A Molecular Biophysical Approach to Diclofenac Topical Gastrointestinal Damage

 ,
,  , ,
, ,  , and
, and 
Abstract
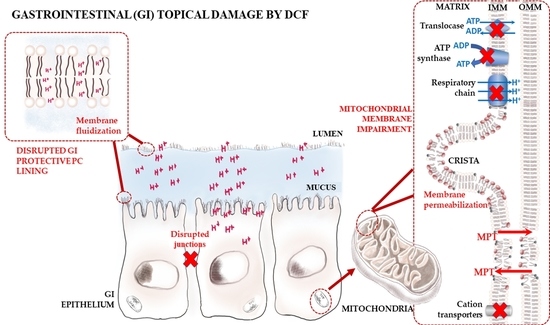
:
1. Introduction
2. Results
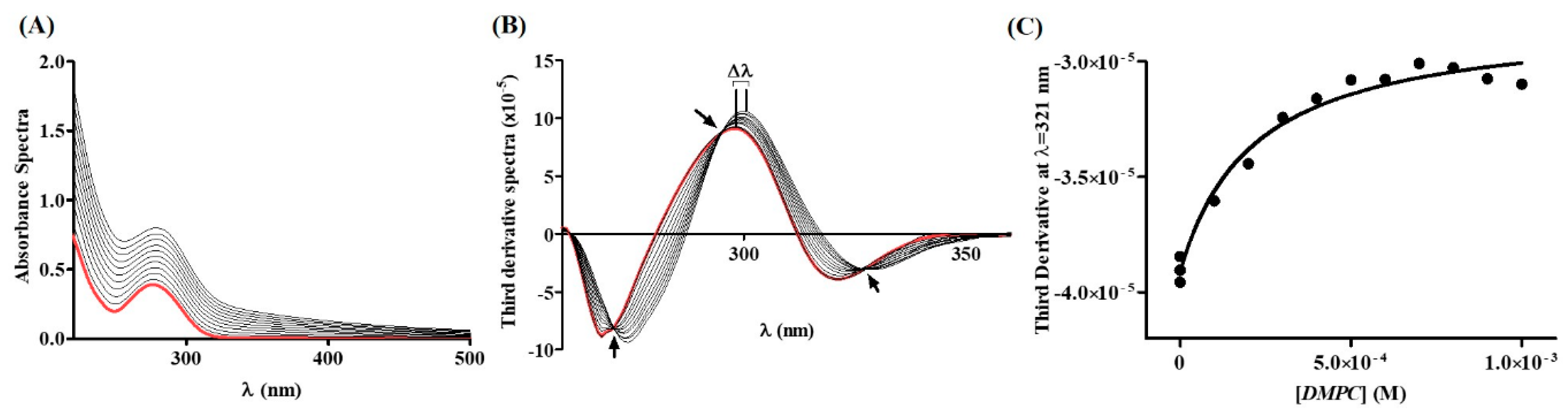
2.1. Diclofenac Distribution Coefficient in a Membrane/Aqueous System
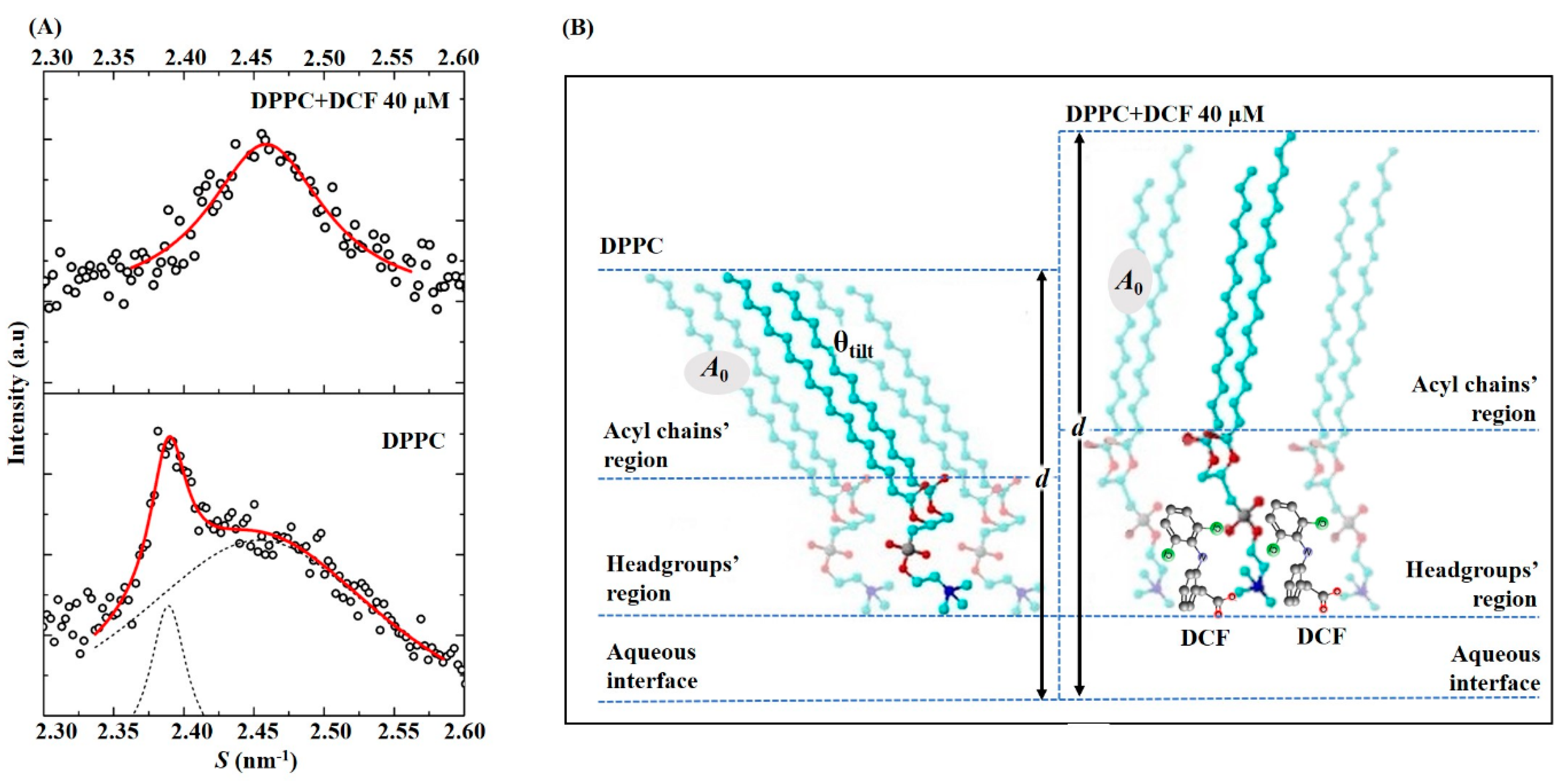
2.2. Diclofenac Location in a Membrane/Aqueous System
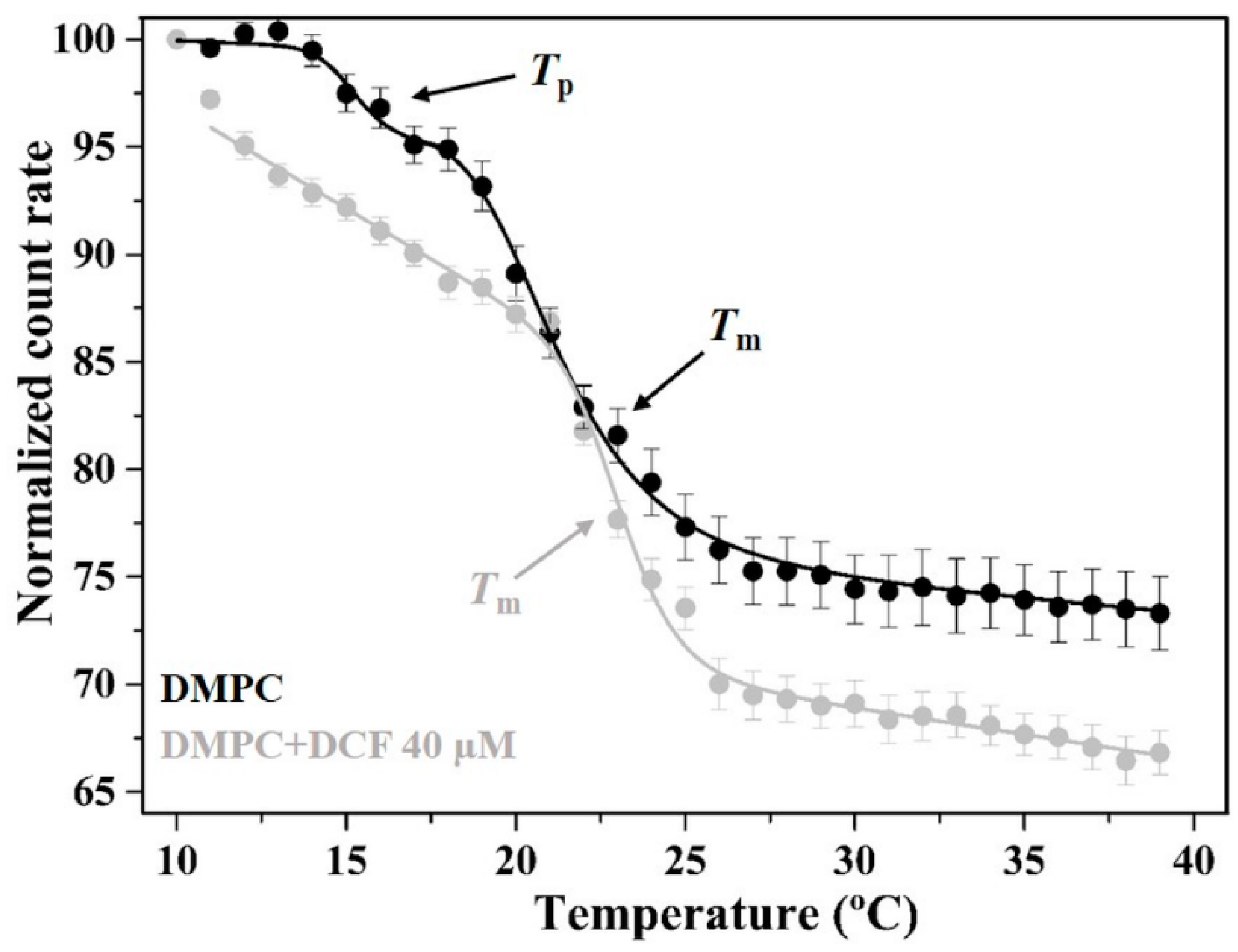
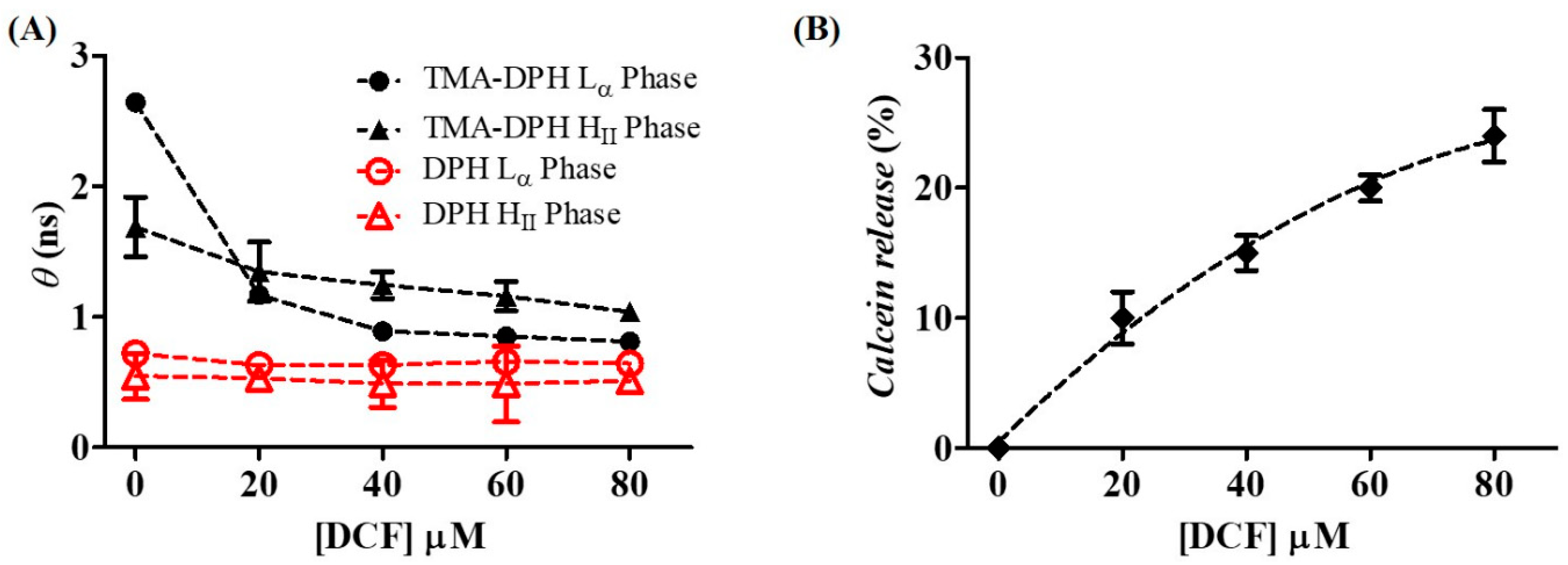
2.3. Effects of Diclofenac in the Microviscosity and Permeability of the Membrane
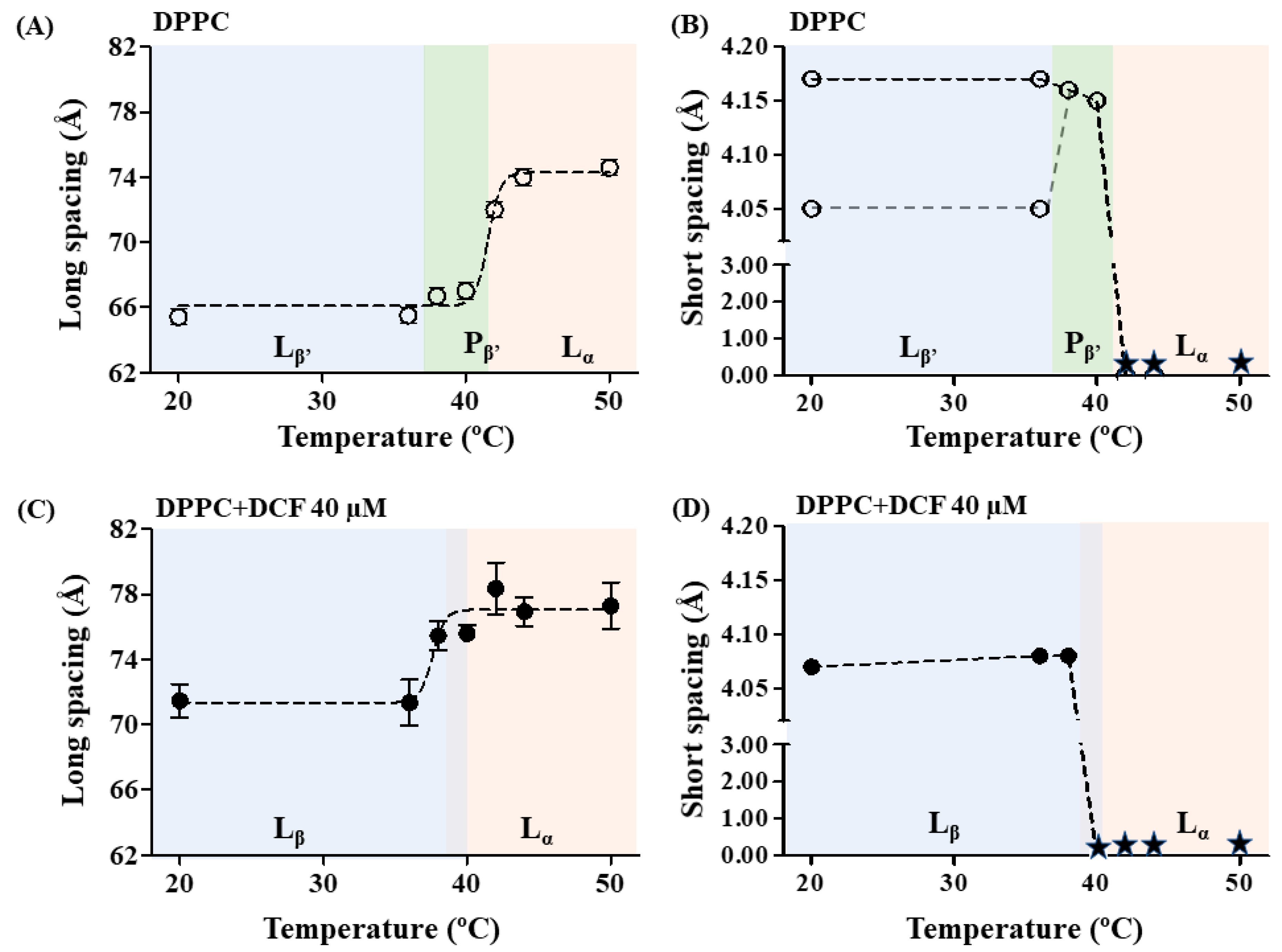
2.4. Effects of Diclofenac in Lipid Packing and Membrane Structure
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Preparation and Labeling of Lipid Model Systems
4.3. Derivative Spectrophotometry Studies
4.4. Fluorescence Quenching Studies
4.5. Fluorescence Anisotropy Studies
4.6. Calcein Leakage Studies
4.7. Differential Scanning Calorimetry Studies
4.8. Dynamic Light Scattering Studies
4.9. Syncrotron Small- and Wide-Angle X-ray Scattering Studies
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| A0 | Cross-sectional area of a single aliphatic chain |
| Absa | Absorbance of the drug in the aqueous media |
| AbsT | Total absorbance of the drug |
| Absl | Absorbance of the drug in the lipid media |
| ATP | Adenosine triphosphate |
| B | Cooperativity of the main phase transition |
| CL | Cardiolipin |
| ξ | Correlation length |
| COX | Cyclooxygenase |
| d | Long or short spacing |
| DCF | Diclofenac or sodium 2-[2-(2,6-dichloroaniline)phenyl]acetate |
| DLS | Dynamic Light Scattering |
| DMPC | 1,2-dimyristoyl-sn-glycero-3-phosphocholine |
| DOPC | 1,2-dioleoyl-sn-glycero-3-phosphocholine |
| DOPE | 1,2-dioleoyl-sn-glycero-3-phosphoethanolamine |
| DPH | 1,6-diphenyl-1,3,5-hexatriene |
| DPPC | 1,2-dipalmitoyl-sn-glycero-3-phosphocholine |
| DSC | Differential Scanning Calorimetry |
| EPC | Egg phosphatidylcholine |
| fwhm | full width of the peaks at one-half of their intensity |
| GI | Gastrointestinal |
| HII | Inverted hexagonal phase |
| Ic | Control fluorescence |
| IMM | Inner mitochondrial membrane |
| IT | Maximum fluorescence value |
| Ix | Fluorescence intensity at a given drug concentration |
| Kd | Distribution coefficient |
| Kq | Bimolecular quenching rate constant |
| KSV | Stern–Volmer constant |
| Lα | Lamellar fluid phase |
| Lβ’ | Lamellar gel phase (tilted chains) |
| Lβ | Lamellar gel phase (untilted chains) |
| LUVs | Large Unilamelar Vesicles |
| MPT | Mitochondrial permeability transition pore |
| NSAID | Non-steroidal anti-inflammatory drug |
| OMM | Outer mitochondrial membrane |
| Pβ’ | Ripple phase |
| PC | Phosphatidylcholine |
| SAXS | Small angle X-ray scattering |
| θ | Rotational correlation time |
| rss | Steady-state fluorescence anisotropy |
| r0 | Fundamental anisotropy |
| τ0 | Lifetime of the excited state |
| Tm | Main phase transition temperature |
| TMA-DPH | N,N,N-Trimethyl-4-(6-phenyl-1,3,5-hexatrien-1-yl)phenylammonium p-toluenesulfonate |
| Tp | Pre-transition temperature |
| TRP | Transient receptor potential |
| Vm | Membrane volume fraction |
| Vl | Lipid molar volume |
| WAXS | Wide Angle X-ray Scattering |
References
- Lonappan, L.; Brar, S.K.; Das, R.K.; Verma, M.; Surampalli, R.Y. Diclofenac and its transformation products: Environmental occurrence and toxicity—A review. Environ. Int. 2016, 96, 127–138. [Google Scholar] [CrossRef] [PubMed]
- Matsui, H.; Murata, Y.; Kobayashi, F.; Shiba, R.; Momo, K.; Kondo, Y.; Nakahara, A.; Muto, H. Diclofenac-induced gastric mucosal fluorescence in rats. Dig. Dis. Sci. 2001, 46, 338–344. [Google Scholar] [CrossRef] [PubMed]
- Yamazaki, T.; Muramoto, M.; Oe, T.; Morikawa, N.; Okitsu, O.; Nagashima, T.; Nishimura, S.; Katayama, Y.; Kita, Y. Diclofenac, a non-steroidal anti-inflammatory drug, suppresses apoptosis induced by endoplasmic reticulum stresses by inhibiting caspase signaling. Neuropharmacology 2006, 50, 558–567. [Google Scholar] [CrossRef] [PubMed]
- Yajima, H.; Yamao, J.; Fukui, H.; Takakura, Y. Up-to-date information on gastric mucosal lesions from long-term NSAID therapy in orthopedic outpatients: A study using logistic regression analysis. J. Orthop. Sci. 2007, 12, 341–346. [Google Scholar] [CrossRef] [PubMed]
- Zien El-Deen, E.E.; El-Mahdy, N.A.; El Rashidy, M.A.; Ghorab, M.M.; Shadeed, G.; Yassin, H.A. Diclofenac-induced gastric ulceration in rats: Protective roles of pantoprazole and misoprostol. Br. J. Pharm. Res. 2016, 11, 1–12. [Google Scholar] [CrossRef]
- Bjarnason, I.; Scarpignato, C.; Holmgren, E.; Olszewski, M.; Rainsford, K.D.; Lanas, A. Mechanisms of damage to the gastrointestinal tract from Nonsteroidal Anti-Inflammatory Drugs. Gastroenterology 2018, 154, 500–514. [Google Scholar] [CrossRef] [PubMed]
- Lúcio, M.; Nunes, C.; Lima, J.L.F.C.; Reis, S. A biophysical approach to the study of the therapeutic and toxic effects of non-steroidal anti-inflammatory drugs. In A Toxicological/Pharmacological Approach to Chemico-Biological Interactions at a Membrane Level; Jurado, A.S., Pedroso de Lima, M.S., Almeida, L., Eds.; Research Signpost, Transworld Research Network: Kerala, India, 2012; pp. 41–70. [Google Scholar]
- Lichtenberger, L.M. Where is the evidence that cyclooxygenase inhibition is the primary cause of nonsteroidal anti-inflammatory drug (NSAID)-induced gastrointestinal injury?: Topical injury revisited. Biochem. Pharmacol. 2001, 61, 631–637. [Google Scholar] [CrossRef]
- Lichtenberger, L.M. Role of Phospholipids in Protection of the GI Mucosa. Dig. Dis. Sci. 2013, 58, 891–893. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lichtenberger, L.M.; Zhou, Y.; Jayaraman, V.; Doyen, J.R.; O’Neil, R.G.; Dial, E.J.; Volk, D.E.; Gorenstein, D.G.; Boggara, M.B.; Krishnamoorti, R. Insight into NSAID-induced membrane alterations, pathogenesis and therapeutics: Characterization of interaction of NSAIDs with phosphatidylcholine. BBA Mol. Cell Biol. Lipids 2012, 1821, 994–1002. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Somasundaram, S.; Rafi, S.; Hayllar, J.; Sigthorsson, G.; Jacob, M.; Price, A.B.; Macpherson, A.; Mahmod, T.; Scott, D.; Wrigglesworth, J.M.; et al. Mitochondrial damage: A possible mechanism of the “topical” phase of NSAID induced injury to the rat intestine. Gut 1997, 41, 344–353. [Google Scholar] [CrossRef] [PubMed]
- Vuda, M.; Kamath, A. Drug induced mitochondrial dysfunction: Mechanisms and adverse clinical consequences. Mitochondrion 2016, 31, 63–74. [Google Scholar] [CrossRef] [PubMed]
- Begriche, K.; Massart, J.; Robin, M.-A.; Borgne-Sanchez, A.; Fromenty, B. Drug-induced toxicity on mitochondria and lipid metabolism: Mechanistic diversity and deleterious consequences for the liver. J. Hepatol. 2011, 54, 773–794. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ng, L.E.; Vincent, A.S.; Halliwell, B.; Wong, K.P. Action of diclofenac on kidney mitochondria and cells. Biochem. Biophys. Res. Commun. 2006, 348, 494–500. [Google Scholar] [CrossRef] [PubMed]
- Altman, R.; Bosch, B.; Brune, K.; Patrignani, P.; Young, C. Advances in NSAID development: Evolution of diclofenac products using pharmaceutical technology. Drugs 2015, 75, 859–877. [Google Scholar] [CrossRef] [PubMed]
- Prince, S.E. Diclofenac-induced renal toxicity in female Wistar albino rats is protected by the pre-treatment of aqueous leaves extract of Madhuca longifolia through suppression of inflammation, oxidative stress and cytokine formation. Biomed Pharmacother 2018, 98, 45–51. [Google Scholar] [CrossRef]
- Galati, G.; Tafazoli, S.; Sabzevari, O.; Chan, T.S.; O’Brien, P.J. Idiosyncratic NSAID drug induced oxidative stress. Chem. Biol. Interact. 2002, 142, 25–41. [Google Scholar] [CrossRef]
- Syed, M.; Skonberg, C.; Hansen, S.H. Mitochondrial toxicity of diclofenac and its metabolites via inhibition of oxidative phosphorylation (ATP synthesis) in rat liver mitochondria: Possible role in drug induced liver injury (DILI). Toxicol. In Vitro 2016, 31, 93–102. [Google Scholar] [CrossRef] [PubMed]
- Masubuchi, Y.; Nakayama, S.; Horie, T. Role of mitochondrial permeability transition in diclofenac-induced hepatocyte injury in rats. Hepatology 2002, 35, 544–551. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dykens, J.A.; Will, Y. The significance of mitochondrial toxicity testing in drug development. Drug Discov. Today 2007, 12, 777–785. [Google Scholar] [CrossRef] [PubMed]
- Giraud, M.-N.; Motta, C.; Romero, J.J.; Bommelaer, G.; Lichtenberger, L.M. Interaction of indomethacin and naproxen with gastric surface-active phospholipids: A possible mechanism for the gastric toxicity of nonsteroidal anti-inflammatory drugs (NSAIDs). Biochem. Pharmacol. 1999, 57, 247–254. [Google Scholar] [CrossRef]
- Treede, I.; Braun, A.; Sparla, R.; Kuhnel, M.; Giese, T.; Turner, J.R.; Anes, E.; Kulaksiz, H.; Fullekrug, J.; Stremmel, W.; et al. Anti-inflammatory effects of phosphatidylcholine. J. Biol. Chem. 2007, 282, 27155–27164. [Google Scholar] [CrossRef] [PubMed]
- Lúcio, M.; Bringezu, F.; Reis, S.; Lima, J.L.; Brezesinski, G. Binding of nonsteroidal anti-inflammatory drugs to DPPC: Structure and thermodynamic aspects. Langmuir 2008, 24, 4132–4139. [Google Scholar] [CrossRef] [PubMed]
- Lúcio, M.; Lima, J.L.F.C.; Reis, S. Drug-membrane interactions: Significance for medicinal chemistry. Curr. Med. Chem. 2010, 17, 1795–1809. [Google Scholar] [CrossRef] [PubMed]
- Manrique-Moreno, M.; Garidel, P.; Suwalsky, M.; Howe, J.; Brandenburg, K. The membrane-activity of Ibuprofen, Diclofenac, and Naproxen: A physico-chemical study with lecithin phospholipids. Biochim. Biophys. Acta 2009, 1788, 1296–1303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nunes, C.; Brezesinski, G.; Lima, J.L.F.C.; Reis, S.; Lúcio, M. Synchrotron SAXS and WAXS study of the interactions of NSAIDs with lipid membranes. J. Phys. Chem. B 2011, 115, 8024–8032. [Google Scholar] [CrossRef] [PubMed]
- Nunes, C.; Brezesinski, G.; Lopes, D.; Lima, J.L.; Reis, S.; Lúcio, M. Lipid-drug interaction: Biophysical effects of tolmetin on membrane mimetic systems of different dimensionality. J. Phys. Chem. B 2011, 115, 12615–12623. [Google Scholar] [CrossRef] [PubMed]
- Nunes, C.; Brezesinski, G.; Pereira-Leite, C.; Lima, J.L.F.C.; Reis, S.; Lúcio, M. NSAIDs interactions with membranes: A biophysical approach. Langmuir 2011, 27, 10847–10858. [Google Scholar] [CrossRef] [PubMed]
- Pereira-Leite, C.; Nunes, C.; Lima, J.L.F.C.; Reis, S.; Lúcio, M. Interaction of celecoxib with membranes: The role of membrane biophysics on its therapeutic and toxic effects. J. Phys. Chem. B 2012, 116, 13608–13617. [Google Scholar] [CrossRef] [PubMed]
- Kanwar, S.S.; Vaiphei, K.; Nehru, B.; Sanyal, S.N. Antioxidative effects of nonsteroidal anti-inflammatory drugs during the initiation stages of experimental colon carcinogenesis in rats. J. Environ. Pathol. Toxicol. Oncol. 2008, 27, 89–100. [Google Scholar] [CrossRef] [PubMed]
- Behal, N.; Kanwar, S.S.; Kaur, J.; Sanyal, S.N. Cyclooxygenase-2 inhibition by etoricoxib modulates plasma membrane fluidity in rat colon. J. Environ. Pathol. Toxicol. Oncol. 2008, 27, 295–302. [Google Scholar] [CrossRef] [PubMed]
- Masubuchi, Y.; Yamada, S.; Horie, T. Diphenylamine as an important structure of nonsteroidal anti-inflammatory drugs to uncouple mitochondrial oxidative phosphorylation. Biochem. Pharmacol. 1999, 58, 861–865. [Google Scholar] [CrossRef]
- Masubuchi, Y.; Yamada, S.; Horie, T. Possible mechanism of hepatocyte injury induced by diphenylamine and its structurally related nonsteroidal anti-inflammatory drugs. J. Pharmacol. Exp. Ther. 2000, 292, 982–987. [Google Scholar] [PubMed]
- Davies, N.M.; Anderson, K.E. Clinical Pharmacokinetics of Diclofenac. Clin. Pharmacokinet. 1997, 33, 184–213. [Google Scholar] [CrossRef] [PubMed]
- Moreno-Sánchez, R.; Bravo, C.; Vásquez, C.; Ayala, G.; Silveira, L.H.; Martínez-Lavín, M. Inhibition and uncoupling of oxidative phosphorylation by nosteroidal anti-inflammatory drugs: Study in mitochondria, submitochondrial particles, cells, and whole heart. Biochem. Pharmacol. 1999, 57, 743–752. [Google Scholar] [CrossRef]
- Hynes, J.; Marroquin, L.D.; Ogurtsov, V.; Christiansen, K.N.; Stevens, G.J.; Papkovsky, D.B.; Will, Y. Investigation of drug-induced mitochondrial toxicity using fluorescence-based oxygen-sensitive probes. Toxicol. Sci. 2006, 92, 186–200. [Google Scholar] [CrossRef] [PubMed]
- Gwanyanya, A.; Macianskiene, R.; Mubagwa, K. Insights into the effects of diclofenac and other non-steroidal anti-inflammatory agents on ion channels. J. Pharm. Pharmacol. 2012, 64, 1359–1375. [Google Scholar] [CrossRef] [PubMed]
- Mazumder, S.; De, R.; Sarkar, S.; Siddiqui, A.A.; Saha, S.J.; Banerjee, C.; Iqbal, M.S.; Nag, S.; Debsharma, S.; Bandyopadhyay, U. Selective scavenging of intra-mitochondrial superoxide corrects diclofenac-induced mitochondrial dysfunction and gastric injury: A novel gastroprotective mechanism independent of gastric acid suppression. Biochem. Pharmacol. 2016, 121, 33–51. [Google Scholar] [CrossRef] [PubMed]
- Ikon, N.; Ryan, R.O. Cardiolipin and mitochondrial cristae organization. Biochim. Biophys. Acta Biomembr. 2017, 1859, 1156–1163. [Google Scholar] [CrossRef] [PubMed]
- Claypool, S.M. Cardiolipin, a critical determinant of mitochondrial carrier protein assembly and function. Biochim. Biophys. Acta 2009, 1788, 2059–2068. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schlattner, U.; Tokarska-Schlattner, M.; Ramirez, S.; Brückner, A.; Kay, L.; Polge, C.; Epand, R.F.; Lee, R.M.; Lacombe, M.L.; Epand, R.M. Mitochondrial kinases and their molecular interaction with cardiolipin. Biochim. Biophys. Acta 2009, 1788, 2032–2047. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dahlberg, M. Polymorphic phase behavior of cardiolipin derivatives studied by coarse-grained molecular dynamics. J. Phys. Chem. B 2007, 111, 7194–7200. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, H.; Lúcio, M.; Lima, J.L.; Matos, C.; Reis, S. Effects of diclofenac on EPC liposome membrane properties. Anal. Bioanal. Chem. 2005, 382, 1256–1264. [Google Scholar] [CrossRef] [PubMed]
- Souza, S.M.; Oliveira, O.N.; Scarpa, M.V.; Oliveira, A.G. Study of the diclofenac/phospholipid interactions with liposomes and monolayers. Colloids Surf. B Biointerfaces 2004, 36, 13–17. [Google Scholar] [CrossRef] [PubMed]
- Suwalsky, M.; Manrique, M.; Villena, F.; Sotomayor, C.P. Structural effects in vitro of the anti-inflammatory drug diclofenac on human erythrocytes and molecular models of cell membranes. Biophys. Chem. 2009, 141, 34–40. [Google Scholar] [CrossRef] [PubMed]
- Alex, A. Physicochemical profiling (solubility, permeability and charge state). Curr. Top. Med. Chem. 2001, 1, 277–351. [Google Scholar] [CrossRef]
- Lúcio, M.; Lima, J.L.F.C.; Reis, S. Chapter 8: Drug-membrane interactions: Molecular mechanisms underlying therapeutic and toxic effects of drugs. In Ideas in Chemistry and Molecular Sciences: Where Chemistry Meets Life; Pignataro, B., Ed.; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2010; pp. 191–203. [Google Scholar]
- Bort, R.; Ponsoda, X.; Jover, R.; Gómez-Lechón, M.J.; Castell, J.V. Diclofenac toxicity to hepatocytes: A role for drug metabolism in cell toxicity. J. Pharmacol. Exp. Ther. 1999, 288, 65–72. [Google Scholar] [PubMed]
- Rainsford, K.D.; Bjarnason, I. NSAIDs: Take with food or after fasting? J. Pharm. Pharmacol. 2012, 64, 465–469. [Google Scholar] [CrossRef] [PubMed]
- Fernandes, E.; Soares, T.B.; Gonçalves, H.; Lúcio, M. Spectroscopic studies as a toolbox for biophysical and chemical characterization of lipid-based nanotherapeutics. Front. Chem. 2018, 6, 1–20. [Google Scholar] [CrossRef] [PubMed]
- Kitamura, K.; Imayoshi, N.; Goto, T.; Shiro, H.; Mano, T.; Nakai, Y. Second derivative spectrophotometric determination of partition coefficients of chlorpromazine and promazine between lecithin bilayer vesicles and water. Anal. Chim. Acta 1995, 304, 101–106. [Google Scholar] [CrossRef]
- Demchenko, A.P.; Mely, Y.; Duportail, G.; Klymchenko, A.S. Monitoring biophysical properties of lipid membranes by environment-sensitive fluorescent probes. Biophys. J. 2009, 96, 3461–3470. [Google Scholar] [CrossRef] [PubMed]
- Lentz, B.R. Membrane “fluidity” as detected by diphenylhexatriene probes. Chem. Phys. Lipids 1989, 50, 171–190. [Google Scholar] [CrossRef]
- Prendergast, F.G.; Haugland, R.P.; Callahan, P.J. 1-[4-(Trimethylamino)phenyl]-6-phenylhexa-1,3,5-triene: Synthesis, fluorescence properties and use as a fluorescence probe of lipid bilayers. Biochemistry 1981, 20, 7333–7338. [Google Scholar] [CrossRef] [PubMed]
- Lakowicz, J.R. Principles of Fluorescence Spectroscopy, 3rd ed.; Springer: New York, NY, USA, 2016. [Google Scholar] [CrossRef]
- Matsui, H.; Shimokawa, O.; Kaneko, T.; Nagano, Y.; Rai, K.; Hyodo, I. The pathophysiology of non-steroidal anti-inflammatory drug (NSAID)-induced mucosal injuries in stomach and small intestine. J. Clin. Biochem. Nutr. 2011, 48, 107–111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chakraborty, H.; Chakraborty, P.K.; Raha, S.; Mandal, P.C.; Sarkar, M. Interaction of piroxicam with mitochondrial membrane and cytochrome c. Biochim. Biophys. Acta 2007, 1768, 1138–1146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Monteiro, J.P.; Martins, A.F.; Lúcio, M.; Reis, S.; Pinheiro, T.J.; Geraldes, C.F.; Oliveira, P.J.; Jurado, A.S. Nimesulide interaction with membrane model systems: Are membrane physical effects involved in nimesulide mitochondrial toxicity? Toxicol. In Vitro 2011, 25, 1215–1223. [Google Scholar] [CrossRef] [PubMed]
- Monteiro, J.P.; Martins, A.F.; Lúcio, M.; Reis, S.; Geraldes, C.F.; Oliveira, P.J.; Jurado, A.S. Interaction of carbonylcyanide p-trifluoromethoxyphenylhydrazone (FCCP) with lipid membrane systems: A biophysical approach with relevance to mitochondrial uncoupling. J. Bioenerg. Biomembr. 2011, 43, 287–298. [Google Scholar] [CrossRef] [PubMed]
- Monteiro, J.P.; Martins, A.F.; Nunes, C.; Morais, C.M.; Lúcio, M.; Reis, S.; Pinheiro, T.J.; Geraldes, C.F.; Oliveira, P.J.; Jurado, A.S. A biophysical approach to menadione membrane interactions: Relevance for menadione-induced mitochondria dysfunction and related deleterious/therapeutic effects. Biochim. Biophys. Acta 2013, 1828, 1899–1908. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kyrikou, I.; Hadjikakou, S.K.; Kovala-Demertzi, D.; Viras, K.; Mavromoustakos, T. Effects of non-steroid anti-inflammatory drugs in membrane bilayers. Chem. Phys. Lipids 2004, 132, 157–169. [Google Scholar] [CrossRef] [PubMed]
- Michel, N.; Fabiano, A.-S.; Polidori, A.; Jack, R.; Pucci, B. Determination of phase transition temperatures of lipids by light scattering. Chem. Phys. Lipids 2006, 139, 11–19. [Google Scholar] [CrossRef] [PubMed]
- Seydel, J.K. Analytical tools for the analysis and quantification of drug-membrane interactions. In Drug-Membrane Interactions; Mannhold, R., Kubinyi, H., Folkers, G., Seydel, J.K., Wiese, M., Eds.; Wiley, VCH: New York, NY, USA, 2013. [Google Scholar] [CrossRef]
- Jain, M.K.; Wu, N.M. Effect of small molecules on the dipalmitoyl lecithin liposomal bilayer: III. Phase transition in lipid bilayer. J. Membr. Biol. 1977, 34, 157–201. [Google Scholar] [CrossRef]
- Kiselev, M.A.; Lombardo, D. Structural characterization in mixed lipid membrane systems by neutron and X-ray scattering. Biochim. Biophys. Acta Gen. Subj. 2017, 1861, 3700–3717. [Google Scholar] [CrossRef] [PubMed]
- Wilson-Ashworth, H.A.; Bahm, Q.; Erickson, J.; Shinkle, A.; Vu, M.P.; Woodbury, D.; Bell, J.D. Differential detection of phospholipid fluidity, order, and spacing by fluorescence spectroscopy of bis-pyrene, prodan, nystatin, and merocyanine 540. Biophys. J. 2006, 91, 4091–4101. [Google Scholar] [CrossRef] [PubMed]
- Jacobson, K.; Papahadjopoulos, D. Phase transitions and phase separations in phospholipid membranes induced by changes in temperature, pH, and concentration of bivalent cations. Biochemistry 1975, 14, 152–161. [Google Scholar] [CrossRef] [PubMed]
- Small, D.M. The Physical Chemistry of Lipids: From Alkanes to Phospholipids; Plenum Press: New York, NY, USA, 1986. [Google Scholar]
- Tristram-Nagle, S.; Zhang, R.; Suter, R.M.; Worthington, C.R.; Sun, W.J.; Nagle, J.F. Measurement of chain tilt angle in fully hydrated bilayers of gel phase lecithins. Biophys. J. 1993, 64, 1097–1109. [Google Scholar] [CrossRef] [Green Version]
- Hauet, N.; Artzner, F.; Boucher, F.; Grabielle-Madelmont, C.; Cloutier, I.; Keller, G.; Lesieur, P.; Durand, D.; Paternostre, M. Interaction between artificial membranes and enflurane, a general volatile anesthetic: DPPC-enflurane Interaction. Biophys. J. 2003, 84, 3123–3137. [Google Scholar] [CrossRef]
- Chakraborty, H.; Roy, S.; Sarkar, M. Interaction of oxicam NSAIDs with DMPC vesicles: Differential partitioning of drugs. Chem. Phys. Lipids 2005, 138, 20–28. [Google Scholar] [CrossRef] [PubMed]
- Lichtenberger, L.M.; Wang, Z.-M.; Romero, J.J.; Ulloa, C.; Perez, J.C.; Giraud, M.-N.; Barreto, J.C. Non-steroidal anti-inflammatory drugs (NSAIDs) associate with zwitterionic phospholipids: Insight into the mechanism and reversal of NSAID-induced gastrointestinal injury. Nat. Med. 1995, 1, 154. [Google Scholar] [CrossRef] [PubMed]
- Lee, A.G. How lipids affect the activities of integral membrane proteins. Biochim. Biophys. Acta 2004, 1666, 62–87. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Zheng, L.X.; Yang, F.Y. Effect of propensity of hexagonal II phase formation on the activity of mitochondrial ubiquinol-cytochrome c reductase and H+-ATPase. Chem. Phys. Lipids 1995, 76, 135–144. [Google Scholar] [CrossRef]
- Krause, M.M.; Brand, M.D.; Krauss, S.; Meisel, C.; Vergin, H.; Burmester, G.R.; Buttgereit, F. Nonsteroidal antiinflammatory drugs and a selective cyclooxygenase 2 inhibitor uncouple mitochondria in intact cells. Arthritis Rheumatol. 2003, 48, 1438–1444. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McCormack, K.; Brune, K. Classical absorption theory and the development of gastric mucosal damage associated with the non-steroidal anti-inflammatory drugs. Arch. Toxicol. 1987, 60, 261–269. [Google Scholar] [CrossRef] [PubMed]
- Różycki, B.; Lipowsky, R. Spontaneous curvature of bilayer membranes from molecular simulations: Asymmetric lipid densities and asymmetric adsorption. J. Chem. Phys. 2015, 142, 054101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shinoda, W. Permeability across lipid membranes. Biochim. Biophys. Acta Biomembr. 2016, 1858, 2254–2265. [Google Scholar] [CrossRef] [PubMed]
- Magalhaes, L.M.; Nunes, C.; Lucio, M.; Segundo, M.A.; Reis, S.; Lima, J.L.F.C. High-throughput microplate assay for the determination of drug partition coefficients. Nat. Protoc. 2010, 5, 1823–1830. [Google Scholar] [CrossRef] [PubMed]
- Lakowicz, J.R.; Prendergast, F.G.; Hogen, D. Fluorescence anisotropy measurements under oxygen quenching conditions as a method to quantify the depolarizing rotations of fluorophores. Application to diphenylhexatriene in isotropic solvents and in lipid bilayers. Biochemistry 1979, 18, 520–527. [Google Scholar] [CrossRef] [PubMed]
- Lúcio, M.; Nunes, C.; Gaspar, D.; Gołębska, K.; Wisniewski, M.; Lima, J.L.F.C.; Brezesinski, G.; Reis, S. Effect of anti-inflammatory drugs in phosphatidylcholine membranes: A fluorescence and calorimetric study. Chem. Phys. Lett. 2009, 471, 300–309. [Google Scholar] [CrossRef]
- Bartlett, G.R. Phosphorus assay in column chromatography. J. Biol. Chem. 1959, 234, 466–468. [Google Scholar] [PubMed]
- Pereira-Leite, C.; Carneiro, C.; Soares, J.X.; Afonso, C.; Nunes, C.; Lucio, M.; Reis, S. Biophysical characterization of the drug-membrane interactions: The case of propranolol and acebutolol. Eur. J. Pharm. Biopharm. 2013, 84, 183–191. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| pH | Δλ | Kd | Log Kd |
|---|---|---|---|
| 3.0 | 26,000 ± 3000 1 | 4.41 ± 0.05 1,*** | |
| 5.0 | 319–328 | 6970 ± 379 2 | 3.84 ± 0.02 2,*** |
| 7.4 | 1200 ± 100 1 | 3.08 ± 0.04 1,*** |
| GI mimicking: PC Membrane Model | ||
| Probe | Lipid Phase | Kq × 109 (M−1·s−1) |
| TMA-DPH | Lβ′ | 2.63 ± 0.03 |
| Lα | 2.30 ± 0.09 *** | |
| DPH | Lβ′ | 0.37 ± 0.02 *** |
| Lα | 1.34 ± 0.04 *** | |
| IMM mimicking: DOPC:DOPE:CL (1:1:1) Membrane Model | ||
| Probe | Lipid Phase | Kq × 109 (M−1·s−1) |
| TMA-DPH | Lα | 1.24 ± 0.06 *** |
| HII | 2.90 ± 0.23 *** | |
| DPH | Lα | 0.73 ± 0.04 * |
| HII | 1.87 ± 0.29 *** | |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fernandes, E.; Soares, T.B.; Gonçalves, H.; Bernstorff, S.; Real Oliveira, M.E.C.D.; Lopes, C.M.; Lúcio, M. A Molecular Biophysical Approach to Diclofenac Topical Gastrointestinal Damage. Int. J. Mol. Sci. 2018, 19, 3411. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms19113411
Fernandes E, Soares TB, Gonçalves H, Bernstorff S, Real Oliveira MECD, Lopes CM, Lúcio M. A Molecular Biophysical Approach to Diclofenac Topical Gastrointestinal Damage. International Journal of Molecular Sciences. 2018; 19(11):3411. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms19113411
Chicago/Turabian StyleFernandes, Eduarda, Telma B. Soares, Hugo Gonçalves, Sigrid Bernstorff, Maria Elisabete C. D. Real Oliveira, Carla M. Lopes, and Marlene Lúcio. 2018. "A Molecular Biophysical Approach to Diclofenac Topical Gastrointestinal Damage" International Journal of Molecular Sciences 19, no. 11: 3411. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms19113411