The Human Microbiota and Obesity: A Literature Systematic Review of In Vivo Models and Technical Approaches

 ,
,  ,
,
Abstract
:1. Introduction
2. Animal Models as Tools to Study the Human Gut Microbiota
2.1. Non-Mammalian Models of the Human Microbiome
2.1.1. Hydra
2.1.2. Honeybee
2.1.3. Zebra Fish
2.2. Mammalian Models of the Human Microbiome
2.2.1. Germ-Free (GF) Mice
2.2.2. Rat
2.2.3. Pigs
3. Methodologies for the Study of Microbiota
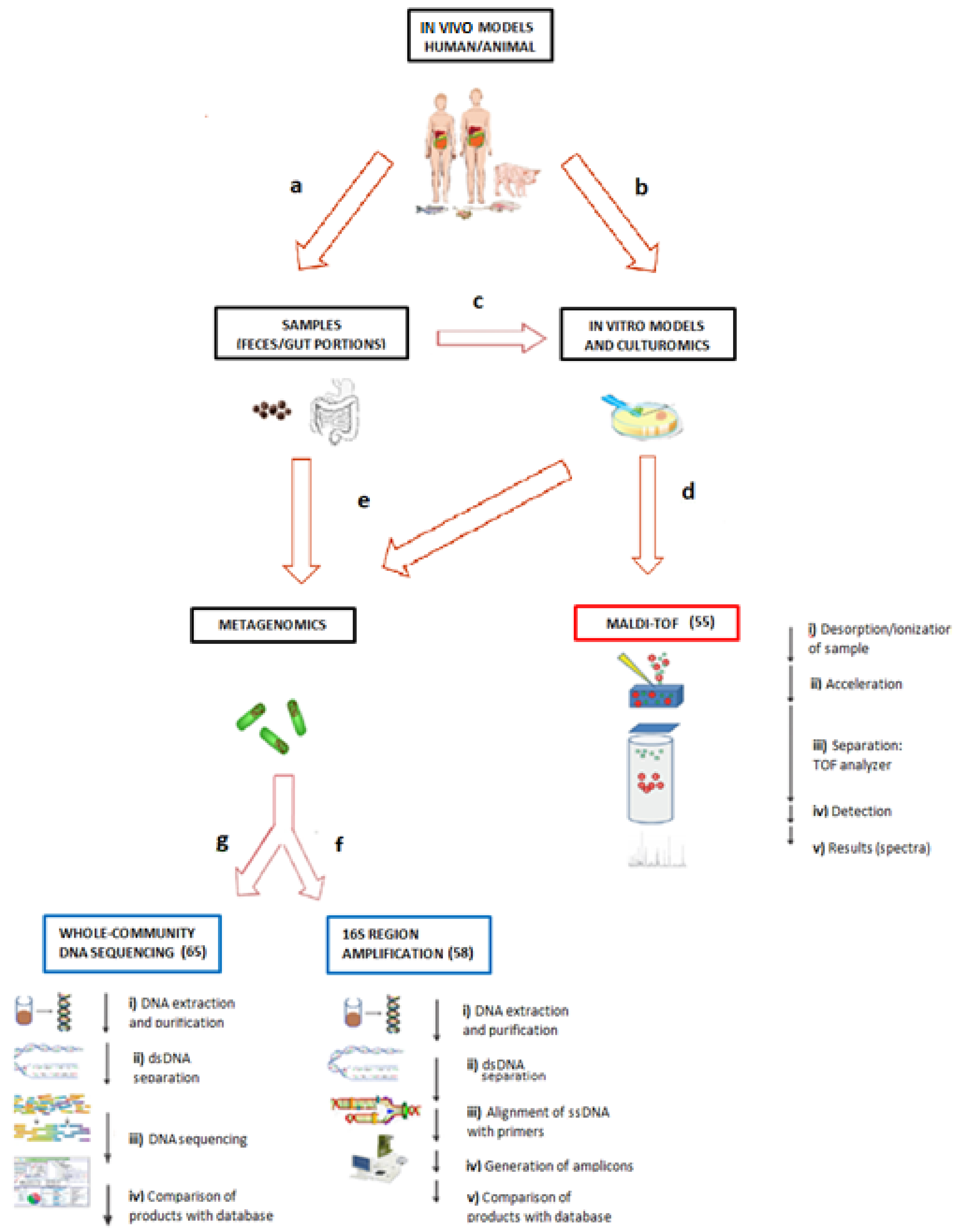
3.1. Culturomics and Matrix-Assisted Laser Desorption/Ionization–Time of Flight (MALDI–TOF)
3.2. Metagenomics
3.3. Database for Microbiota Genomic Data
4. Fecal Microbiota Transplantation as a New Therapeutic Approach for Obesity
5. Concluding Remarks and Perspectives
6. Search Strategy and Selection Criteria
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- World Health Organ. Obesity and Overweight. Available online: http://www.who.int/news-room/fact-sheets/detail/obesity-and-overweight (accessed on 21 August 2018).
- Hruby, A.; Hu, F.B. The Epidemiology of Obesity: A Big Picture. PharmacoEconomics 2015, 33, 673–689. [Google Scholar] [CrossRef] [PubMed]
- Duranti, S.; Ferrario, C.; van Sinderen, D.; Ventura, M.; Turroni, F. Obesity and microbiota: An example of an intricate relationship. Genes Nutr. 2017, 12, 18. [Google Scholar] [CrossRef] [PubMed]
- Carrera-Quintanar, L.; López Roa, R.I.; Quintero-Fabián, S.; Sánchez-Sánchez, M.A.; Vizmanos, B.; Ortuño-Sahagún, D. Phytochemicals That Influence Gut Microbiota as Prophylactics and for the Treatment of Obesity and Inflammatory Diseases. Mediat. Inflamm. 2018, 2018, 9734845. [Google Scholar] [CrossRef] [PubMed]
- Goodman, A.L. In vivo and animal models of the human gut microbiome. In Humman Microbiota Microbiome; Marchesi, J.R., Ed.; CABI: Wallingford, UK, 2014; pp. 124–135. [Google Scholar]
- Ellegaard, K.M.; Engel, P. Beyond 16S rRNA Community Profiling: Intra-Species Diversity in the Gut Microbiota. Front. Microbiol. 2016, 7, 1475. [Google Scholar] [CrossRef] [PubMed]
- Murillo-Rincon, A.P.; Klimovich, A.; Pemöller, E.; Taubenheim, J.; Mortzfeld, B.; Augustin, R.; Bosch, T.C.G. Spontaneous body contractions are modulated by the microbiome of Hydra. Sci. Rep. 2017, 7, 15937. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buzgariu, W.; Al Haddad, S.; Tomczyk, S.; Wenger, Y.; Galliot, B. Multi-functionality and plasticity characterize epithelial cells in Hydra. Tissue Barriers 2015, 3, e1068908. [Google Scholar] [CrossRef] [PubMed]
- Tomczyk, S.; Fischer, K.; Austad, S.; Galliot, B. Hydra, a powerful model for aging studies. Invertebr. Reprod. Dev. 2015, 59, 11–16. [Google Scholar] [CrossRef] [PubMed]
- Augustin, R.; Schröder, K.; Rincón, A.P.M.; Fraune, S.; Anton-Erxleben, F.; Herbst, E.M.; Wittlieb, J.; Schwentner, M.; Grötzinger, J.; Wassenaar, T.M.; et al. A secreted antibacterial neuropeptide shapes the microbiome of Hydra. Nat. Commun. 2017, 8, 698. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deines, P.; Bosch, T.C.G. Transitioning from Microbiome Composition to Microbial Community Interactions: The Potential of the Metaorganism Hydra as an Experimental Model. Front. Microbiol. 2016, 7, 1610. [Google Scholar] [CrossRef] [PubMed]
- Greenblum, S.; Carr, R.; Borenstein, E. Extensive strain-level copy-number variation across human gut microbiome species. Cell 2015, 160, 583–594. [Google Scholar] [CrossRef] [PubMed]
- Raymann, K.; Bobay, L.-M.; Moran, N.A. Antibiotics reduce genetic diversity of core species in the honeybee gut microbiome. Mol. Ecol. 2018, 27, 2057–2066. [Google Scholar] [CrossRef] [PubMed]
- Powell, E.; Ratnayeke, N.; Moran, N.A. Strain diversity and host specificity in a specialized gut symbiont of honeybees and bumblebees. Mol. Ecol. 2016, 25, 4461–4471. [Google Scholar] [CrossRef] [PubMed]
- Ellegaard, K.M.; Tamarit, D.; Javelind, E.; Olofsson, T.C.; Andersson, S.G.E.; Vásquez, A. Extensive intra-phylotype diversity in lactobacilli and bifidobacteria from the honeybee gut. BMC Genom. 2015, 16, 284. [Google Scholar] [CrossRef] [PubMed]
- Stones, D.H.; Fehr, A.G.J.; Thompson, L.; Rocha, J.; Perez-Soto, N.; Madhavan, V.T.P.; Voelz, K.; Krachler, A.M. Zebrafish (Danio rerio) as a Vertebrate Model Host to Study Colonization, Pathogenesis, and Transmission of Foodborne Escherichia coli O157. mSphere 2017, 2, e00365–17. [Google Scholar] [CrossRef] [PubMed]
- Sheng, Y.; Ren, H.; Limbu, S.M.; Sun, Y.; Qiao, F.; Zhai, W.; Du, Z.-Y.; Zhang, M. The Presence or Absence of Intestinal Microbiota Affects Lipid Deposition and Related Genes Expression in Zebrafish (Danio rerio). Front. Microbiol. 2018, 9, 1124. [Google Scholar] [CrossRef] [PubMed]
- Zac Stephens, W.; Burns, A.R.; Stagaman, K.; Wong, S.; Rawls, J.F.; Guillemin, K.; Bohannan, B.J.M. The composition of the zebrafish intestinal microbial community varies across development. ISME J. 2016, 10, 644–654. [Google Scholar] [CrossRef] [PubMed]
- David, L.A.; Maurice, C.F.; Carmody, R.N.; Gootenberg, D.B.; Button, J.E.; Wolfe, B.E.; Ling, A.V.; Devlin, A.S.; Varma, Y.; Fischbach, M.A.; et al. Diet rapidly and reproducibly alters the human gut microbiome. Nature 2014, 505, 559–563. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wong, S.; Stephens, W.Z.; Burns, A.R.; Stagaman, K.; David, L.A.; Bohannan, B.J.M.; Guillemin, K.; Rawls, J.F. Ontogenetic Differences in Dietary Fat Influence Microbiota Assembly in the Zebrafish Gut. mBio 2015, 6, e00687-15. [Google Scholar] [CrossRef] [PubMed]
- Valenzuela, M.-J.; Caruffo, M.; Herrera, Y.; Medina, D.A.; Coronado, M.; Feijóo, C.G.; Muñoz, S.; Garrido, D.; Troncoso, M.; Figueroa, G.; et al. Evaluating the Capacity of Human Gut Microorganisms to Colonize the Zebrafish Larvae (Danio rerio). Front. Microbiol. 2018, 9, 1032. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Ma, L.; Ma, Y.; Zhang, F.; Zhao, C.; Nie, Y. Insights into the role of gut microbiota in obesity: Pathogenesis, mechanisms, and therapeutic perspectives. Protein Cell 2018, 9, 397–403. [Google Scholar] [CrossRef] [PubMed]
- Bäckhed, F.; Ding, H.; Wang, T.; Hooper, L.V.; Koh, G.Y.; Nagy, A.; Semenkovich, C.F.; Gordon, J.I. The gut microbiota as an environmental factor that regulates fat storage. Proc. Natl. Acad. Sci. USA 2004, 101, 15718–15723. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boulangé, C.L.; Neves, A.L.; Chilloux, J.; Nicholson, J.K.; Dumas, M.-E. Impact of the gut microbiota on inflammation, obesity, and metabolic disease. Genome Med. 2016, 8, 42. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Kuang, Z.; Yu, X.; Ruhn, K.A.; Kubo, M.; Hooper, L.V. The intestinal microbiota regulates body composition through NFIL3 and the circadian clock. Science 2017, 357, 912–916. [Google Scholar] [CrossRef] [PubMed]
- Orland, F.J.; Blayney, J.R.; Harrison, R.W.; Reyniers, J.A.; Trexler, P.C.; Ervin, R.F.; Gordon, H.A.; Wagner, M. Experimental caries in germfree rats inoculated with enterococci. J. Am. Dent. Assoc. 1955, 50, 259–272. [Google Scholar] [CrossRef] [PubMed]
- Martín, R.; Bermúdez-Humarán, L.G.; Langella, P. Gnotobiotic Rodents: An In Vivo Model for the Study of Microbe-Microbe Interactions. Front. Microbiol. 2016, 7, 409. [Google Scholar] [CrossRef] [PubMed]
- Sugahara, H.; Odamaki, T.; Fukuda, S.; Kato, T.; Xiao, J.; Abe, F.; Kikuchi, J.; Ohno, H. Probiotic Bifidobacterium longum alters gut luminal metabolism through modification of the gut microbial community. Sci. Rep. 2015, 5, 13548. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aron-Wisnewsky, J.; Clément, K. The gut microbiome, diet, and links to cardiometabolic and chronic disorders. Nat. Rev. Nephrol. 2016, 12, 169–181. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Chen, H.; Mao, B.; Yang, Q.; Zhao, J.; Gu, Z.; Zhang, H.; Chen, Y.Q.; Chen, W. Microbial Biogeography and Core Microbiota of the Rat Digestive Tract. Sci. Rep. 2017, 8, 45840. [Google Scholar] [CrossRef] [PubMed]
- Lecomte, V.; Kaakoush, N.O.; Maloney, C.A.; Raipuria, M.; Huinao, K.D.; Mitchell, H.M.; Morris, M.J. Changes in Gut Microbiota in Rats Fed a High Fat Diet Correlate with Obesity-Associated Metabolic Parameters. PLoS ONE 2015, 10, e0126931. [Google Scholar] [CrossRef] [PubMed]
- Flemer, B.; Gaci, N.; Borrel, G.; Sanderson, I.R.; Chaudhary, P.P.; Tottey, W.; O’Toole, P.W.; Brugère, J.F. Fecal microbiota variation across the lifespan of the healthy laboratory rat. Gut Microbes 2017, 8, 428–439. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Al-awar, A.; Kupai, K.; Veszelka, M.; Szűcs, G.; Attieh, Z.; Murlasits, Z.; Török, S.; Pósa, A.; Varga, C. Experimental Diabetes Mellitus in Different Animal Models. J. Diabetes Res. 2016, 2016, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Tulstrup, M.V.-L.; Christensen, E.G.; Carvalho, V.; Linninge, C.; Ahrné, S.; Højberg, O.; Licht, T.R.; Bahl, M.I. Antibiotic Treatment Affects Intestinal Permeability and Gut Microbial Composition in Wistar Rats Dependent on Antibiotic Class. PLoS ONE 2015, 10, e0144854. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ericsson, A.C.; Akter, S.; Hanson, M.M.; Busi, S.B.; Parker, T.W.; Schehr, R.J.; Hankins, M.A.; Ahner, C.E.; Davis, J.W.; Franklin, C.L.; et al. Differential susceptibility to colorectal cancer due to naturally occurring gut microbiota. Oncotarget 2015, 6, 33689–33704. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rabot, S.; Jaglin, M.; Daugé, V.; Naudon, L. Impact of the gut microbiota on the neuroendocrine and behavioural responses to stress in rodents. OCL 2016, 23, D116. [Google Scholar] [CrossRef]
- Chen, M.; Lu, B.; Li, Y.; Wang, Y.; Zheng, H.; Zhong, D.; Liao, Z.; Wang, M.; Ma, F.; Liao, Q.; et al. Metabolomics insights into the modulatory effects of long-term compound polysaccharide intake in high-fat diet-induced obese rats. Nutr. Metab. 2018, 15, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marques, C.; Meireles, M.; Norberto, S.; Leite, J.; Freitas, J.; Pestana, D.; Faria, A.; Calhau, C. High-fat diet-induced obesity Rat model: A comparison between Wistar and Sprague-Dawley Rat. Adipocyte 2015, 5, 11–21. [Google Scholar] [CrossRef] [PubMed]
- Sciascia, Q.; Daş, G.; Metges, C.C. REVIEW: The pig as a model for humans: Effects of nutritional factors on intestinal function and health. J. Anim. Sci. 2016, 94, 441–452. [Google Scholar] [CrossRef]
- Yang, H.; Huang, X.; Fang, S.; Xin, W.; Huang, L.; Chen, C. Uncovering the composition of microbial community structure and metagenomics among three gut locations in pigs with distinct fatness. Sci. Rep. 2016, 6, 27427. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rérat, A.; Fiszlewicz, M.; Giusi, A.; Vaugelade, P. Influence of meal frequency on postprandial variations in the production and absorption of volatile fatty acids in the digestive tract of conscious pigs. J. Anim. Sci. 1987, 64, 448–456. [Google Scholar] [CrossRef] [PubMed]
- Von Engelhardt, W.; Bartels, J.; Kirschberger, S.; Meyer zu Düttingdorf, H.D.; Busche, R. Role of short-chain fatty acids in the hind gut. Vet. Q. 1998, 20 (Suppl. 3), S52–S59. [Google Scholar] [CrossRef] [PubMed]
- Fan, P.; Liu, P.; Song, P.; Chen, X.; Ma, X. Moderate dietary protein restriction alters the composition of gut microbiota and improves ileal barrier function in adult pig model. Sci. Rep. 2017, 7, 43412. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heinritz, S.N.; Weiss, E.; Eklund, M.; Aumiller, T.; Heyer, C.M.E.; Messner, S.; Rings, A.; Louis, S.; Bischoff, S.C.; Mosenthin, R. Impact of a High-Fat or High-Fiber Diet on Intestinal Microbiota and Metabolic Markers in a Pig Model. Nutrients 2016, 8, 317. [Google Scholar] [CrossRef] [PubMed]
- Franzenburg, S.; Fraune, S.; Altrock, P.M.; Künzel, S.; Baines, J.F.; Traulsen, A.; Bosch, T.C. Bacterial colonization of Hydra hatchlings follows a robust temporal pattern. ISME J. 2013, 7, 781–790. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kešnerová, L.; Mars, R.A.T.; Ellegaard, K.M.; Troilo, M.; Sauer, U.; Engel, P. Disentangling metabolic functions of bacteria in the honey bee gut. PLoS Biol. 2017, 15, e2003467. [Google Scholar] [CrossRef] [PubMed]
- Marcobal, A.; Yusufaly, T.; Higginbottom, S.; Snyder, M.; Sonnenburg, J.L.; Mias, G.I. Metabolome progression during early gut microbial colonization of gnotobiotic mice. Sci. Rep. 2015, 5, 11589. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heinritz, S.N.; Weiss, E.; Eklund, M.; Aumiller, T.; Louis, S.; Rings, A.; Messner, S.; Camarinha-Silva, A.; Seifert, J.; Bischoff, S.C.; et al. Intestinal Microbiota and Microbial Metabolites Are Changed in a Pig Model Fed a High-Fat/Low-Fiber or a Low-Fat/High-Fiber Diet. PLoS ONE 2016, 11, e0154329. [Google Scholar] [CrossRef] [PubMed]
- Tanner, S.A.; Zihler Berner, A.; Rigozzi, E.; Grattepanche, F.; Chassard, C.; Lacroix, C. In Vitro Continuous Fermentation Model (PolyFermS) of the Swine Proximal Colon for Simultaneous Testing on the Same Gut Microbiota. PLoS ONE 2014, 9, e94123. [Google Scholar] [CrossRef] [PubMed]
- Poeker, S.A.; Geirnaert, A.; Berchtold, L.; Greppi, A.; Krych, L.; Steinert, R.E.; de Wouters, T.; Lacroix, C. Understanding the prebiotic potential of different dietary fibers using an in vitro continuous adult fermentation model (PolyFermS). Sci. Rep. 2018, 8, 4318. [Google Scholar] [CrossRef] [PubMed]
- Finegold, S.M.; Sutter, V.L.; Sugihara, P.T.; Elder, H.A.; Lehmann, S.M.; Phillips, R.L. Fecal microbial flora in Seventh Day Adventist populations and control subjects. Am. J. Clin. Nutr. 1977, 30, 1781–1792. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hiergeist, A.; Gläsner, J.; Reischl, U.; Gessner, A. Analyses of Intestinal Microbiota: Culture versus Sequencing. ILAR J. 2015, 56, 228–240. [Google Scholar] [CrossRef] [PubMed]
- Lok, C. Mining the microbial dark matter. Nature 2015, 522, 270–273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lagier, J.-C.; Khelaifia, S.; Alou, M.T.; Ndongo, S.; Dione, N.; Hugon, P.; Caputo, A.; Cadoret, F.; Traore, S.I.; Dubourg, G.; et al. Culture of previously uncultured members of the human gut microbiota by culturomics. Nat. Microbiol. 2016, 1, 16203. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rahi, P.; Prakash, O.; Shouche, Y.S. Matrix-Assisted Laser Desorption/Ionization Time-of-Flight Mass-Spectrometry (MALDI-TOF MS) Based Microbial Identifications: Challenges and Scopes for Microbial Ecologists. Front. Microbiol. 2016, 7, 1359. [Google Scholar] [CrossRef] [PubMed]
- Singhal, N.; Kumar, M.; Kanaujia, P.K.; Virdi, J.S. MALDI-TOF mass spectrometry: An emerging technology for microbial identification and diagnosis. Front. Microbiol. 2015, 6, 791. [Google Scholar] [CrossRef] [PubMed]
- Tseng, C.-H.; Wu, C.-Y. The gut microbiome in obesity. J. Formos. Med. Assoc. Taiwan Yi Zhi 2018. [Google Scholar] [CrossRef] [PubMed]
- Srinivasan, R.; Karaoz, U.; Volegova, M.; MacKichan, J.; Kato-Maeda, M.; Miller, S.; Nadarajan, R.; Brodie, E.L.; Lynch, S.V. Use of 16S rRNA Gene for Identification of a Broad Range of Clinically Relevant Bacterial Pathogens. PLoS ONE 2015, 10, e0117617. [Google Scholar] [CrossRef] [PubMed]
- Angelakis, E.; Lagier, J.-C. Samples and techniques highlighting the links between obesity and microbiota. Microb. Pathog. 2017, 106, 119–126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, C.; Zhao, L. Strain-level dissection of the contribution of the gut microbiome to human metabolic disease. Genome Med. 2016, 8, 41. [Google Scholar] [CrossRef] [PubMed]
- Venter, J.C.; Remington, K.; Heidelberg, J.F.; Halpern, A.L.; Rusch, D.; Eisen, J.A.; Wu, D.; Paulsen, I.; Nelson, K.E.; Nelson, W.; et al. Environmental genome shotgun sequencing of the Sargasso Sea. Science 2004, 304, 66–74. [Google Scholar] [CrossRef] [PubMed]
- Hakovirta, J.R.; Prezioso, S.; Hodge, D.; Pillai, S.P.; Weigel, L.M. Identification and Analysis of Informative Single Nucleotide Polymorphisms in 16S rRNA Gene Sequences of the Bacillus cereus Group. J. Clin. Microbiol. 2016, 54, 2749–2756. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bacci, G.; Bani, A.; Bazzicalupo, M.; Ceccherini, M.T.; Galardini, M.; Nannipieri, P.; Pietramellara, G.; Mengoni, A. Evaluation of the Performances of Ribosomal Database Project (RDP) Classifier for Taxonomic Assignment of 16S rRNA Metabarcoding Sequences Generated from Illumina-Solexa NGS. J. Genom. 2015, 3, 36–39. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yilmaz, P.; Parfrey, L.W.; Yarza, P.; Gerken, J.; Pruesse, E.; Quast, C.; Schweer, T.; Peplies, J.; Ludwig, W.; Glöckner, F.O. The SILVA and “All-species Living Tree Project (LTP)” taxonomic frameworks. Nucleic Acids Res. 2014, 42, D643–D648. [Google Scholar] [CrossRef] [PubMed]
- Vollmers, J.; Wiegand, S.; Kaster, A.-K. Comparing and Evaluating Metagenome Assembly Tools from a Microbiologist’s Perspective—Not Only Size Matters! PLoS ONE 2017, 12, e0169662. [Google Scholar] [CrossRef] [PubMed]
- Meisel, J.S.; Grice, E.A. The Human Microbiome. In Genomic Precision Medicine, 3rd ed.; Ginsburg, G.S., Willard, H.F., Eds.; Academic Press: Boston, MA, USA, 2017; Chapter 4; pp. 63–77. [Google Scholar]
- Hugon, P.; Lagier, J.-C.; Robert, C.; Lepolard, C.; Papazian, L.; Musso, D.; Vialettes, B.; Raoult, D. Molecular Studies Neglect Apparently Gram-Negative Populations in the Human Gut Microbiota. J. Clin. Microbiol. 2013, 51, 3286–3293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Angelakis, E.; Bachar, D.; Henrissat, B.; Armougom, F.; Audoly, G.; Lagier, J.-C.; Robert, C.; Raoult, D. Glycans affect DNA extraction and induce substantial differences in gut metagenomic studies. Sci. Rep. 2016, 6, 26276. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schrader, C.; Schielke, A.; Ellerbroek, L.; Johne, R. PCR inhibitors—Occurrence, properties and removal. J. Appl. Microbiol. 2012, 113, 1014–1026. [Google Scholar] [CrossRef] [PubMed]
- Wesolowska-Andersen, A.; Bahl, M.I.; Carvalho, V.; Kristiansen, K.; Sicheritz-Pontén, T.; Gupta, R.; Licht, T.R. Choice of bacterial DNA extraction method from fecal material influences community structure as evaluated by metagenomic analysis. Microbiome 2014, 2, 19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dilhari, A.; Sampath, A.; Gunasekara, C.; Fernando, N.; Weerasekara, D.; Sissons, C.; McBain, A.; Weerasekera, M. Evaluation of the impact of six different DNA extraction methods for the representation of the microbial community associated with human chronic wound infections using a gel-based DNA profiling method. AMB Express 2017, 7, 179. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Huang, H.; Wu, C.H. Protein Bioinformatics Databases and Resources. In Methods Molecular Biology; Springer: Clifton, NJ, USA, 2017; Volume 1558, pp. 3–39. [Google Scholar]
- Krishnan, S.; Alden, N.; Lee, K. Pathways and Functions of Gut Microbiota Metabolism Impacting Host Physiology. Curr. Opin. Biotechnol. 2015, 36, 137–145. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Hua, Q. Applications of Genome-Scale Metabolic Models in Biotechnology and Systems Medicine. Front. Physiol. 2016, 6, 413. [Google Scholar] [CrossRef] [PubMed]
- Ha, C.W.; Lam, Y.Y.; Holmes, A.J. Mechanistic links between gut microbial community dynamics, microbial functions and metabolic health. World J. Gastroenterol. WJG 2014, 20, 16498–16517. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Furumichi, M.; Tanabe, M.; Sato, Y.; Morishima, K. KEGG: New perspectives on genomes, pathways, diseases and drugs. Nucleic Acids Res. 2017, 45, D353–D361. [Google Scholar] [CrossRef] [PubMed]
- Integrative HMP (iHMP) Research Network Consortium. The Integrative Human Microbiome Project: Dynamic Analysis of Microbiome-Host Omics Profiles during Periods of Human Health and Disease. Cell Host Microbe 2014, 16, 276–289. [Google Scholar] [CrossRef] [PubMed]
- Blanco-Míguez, A.; Gutiérrez-Jácome, A.; Fdez-Riverola, F.; Lourenço, A.; Sánchez, B. MAHMI database: A comprehensive MetaHit-based resource for the study of the mechanism of action of the human microbiota. Database 2017, 2017. [Google Scholar] [CrossRef] [PubMed]
- Davis, S.C.; Yadav, J.S.; Barrow, S.D.; Robertson, B.K. Gut microbiome diversity influenced more by the Westernized dietary regime than the body mass index as assessed using effect size statistic. MicrobiologyOpen 2017. [Google Scholar] [CrossRef] [PubMed]
- Shang, Y.; Khafipour, E.; Derakhshani, H.; Sarna, L.K.; Woo, C.W.; Siow, Y.L.; Karmin, O. Short Term High Fat Diet Induces Obesity-Enhancing Changes in Mouse Gut Microbiota That are Partially Reversed by Cessation of the High Fat Diet. Lipids 2017, 52, 499–511. [Google Scholar] [CrossRef] [PubMed]
- Miele, L.; Giorgio, V.; Alberelli, M.A.; De Candia, E.; Gasbarrini, A.; Grieco, A. Impact of Gut Microbiota on Obesity, Diabetes, and Cardiovascular Disease Risk. Curr. Cardiol. Rep. 2015, 17, 120. [Google Scholar] [CrossRef] [PubMed]
- Vrieze, A.; Van Nood, E.; Holleman, F.; Salojärvi, J.; Kootte, R.S.; Bartelsman, J.F.; Dallinga–Thie, G.M.; Ackermans, M.T.; Serlie, M.J.; Oozeer, R.; et al. Transfer of intestinal microbiota from lean donors increases insulin sensitivity in individuals with metabolic syndrome. Gastroenterology 2012, 143, 913–916.e7. [Google Scholar] [CrossRef] [PubMed]
- Jayasinghe, T.N.; Chiavaroli, V.; Holland, D.J.; Cutfield, W.S.; O’Sullivan, J.M. The New Era of Treatment for Obesity and Metabolic Disorders: Evidence and Expectations for Gut Microbiome Transplantation. Front. Cell. Infect. Microbiol. 2016, 6, 15. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| Complications in Human Host Models | Solutions |
|---|---|
| Variation in host genome |
|
| Environmental exposures (toxins, antibiotics, diet) |
|
| Tractability |
|
| Difficult-to-replicate experiments due to unique microbiota of each individual |
|
| Animal Model | Main Characteristics of the Model | Aspect of the Microbiota to Study | Methodology Employed | Reference |
|---|---|---|---|---|
(A) Hydra (Hydra spp.) |
|
|
| [45] |
(B) Honeybee (Apis mellifera) |
|
|
| [46] |
(C) Zebrafish (Danio rerio) |
|
|
| [20] |
(D) Mice (Mus musculus) |
|
|
| [47] |
(E) Rat (Rattus novergicus) |
|
|
| [30] |
(F) Pig (Sus scrofa) |
|
|
| [48] |
| Technique/Process | Biases | Reference |
|---|---|---|
| Pyrosequencing |
| [67] |
| PCR amplification |
| [68] |
| [69] | |
| Disruption of bacterial membranes |
| [70] |
| DNA extraction methods |
| [71] |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Carrera-Quintanar, L.; Ortuño-Sahagún, D.; Franco-Arroyo, N.N.; Viveros-Paredes, J.M.; Zepeda-Morales, A.S.; Lopez-Roa, R.I. The Human Microbiota and Obesity: A Literature Systematic Review of In Vivo Models and Technical Approaches. Int. J. Mol. Sci. 2018, 19, 3827. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms19123827
Carrera-Quintanar L, Ortuño-Sahagún D, Franco-Arroyo NN, Viveros-Paredes JM, Zepeda-Morales AS, Lopez-Roa RI. The Human Microbiota and Obesity: A Literature Systematic Review of In Vivo Models and Technical Approaches. International Journal of Molecular Sciences. 2018; 19(12):3827. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms19123827
Chicago/Turabian StyleCarrera-Quintanar, Lucrecia, Daniel Ortuño-Sahagún, Noel N. Franco-Arroyo, Juan M. Viveros-Paredes, Adelaida S. Zepeda-Morales, and Rocio I. Lopez-Roa. 2018. "The Human Microbiota and Obesity: A Literature Systematic Review of In Vivo Models and Technical Approaches" International Journal of Molecular Sciences 19, no. 12: 3827. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms19123827