Partridge, L.; Longo, V. Extending healthy life span--from yeast to humans. Science 2010, 328, 321–326. [Google Scholar] [CrossRef] [PubMed]
- Bhushan, A.; Fondell, E.; Ascherio, A.; Yuan, C.; Grodstein, F.; Willett, W. Adherence to Mediterranean diet and subjective cognitive function in men. Eur. J. Epidemiol. 2018, 33, 223–234. [Google Scholar] [CrossRef] [PubMed]
- Schwingshackl, L.; Schwedhelm, C.; Galbete, C.; Hoffmann, G. Adherence to Mediterranean Diet and Risk of Cancer: An Updated Systematic Review and Meta-Analysis. Nutrients 2017, 9, 1063. [Google Scholar] [CrossRef] [PubMed]
- Schwingshackl, L.; Hoffmann, G. Does a Mediterranean-Type Diet Reduce Cancer Risk? Curr. Nutr. Rep. 2016, 5, 9–17. [Google Scholar] [CrossRef] [PubMed]
- Moore, S.C.; Lee, I.M.; Weiderpass, E.; Campbell, P.T.; Sampson, J.N.; Kitahara, C.M.; Keadle, S.K.; Arem, H.; Berrington de Gonzalez, A.; Hartge, P.; et al. Association of Leisure-Time Physical Activity With Risk of 26 Types of Cancer in 1.44 Million Adults. JAMA Int. Med. 2016, 176, 816–825. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tolppanen, A.M.; Solomon, A.; Kulmala, J.; Kåreholt, I.; Ngandu, T.; Rusanen, M.; Laatikainen, T.; Soininen, H.; Kivipelto, M. Leisure-time physical activity from mid- to late life, body mass index, and risk of dementia. Alzheimers Dement. 2015, 11, 434–443. [Google Scholar] [CrossRef] [PubMed]
- Schairer, C.; Lubin, J.; Troisi, R.; Sturgeon, S.; Brinton, L.; Hoover, R. Menopausal estrogen and estrogen-progestin replacement therapy and breast cancer risk. JAMA 2000, 283, 485–491. [Google Scholar] [CrossRef] [PubMed]
- Pike, M.C.; Spicer, D.V.; Dahmoush, L.; Press, M.F. Estrogens progestogens normal breast cell proliferation and breast cancer risk. Epidemiol. Rev. 1993, 15, 17–35. [Google Scholar] [CrossRef] [PubMed]
- Paganini-Hill, A.; Henderson, V.W. Estrogen replacement therapy and risk of Alzheimer disease. Arch. Int. Med. 1996, 156, 2213–2217. [Google Scholar] [CrossRef]


{kind=link}
| Mechanism of Apoptosis | S. cerevisiae | Homo Sapiens |
|---|---|---|
| Caspase | − | + |
| Metacaspase | + | − |
| Cytochrome c induced apoptosis | + | + |
| Mitochondrial permeability transition | + | + |
| EndoG | + | + |
| Parkinson-like activation of EndoG apoptotic pathway by α-synuclein aggregates | + | + |
| AIFs (Apoptotic Induction Factors) | + | + |
| OMI/HTRA apoptotic protease | + | + |
| Suppression of apoptotic activity causes co-suppression of aerobic respiration (Warburg effect) | + | + |
| Aerobic respiration stimulates apoptotic activity (inverse Warburg effect) | + | + |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kaczanowski, S.; Klim, J.; Zielenkiewicz, U. An Apoptotic and Endosymbiotic Explanation of the Warburg and the Inverse Warburg Hypotheses. Int. J. Mol. Sci. 2018, 19, 3100. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms19103100
Kaczanowski S, Klim J, Zielenkiewicz U. An Apoptotic and Endosymbiotic Explanation of the Warburg and the Inverse Warburg Hypotheses. International Journal of Molecular Sciences. 2018; 19(10):3100. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms19103100
Chicago/Turabian StyleKaczanowski, Szymon, Joanna Klim, and Urszula Zielenkiewicz. 2018. "An Apoptotic and Endosymbiotic Explanation of the Warburg and the Inverse Warburg Hypotheses" International Journal of Molecular Sciences 19, no. 10: 3100. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms19103100