Exploiting PLGA-Based Biocompatible Nanoparticles for Next-Generation Tolerogenic Vaccines against Autoimmune Disease


 ,
, 
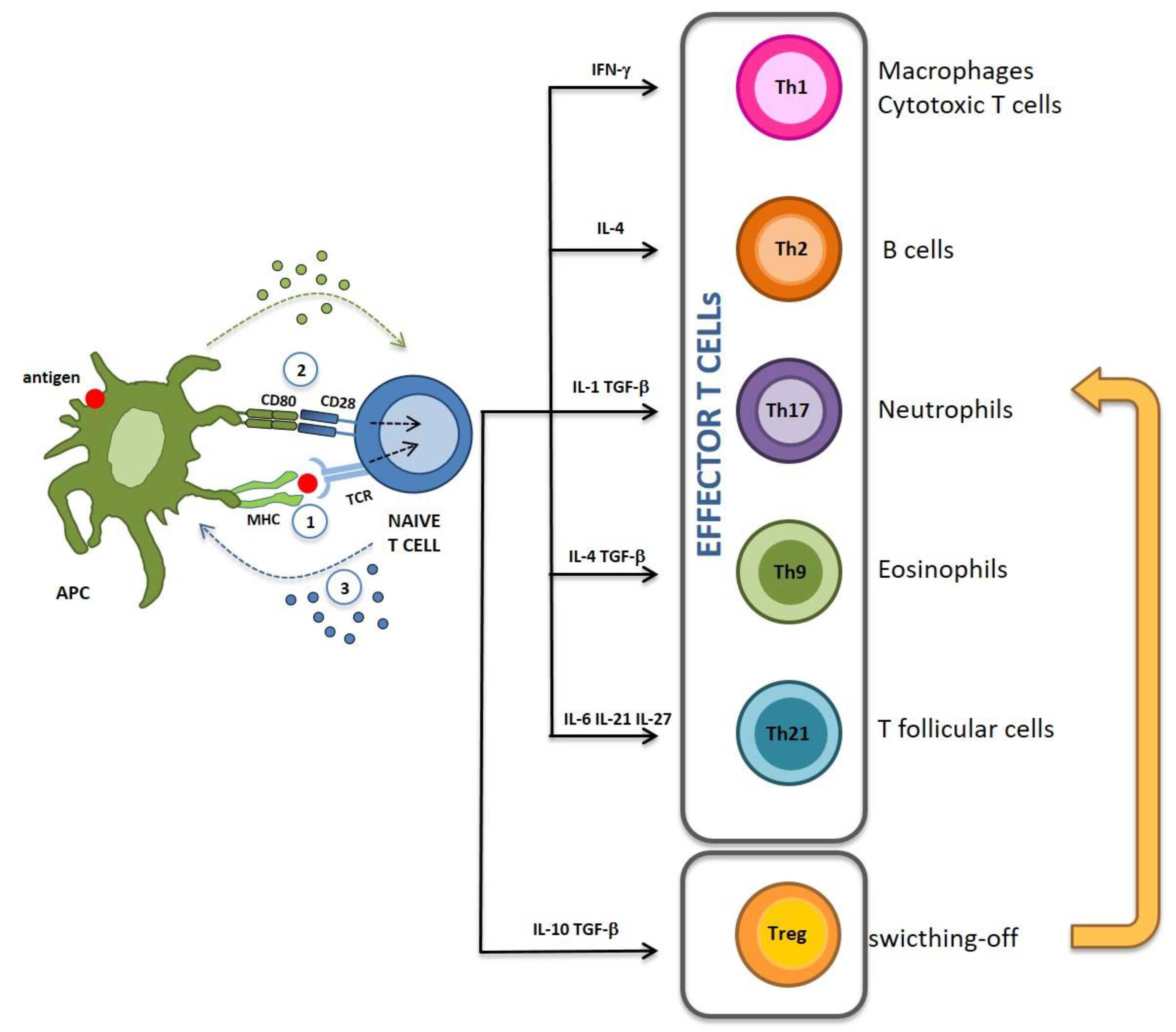{kind=link}
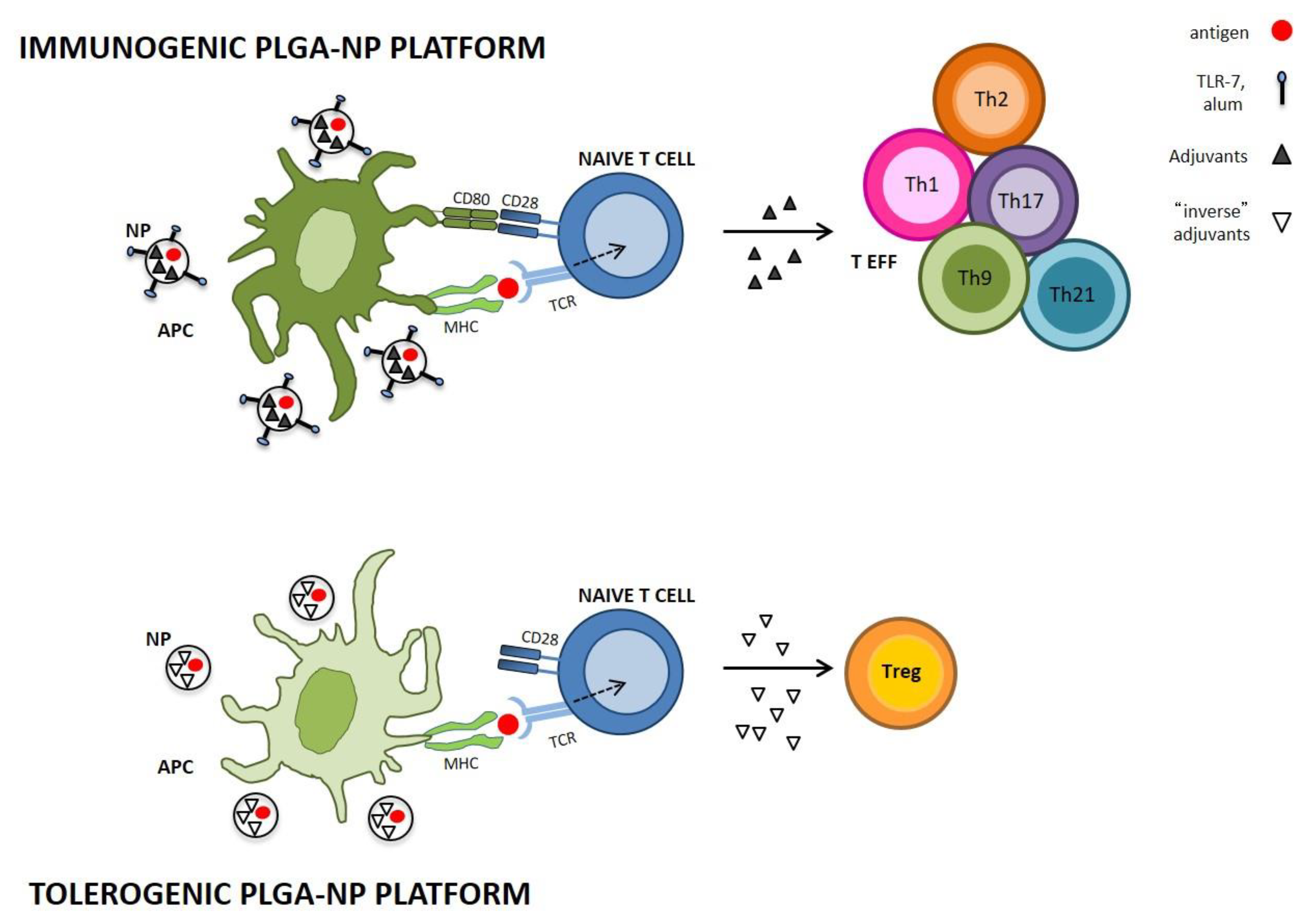{kind=link}
Abstract
:1. Immunogenic and Tolerogenic Vaccinations
2. Nanoparticles: An Overview
3. PLGA-NPs: In Vitro and In Vivo Antigen Release
3.1. PLGA-NPs in Immunogenic Vaccination
3.2. PLGA-NPs in Tolerogenic Vaccination
3.2.1. Experimental Autoimmune Encephalomyelitis
3.2.2. Rheumatoid Arthritis
3.2.3. Type 1 Diabetes
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| AFM | Atomic force microscope |
| APC | Antigen-presenting cell |
| BSA | Bovine serum albumin |
| CFA | Complete Freund’s adjuvant |
| CIA | Collagen-induced arthritis |
| CII | Collagen type II |
| CNS | Central nervous system |
| DC | Dendritic cells |
| EAE | Experimental autoimmune encephalomyelitis |
| EAN | Experimental autoimmune neuritis |
| EPR | Enhanced permeability and retention |
| FDA | Food and drug Administration |
| GM-CSF | Granulocyte-macrophage colony-stimulating factor |
| HSP | Heat shock protein |
| i.v. | intravenous |
| IFN | interferon |
| Ig | immunoglobulin |
| IL | interleukin |
| iTreg | Induced Treg |
| MBP | Myelin basic protein |
| MHC | Major histocompatibility complex |
| MOG | Myelin-oligodendrocyte glycoprotein |
| MS | Multiple sclerosis |
| NIR | Near-infrared |
| NK | Natural killer |
| NOD | Non Obese Diabetic |
| NPs | nanoparticles |
| nTreg | Natural Treg |
| OVA | Ovalbumin |
| PEG | Polyethylene glycol |
| PGIA | Proteoglycan-induced arthritis |
| PLGA | Poly(lactic-co-glycolic acid) |
| PLP | Proteolipid protein |
| RA | rheumatoid arthritis |
| RR | Relapse remitting |
| s.c. | subcutaneous |
| SEM | Scanning electron microscopy |
| T1D | Type 1 diabetes |
| TCR | T-cell receptor |
| TEM | transmission electron microscope |
| TFH | T follicular helper |
| TGF | Transforming growth factor |
| Th | T helper |
| TLR | Toll like receptor |
| Treg | T regulatory |
References
- Zhu, J.; Paul, W.E. Heterogeneity and plasticity of T helper cells. Cell Res. 2010, 20, 4–12. [Google Scholar] [CrossRef] [PubMed]
- Deenick, E.K.; Ma, C.S. The regulation and role of T follicular helper cells in immunity. Immunology 2011, 134, 361–367. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bradley, L.M.; Dalton, D.K.; Croft, M. A direct role for IFN-γ in regulation of Th1 cell development. J. Immunol. 1996, 157, 1350. [Google Scholar] [PubMed]
- Swain, S.L.; Weinberg, A.D.; English, M.; Huston, G. IL-4 directs the development of Th2-like helper effectors. J. Immunol. 1990, 145, 3796. [Google Scholar] [PubMed]
- Chung, Y.; Chang, S.H.; Martinez, G.J.; Yang, X.O.; Nurieva, R.; Kang, H.S.; Ma, L.; Watowich, S.S.; Jetten, A.M.; Tian, Q.; et al. Critical regulation of early Th17 cell differentiation by interleukin-1 signaling. Immunity 2009, 30, 576–587. [Google Scholar] [CrossRef]
- Mesturini, R.; Gigliotti, C.L.; Orilieri, E.; Cappellano, G.; Soluri, M.F.; Boggio, E.; Woldetsadik, A.; Dianzani, C.; Sblattero, D.; Chiocchetti, A.; et al. Differential induction of IL-17, IL-10, and IL-9 in human T helper cells by B7h and B7.1. Cytokine 2013, 64, 322–330. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaplan, M.H. Th9 cells: Differentiation and disease. Immunol Rev. 2013, 252, 104–115. [Google Scholar] [CrossRef]
- Jutel, M.; Akdis, M.; Budak, F.; Aebischer-Casaulta, C.; Wrzyszcz, M.; Blaser, K.; Akdis, C.A. IL-10 and TGF-beta cooperate in the regulatory T cell response to mucosal allergens in normal immunity and specific immunotherapy. Eur. J. Immunol. 2003, 33, 1205–1214. [Google Scholar] [CrossRef]
- Asseman, C.; Mauze, S.; Leach, M.W.; Coffman, R.L.; Powrie, F. An essential role for interleukin 10 in the function of regulatory T cells that inhibit intestinal inflammation. J. Exp. Med. 1999, 190, 995–1004. [Google Scholar] [CrossRef]
- Nakamura, K.; Kitani, A.; Fuss, I.; Pedersen, A.; Harada, N.; Nawata, H.; Strober, W. TGF-beta 1 plays an important role in the mechanism of CD4+CD25+ regulatory T cell activity in both humans and mice. J. Immunol. 2004, 172, 834–842. [Google Scholar] [CrossRef]
- Vignali, D.A.A.; Collison, L.W.; Workman, C.J. How regulatory T cells work. Nature Rev. Immunol. 2008, 8, 523–532. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dejaco, C.; Duftner, C.; Grubeck-Loebenstein, B.; Schirmer, M. Imbalance of regulatory T cells in human autoimmune diseases. Immunology 2006, 117, 289–300. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, Q.; Adams, J.Y.; Tooley, A.J.; Bi, M.; Fife, B.T.; Serra, P.; Santamaria, P.; Locksley, R.M.; Krummel, M.F.; Bluestone, J.A. Visualizing regulatory T cell control of autoimmune responses in nonobese diabetic mice. Nat. Immunol. 2006, 7, 83–92. [Google Scholar] [CrossRef] [PubMed]
- Lechler, R.; Chai, J.G.; Marelli-Berg, F.; Lombardi, G. The contributions of T-cell anergy to peripheral T-cell tolerance. Immunology 2001, 103, 262–269. [Google Scholar] [CrossRef] [PubMed]
- Weiner, H.L. Induction and mechanism of action of transforming growth factor-beta-secreting Th3 regulatory cells. Immunol. Rev. 2001, 182, 207–214. [Google Scholar] [CrossRef] [PubMed]
- Weiner, H.L. Oral tolerance: Immune mechanisms and the generation of Th3-type TGF-beta-secreting regulatory cells. Microbes Infect. 2001, 3, 947–954. [Google Scholar] [CrossRef]
- Marelli-Berg, F.M.; Lechler, R.I. Antigen presentation by parenchymal cells: A route to peripheral tolerance? Immunol. Rev. 1999, 172, 297–314. [Google Scholar] [CrossRef]
- Garza, K.M.; Agersborg, S.S.; Baker, E.; Tung, K.S. Persistence of physiological self antigen is required for the regulation of self tolerance. J. Immunol. 2000, 164, 3982–3989. [Google Scholar] [CrossRef]
- Wu, J.; Xie, A.; Chen, W. Cytokine regulation of immune tolerance. Burns Trauma 2014, 2, 11–17. [Google Scholar]
- LaMothe, R.A.; Kolte, P.N.; Vo, T.; Ferrari, J.D.; Gelsinger, T.C.; Wong, J.; Chan, V.T.; Ahmed, S.; Srinivasan, A.; Deitemeyer, P.; et al. Tolerogenic Nanoparticles Induce Antigen-Specific Regulatory T Cells and Provide Therapeutic Efficacy and Transferrable Tolerance against Experimental Autoimmune Encephalomyelitis. Front. Immunol. 2018, 9, 281. [Google Scholar] [CrossRef]
- Emerich, D.F.; Thanos, G.C. Nanotechnology and medicine. Expert Opin. Biol. Therapy 2003, 3, 655–663. [Google Scholar] [CrossRef] [PubMed]
- Marcato, P.D.; Durán, N. New aspects of nanopharmaceutical delivery systems. J. Nanosci. Nanotechnol. 2008, 8, 2216–2229. [Google Scholar] [CrossRef] [PubMed]
- Stella, B.; Peira, E.; Dianzani, C.; Gallarate, M.; Battaglia, L.; Gigliotti, C.L.; Boggio, E.; Dianzani, U.; Dosio, F. Development and Characterization of Solid Lipid Nanoparticles Loaded with a Highly Active Doxorubicin Derivative. Nanomaterials (Basel) 2018, 8, 110. [Google Scholar] [CrossRef] [PubMed]
- Clemente, N.; Ferrara, B.; Gigliotti, C.L.; Boggio, E.; Capucchio, M.T.; Biasibetti, E.; Schiffer, D.; Mellai, M.; Annovazzi, L.; Cangemi, L.; et al. Solid Lipid Nanoparticles Carrying Temozolomide for Melanoma Treatment. Preliminary In Vitro and In Vivo Studies. Int J. Mol. Sci. 2018, 19, 255. [Google Scholar] [CrossRef] [PubMed]
- Minelli, R.; Occhipinti, S.; Gigliotti, C.L.; Barrera, G.; Gasco, P.; Conti, L.; Chiocchetti, A.; Zara, G.P.; Fantozzi, R.; Giovarelli, M.; et al. Solid lipid nanoparticles of cholesteryl butyrate inhibit the proliferation of cancer cells in vitro and in vivo models. Br. J. Pharmacol. 2013, 170, 233–244. [Google Scholar] [CrossRef] [PubMed]
- Gigliotti, C.L.; Minelli, R.; Cavalli, R.; Occhipinti, S.; Barrera, G.; Pizzimenti, S.; Cappellano, G.; Boggio, E.; Conti, L.; Fantozzi, R.; et al. In Vitro and In Vivo Therapeutic Evaluation of Camptothecin-Encapsulated β-Cyclodextrin Nanosponges in Prostate Cancer. J. Biomed. Nanotechnol. 2016, 12, 114–127. [Google Scholar] [CrossRef]
- Gigliotti, C.L.; Ferrara, B.; Occhipinti, S.; Boggio, E.; Barrera, G.; Pizzimenti, S.; Giovarelli, M.; Fantozzi, R.; Chiocchetti, A.; Argenziano, M.; et al. Enhanced cytotoxic effect of camptothecin nanosponges in anaplastic thyroid cancer cells in vitro and in vivo on orthotopic xenograft tumors. Drug Deliv. 2017, 24, 670–680. [Google Scholar] [CrossRef] [Green Version]
- Dianzani, C.; Foglietta, F.; Ferrara, B.; Rosa, A.C.; Muntoni, E.; Gasco, P.; Della Pepa, C.; Canaparo, R.; Serpe, L. Solid lipid nanoparticles delivering anti-inflammatory drugs to treat inflammatory bowel disease: Effects in an in vivo model. World J. Gastroenterol. 2017, 23, 4200–4210. [Google Scholar] [CrossRef]
- Kermanizadeh, A.; Powell, L.G.; Stone, V.; Møller, P. Nanodelivery systems and stabilized solid-drug nanoparticles for orally administered medicine: Current landscape. Int. J. Nanomed. 2018, 13, 7575–7605. [Google Scholar] [CrossRef]
- Dianzani, C.; Zara, G.P.; Maina, G.; Pettazzoni, P.; Pizzimenti, S.; Rossi, F.; Gigliotti, C.L.; Ciamporcero, E.S.; Daga, M.; Barrera, G. Drug delivery nanoparticles in skin cancers. Biomed. Res. Int. 2014, 2014, 895986. [Google Scholar] [CrossRef]
- Xiang, S.D.; Selomulya, C.; Ho, J.; Apostolopoulos, V.; Plebanski, M. Delivery of DNA vaccines: An overview on the use of biodegradable polymeric and magnetic nanoparticles. Wiley Interdiscip. Rev. Nanomed. Nanobiotechnol. 2010, 2, 205–218. [Google Scholar] [CrossRef] [PubMed]
- Samanta, A.; Medintz, I.L. Nanoparticles and DNA—A powerful and growing functional combination in bionanotechnology. Nanoscale 2016, 8, 9037–9095. [Google Scholar] [CrossRef] [PubMed]
- Baetke, S.C.; Lammers, T.; Kiessling, F. Applications of nanoparticles for diagnosis and therapy of cancer. Br. J. Radiol. 2015, 88, 20150207. [Google Scholar] [CrossRef] [PubMed]
- Leleux, J.; Roy, K. Micro and nanoparticle-based delivery systems for vaccine immunotherapy: An immunological and materials perspective. Adv. Healthc. Mater. 2013, 2, 72–94. [Google Scholar] [CrossRef] [PubMed]
- Dobrovolskaia, M.A.; McNeil, S.E. Immunological properties of engineered nanomaterials. Nat. Nanotechnol. 2007, 2, 469–478. [Google Scholar] [CrossRef] [PubMed]
- Kishimoto, T.K.; Maldonado, R.A. Nanoparticles for the Induction of Antigen-Specific Immunological Tolerance. Front. Immunol. 2018, 9, 230. [Google Scholar] [CrossRef]
- Ilinskaya, A.N.; Dobrovolskaia, M.A. Immunosuppressive and anti-inflammatory properties of engineered nanomaterials. Br. J. Pharmacol. 2014, 171, 3988–4000. [Google Scholar] [CrossRef] [Green Version]
- Oyewumi, M.O.; Kumar, A.; Cui, Z. Nano-microparticles as immune adjuvants: Correlating particle sizes and the resultant immune responses. Expert Rev. Vaccines 2010, 9, 1095–1107. [Google Scholar] [CrossRef]
- Kadengodlu, P.A.; Hebishima, T.; Takeshima, S.N.; Ito, M.; Liu, M.; Abe, H.; Aida, Y.; Aigaki, T.; Ito, Y. Positively charged cholesterol-recombinant human gelatins foster the cellular uptake of proteins and murine immune reactions. Int. J. Nanomed. 2012, 7, 5437–5450. [Google Scholar]
- Fromen, C.A.; Rahhal, T.B.; Robbins, G.R.; Kai, M.P.; Shen, T.W.; Luft, J.C.; DeSimone, J.M. Nanoparticle surface charge impacts distribution, uptake and lymph node trafficking by pulmonary antigen-presenting cells. Nanomedicine 2015, 12, 677–687. [Google Scholar] [CrossRef]
- Manolova, V.; Flace, A.; Bauer, M.; Schwarz, K.; Saudan, P.; Bachmann, M.F. Nanoparticles target distinct dendritic cell populations according to their size. Eur. J. Immunol. 2008, 38, 1404–1413. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blank, F.; Stumbles, P.A.; Seydoux, E.; Holt, P.G.; Fink, A.; Rothen-Rutishauser, B.; Strickland, D.H.; von Garnier, C. Size-dependent uptake of particles by pulmonary antigen-presenting cell populations and trafficking to regional lymph nodes. Am. J. Respir Cell. Mol. Biol. 2013, 49, 67–77. [Google Scholar] [CrossRef] [PubMed]
- Xiang, S.D.; Scholzen, A.; Minigo, G.; David, C.; Apostolopoulos, V.; Mottram, P.L.; Plebanski, M. Pathogen recognition and development of particulate vaccines: Doessize matter? Methods 2006, 40, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Makadia, H.K.; Siegel, S.J. Poly Lactic-co-Glycolic Acid (PLGA) as Biodegradable Controlled DrugDelivery Carrier. Polymers (Basel) 2011, 3, 1377–1397. [Google Scholar] [CrossRef] [PubMed]
- Gentile, P.; Chiono, V.; Carmagnola, I.; Hatton, P.V. An overview of poly(lactic-co-glycolic) acid (PLGA)-based biomaterials for bone tissue engineering. Int. J. Mol. Sci. 2014, 15, 3640–3659. [Google Scholar] [CrossRef]
- Samavedi, S.; Poindexter, L.K.; Van Dyke, M.; Goldstein, A.S. Synthetic biomaterials for regenerative medicine applications. Regen. Med. Appl. Organ. Transpl. 2014, 81–99. [Google Scholar] [CrossRef]
- Houchin, M.L.; Topp, E.M. Physical properties of PLGA films during polymer degradation. J. Appl. Polym. Sci. 2009, 114, 2848–2854. [Google Scholar] [CrossRef]
- Passerini, N.; Craig, D.Q.M. An investigation into the effects of residual water on the glass transition temperature of polylactide microspheres using modulated temperature DSC. J. Control. Release 2001, 73, 111–115. [Google Scholar] [CrossRef]
- Park, P.I.P.; Jonnalagadda, S. Predictors of glass transition in the biodegradable polylactide and poly-lactide-co-glycolide polymers. J. Appl. Polym. Sci. 2006, 100, 1983–1987. [Google Scholar] [CrossRef]
- Carvalho, P.M.; Felício, M.R.; Santos, N.C.; Gonçalves, S.; Domingues, M.M. Application of Light Scattering Techniques to Nanoparticle Characterization and Development. Front. Chem. 2018, 6, 237. [Google Scholar] [CrossRef]
- Balasubramanian, S.; Girija, A.R.; Nagaoka, Y.; Iwai, S.; Suzuki, M.; Kizhikkilot, V.; Yoshida, Y.; Maekawa, T.; Nair, S.D. Curcumin and 5-fluorouracil-loaded, folate- and transferrin-decorated polymeric magnetic nanoformulation: A synergistic cancer therapeutic approach, accelerated by magnetic hyperthermia. Int. J. Nanomed. 2014, 9, 437–459. [Google Scholar]
- Swider, E.; Koshkina, O.; Tel, J.; Cruz, L.J.; de Vries, I.J.M.; Srinivas, M. Customizing poly(lactic-co-glycolic acid) particles for biomedical applications. Acta Biomater. 2018, 73, 38–51. [Google Scholar] [CrossRef] [PubMed]
- Boix-Garriga, E.; Acedo, P.; Casadó, A.; Villanueva, A.; Stockert, J.C.; Cañete, M.; Mora, M.; Sagristá, M.L.; Nonell, S. Poly(D,L-lactide-co-glycolide) nanoparticles as delivery agents for photodynamic therapy: Enhancing singlet oxygen release and photototoxicity by surface PEG coating. Nanotechnology 2015, 26, 365104. [Google Scholar] [CrossRef] [PubMed]
- Trofymchuk, K.; Reisch, A.; Shulov, I.; Mély, Y.; Klymchenko, A.S. Tuning the color and photostability of perylene diimides inside polymer nanoparticles: Towards biodegradable substitutes of quantum dots. Nanoscale 2014, 6, 12934–12942. [Google Scholar] [CrossRef]
- Hussein, A.S.; Abdullah, N.; Ahmadun, F.R. In vitro degradation of poly (D,L-lactide-co-glycolide) nanoparticles loaded with linamarin. IET Nanobiotechnol. 2013, 7, 33–41. [Google Scholar] [CrossRef] [PubMed]
- Capan, Y.; Woo, B.H.; Gebrekidan, S.; Ahmed, S.; DeLuca, P.P. Influence of formulation parameters on the characteristics of poly(D,L-lactide-co-glycolide) microspheres containing poly(L-lysine) complexed plasmid DNA. J. Control. Release 1999, 60, 279–286. [Google Scholar] [CrossRef]
- Chaisri, W.; Hennink, W.E.; Okonogi, S. Preparation and characterization of cephalexin loaded PLGA microspheres. Curr. Drug Deliv. 2009, 6, 69–75. [Google Scholar] [CrossRef] [PubMed]
- Cappellano, G.; Woldetsadik, A.D.; Orilieri, E.; Shivakumar, Y.; Rizzi, M.; Carniato, F.; Gigliotti, C.L.; Boggio, E.; Clemente, N.; Comi, C.; et al. Subcutaneous inverse vaccination with PLGA particles loaded with a MOG peptide and IL-10 decreases the severity of experimental autoimmune encephalomyelitis. Vaccine 2014, 32, 5681–5689. [Google Scholar] [CrossRef] [Green Version]
- Hines, D.J.; Kaplan, D.L. Poly(lactic-co-glycolic) acid-controlled-release systems: Experimental and modeling insights. Crit. Rev. Ther. Drug Carrier Syst. 2013, 30, 257–276. [Google Scholar] [CrossRef]
- Wang, J.; Wang, B.M.; Schwendeman, S.P. Characterization of the initial burst release of a model peptide from poly(D,L-lactide-co-glycolide) microspheres. J. Control. Release 2002, 82, 289–307. [Google Scholar] [CrossRef]
- Panyam, J.; Dali, M.M.; Sahoo, S.K.; Ma, W.; Chakravarthi, S.S.; Amidon, G.L.; Levy, R.J.; Labhasetwar, V. Polymer degradation and in vitro release of a model protein from poly(D,L-lactide-co-glycolide) nano- and microparticles. J. Control. Release 2003, 92, 173–187. [Google Scholar] [CrossRef]
- Körber, M. PLGA erosion: Solubility- or diffusion-controlled? Pharm Res. 2010, 27, 2414–2420. [Google Scholar] [CrossRef] [PubMed]
- Klose, D.; Siepmann, F.; Elkharraz, K.; Krenzlin, S.; Siepmann, J. How porosity and size affect the drug release mechanisms from PLGA-based microparticles. Int. J. Pharm. 2006, 314, 198–206. [Google Scholar] [CrossRef] [PubMed]
- Pitt, C.G.; Gratzl, M.M.; Kimmel, G.L.; Surles, J.; Schindler, A. Aliphatic polyesters II. The degradation of poly (DL-lactide), poly (epsilon-caprolactone), and theircopolymers in vivo. Biomaterials 1981, 2, 215–220. [Google Scholar] [CrossRef]
- Park, T.G. Degradation of poly(lactic-co-glycolic acid) microspheres: Effect of copolymer composition. Biomaterials 1995, 16, 1123–1130. [Google Scholar] [CrossRef]
- Williams, D.F. Biodegradation of surgical polymers. J. Mater. Sci. 1982, 17, 1233–1246. [Google Scholar] [CrossRef]
- Vert, M. Polyglycolide and copolyesters with lactides. In Biopolymers: Biology, Chemistry, Biotechnology, Applications; Doi, Y., Steinbuchel, A., Eds.; Wiley-VCH Verlag GmbH: Weinheim, Germany, 2002; Volume 4, pp. 179–202. [Google Scholar]
- Guo, L.Y.; Yan, S.Z.; Li, Q.; Xu, Q.; Lin, X.; Qi, S.S.; Yu, S.Q.; Chen, S.L. Poly(lactic-co-glycolic) acid nanoparticles improve oral bioavailability of hypocrellin A in rat. RSC Adv. 2017, 7, 42073. [Google Scholar] [CrossRef]
- Anderson, J.M.; Shive, S.M. Biodegradation and biocompatibility of PLA and PLGA microspheres. Adv. Drug. Deliv. Rev. 1997, 28, 5–24. [Google Scholar] [CrossRef]
- Sasaki, K.; Igarashi, M.; Hinata, M.; Komori, Y.; Fukushima, K. Simulation of Drug Release from PLGA Particles In Vivo. J. Drug Deliv. 2013, 2013, 513950. [Google Scholar] [CrossRef] [PubMed]
- Mohammad, A.K.; Reineke, J.J. Quantitative detection of PLGA nanoparticle degradation in tissues following intravenous administration. Mol. Pharm. 2013, 10, 2183–2189. [Google Scholar] [CrossRef]
- Lu, L.; Peter, S.J.; Lyman, M.D.; Lai, H.L.; Leite, S.M.; Tamada, J.A.; Uyama, S.; Vacanti, J.P.; Langer, R.; Mikos, A.G. In vitro and in vivo degradation of porous poly(D,L-lactic-co-glycolic acid) foams. Biomaterials 2000, 21, 1837–1845. [Google Scholar] [CrossRef]
- Diwan, M.; Elamanchili, P.; Cao, M.; Samuel, J. Dose sparing of CpG oligodeoxynucleotide vaccine adjuvants by nanoparticle delivery. Curr. Drug Deliv. 2004, 1, 405–412. [Google Scholar] [CrossRef] [PubMed]
- O’Hagan, D.T.; Jeffery, H.; Davis, S.S. Long-term antibody responses in mice following subcutaneous immunization with ovalbumin entrapped in biodegradable microparticles. Vaccine 1993, 11, 965–969. [Google Scholar] [CrossRef]
- Uchida, T.; Martin, S.; Foster, T.P.; Wardley, R.C.; Grimm, S. Dose and Load Studies for Subcutaneous and Oral Delivery of Poly(lactide-co-glycolide) Microspheres Containing Ovalbumin. Pharm Res. 1994, 11, 1009–1015. [Google Scholar] [CrossRef] [PubMed]
- Igartua, M.; Hernández, R.M.; Esquisabel, A.; Gascón, A.R.; Calvo, M.B.; Pedraz, J.L. Enhanced immune response after subcutaneous and oral immunization with biodegradable PLGA microspheres. J. Control. Release 1998, 56, 63–73. [Google Scholar] [CrossRef]
- Ilyinskii, P.O.; Roy, C.J.; O’Neil, C.P.; Browning, E.A.; Pittet, L.A.; Altreuter, D.H.; Alexis, F.; Tonti, E.; Shi, J.; Basto, P.A.; et al. Adjuvant-carrying synthetic vaccine particles augment the immune response to encapsulated antigen and exhibit strong local immune activation without inducing systemic cytokine release. Vaccine 2014, 32, 2882–2895. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, J.; Babensee, J.E. Differential functional effects of biomaterials on dendritic cell maturation. Acta Biomater. 2012, 8, 3606–3617. [Google Scholar] [CrossRef] [Green Version]
- Elamanchili, P.; Diwan, M.; Cao, M.; Samuel, J. Characterization of poly(D,L-lactic-co-glycolic acid) based nanoparticulate system for enhanced delivery of antigens to dendritic cells. Vaccine 2004, 22, 2406–2412. [Google Scholar] [CrossRef] [PubMed]
- Elamanchili, P.; Lutsiak, C.M.; Hamdy, S.; Diwan, M.; Samuel, J. “Pathogen-mimicking” nanoparticles for vaccine delivery to dendritic cells. J. Immunother. 2007, 4, 378–395. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, M.; Babensee, J.E. Poly(lactic-co-glycolic acid) enhances maturation of human monocyte-derived dendritic cells. J. Biomed. Mater. Res. A 2004, 71, 45–54. [Google Scholar] [CrossRef] [PubMed]
- Raghuvanshi, R.S.; Katare, Y.K.; Lalwani, K.; Ali, M.M.; Singh, O.; Panda, A.K. Improved immune response from biodegradable polymer particles entrapping tetanus toxoid by use of different immunization protocol and adjuvants. Int. J. Pharm. 2002, 245, 109–121. [Google Scholar] [CrossRef]
- Hamdy, S.; Molavi, O.; Ma, Z.; Haddadi, A.; Alshamsan, A.; Gobti, Z.; Elhasi, S.; Samuel, J.; Lavasanifar, A. Co-delivery of cancer-associated antigen and Toll-like receptor 4 ligand in PLGA nanoparticles induces potent CD8+ T cell-mediated anti-tumor immunity. Vaccine 2008, 26, 5046–5057. [Google Scholar] [CrossRef] [PubMed]
- Ding, T.; Sun, J.; Zhang, P. Immune evaluation of biomaterials in TNF-α and IL-β at mRNA level. J. Mater. Sci. Mater. Med. 2007, 18, 2233–2236. [Google Scholar] [CrossRef] [PubMed]
- Sharp, F.A.; Ruane, D.; Claass, B.; Creagh, E.; Harris, J.; Malyala, P.; Singh, M.; O’Hagan, D.T.; Pétrilli, V.; Tschopp, J.; et al. Uptake of particulate vaccine adjuvants by dendritic cells activates the NALP3 inflammasome. Proc. Natl. Acad. Sci. USA 2009, 106, 870–875. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raghuvanshi, R.J.; Mistra, A.; Talwar, G.P.; Levy, R.J.; Labhasetwar, V. Enhanced immune response with a combination of alum and biodegradable nanoparticles containing tetanus toxoid. J. Microencapsul. 2001, 18, 723–732. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.; Xu, L.; Liang, C.; Wang, C.; Peng, R.; Liu, Z. Photothermal therapy with immune-adjuvant nanoparticles together with checkpoint blockade for effective cancer immunotherapy. Nat. Commun. 2016, 7, 13193. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ebrahimian, M.; Hashemi, M.; Maleki, M.; Hashemitabar, G.; Abnous, K.; Ramezani, M.; Haghparast, A. Co-delivery of Dual Toll-Like Receptor Agonists and Antigen in Poly(Lactic-Co-Glycolic) Acid/Polyethylenimine Cationic Hybrid Nanoparticles Promote Efficient In Vivo Immune Responses. Front. Immunol. 2017, 8, 1077. [Google Scholar] [CrossRef] [PubMed]
- Su, L.F.; Del Alcazar, D.; Stelekati, E.; Wherry, E.J.; Davis, M.M. Antigen exposure shapes the ratio between antigen-specific Tregs and conventional T cells in human peripheral blood. Proc. Natl. Acad. Sci. USA 2016, 113, e6192–e6198. [Google Scholar] [CrossRef] [PubMed]
- Maldonado, R.A.; LaMothe, R.A.; Ferrari, J.D.; Zhang, A.H.; Rossi, R.J.; Kolte, P.N.; Griset, A.P.; O’Neil, C.; Altreuter, D.H.; Browning, E.; et al. Polymeric synthetic nanoparticles for the induction of antigen-specific immunological tolerance. Proc. Natl. Acad. Sci. USA 2015, 112, e156–e165. [Google Scholar] [CrossRef]
- Getts, D.R.; Martin, A.J.; McCarthy, D.P.; Terry, R.L.; Hunter, Z.N.; Yap, W.T.; Getts, M.T.; Pleiss, M.; Luo, X.; King, N.J.; et al. Microparticles bearing encephalitogenic peptides induce T-cell toleranceand ameliorate experimental autoimmune encephalomyelitis. Nat. Biotechnol. 2012, 30, 1217–1224. [Google Scholar] [CrossRef]
- Casey, L.M.; Pearson, R.M.; Hughes, K.R.; Liu, J.M.H.; Rose, J.A.; North, M.G.; Wang, L.Z.; Lei, M.; Miller, S.D.; Shea, L.D. Conjugation of Transforming Growth Factor Beta to Antigen-Loaded Poly(lactide- co-glycolide) Nanoparticles Enhances Efficiency of Antigen-Specific Tolerance. Bioconjug Chem. 2018, 29, 813–823. [Google Scholar] [CrossRef] [PubMed]
- Kuo, R.; Saito, E.; Miller, S.D.; Shea, L.D. Peptide-Conjugated Nanoparticles Reduce Positive Co-stimulatory Expression and T Cell Activity to Induce Tolerance. Mol. Ther. 2017, 25, 1676–1685. [Google Scholar] [CrossRef] [PubMed]
- Gammon, J.M.; Tostanoski, L.H.; Adapa, A.R.; Chiu, Y.C.; Jewell, C.M. Controlled delivery of a metabolic modulator promotes regulatory T cells and restrains autoimmunity. J. Control. Release 2015, 210, 169–178. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cho, J.J.; Stewart, J.M.; Drashansky, T.T.; Brusko, M.A.; Zuniga, A.N.; Lorentsen, K.J.; Keselowsky, B.G.; Avram, D. An antigen-specific semi-therapeutic treatment with local delivery of tolerogenic factors through a dual-sized microparticle system blocks experimental autoimmune encephalomyelitis. Biomaterials 2017, 143, 79–92. [Google Scholar] [CrossRef] [PubMed]
- Pei, W.; Wan, X.; Shahzad, K.A.; Zhang, L.; Song, S.; Jin, X.; Wang, L.; Zhao, C.; Shen, C. Direct modulation of myelin-autoreactive CD4(+) and CD8(+) T cells in EAE mice by a tolerogenic nanoparticle co-carrying myelin peptide-loaded major histocompatibility complexes, CD47 and multiple regulatory molecules. Int. J. Nanomedicine. 2018, 13, 3731–3750. [Google Scholar] [CrossRef]
- Kim, W.U.; Lee, W.K.; Ryoo, J.W.; Kim, S.H.; Kim, J.; Youn, J.; Min, S.Y.; Bae, E.Y.; Hwang, S.Y.; Park, S.H.; et al. Suppression of collagen-induced arthritis by single administration of poly(lactic-co-glycolic acid) nanoparticles entrapping type II collagen: A novel treatment strategy for induction of oral tolerance. Arthritis Rheum. 2002, 46, 1109–1120. [Google Scholar] [CrossRef] [Green Version]
- Keijzer, C.; Slütter, B.; van der Zee, R.; Jiskoot, W.; van Eden, W.; Broere, F. PLGA, PLGA-TMC and TMC-TPP nanoparticles differentially modulate the outcome of nasal vaccination by inducing tolerance or enhancing humoral immunity. PLoS ONE 2011, 6, e26684. [Google Scholar] [CrossRef]
- Yoon, Y.M.; Lewis, J.S.; Carstens, M.R.; Campbell-Thompson, M.; Wasserfall, C.H.; Atkinson, M.A.; Keselowsky, B.G. A combination hydrogel microparticle-based vaccine prevents type 1 diabetes in non-obese diabetic mice. Sci. Rep. 2015, 5, 13155. [Google Scholar] [CrossRef] [Green Version]
- Verbeke, C.S.; Gordo, S.; Schubert, D.A.; Lewin, S.A.; Desai, R.M.; Dobbins, J.; Wucherpfennig, K.W.; Mooney, D.J. Multicomponent Injectable Hydrogels for Antigen-Specific Tolerogenic Immune Modulation. Adv. Healthc. Mater. 2017, 6, 1600773. [Google Scholar] [CrossRef] [Green Version]
- Lewis, J.S.; Dolgova, N.V.; Zhang, Y.; Xia, C.Q.; Wasserfall, C.H.; Atkinson, M.A.; Clare-Salzler, M.J.; Keselowsky, B.G. A combination dual-sized microparticle system modulates dendritic cells and prevents type 1 diabetes in prediabetic NOD mice. Clin. Immunol. 2015, 160, 90–102. [Google Scholar] [CrossRef] [Green Version]
- Goverman, J. Autoimmune T cell responses in the central nervous system. Nat. Rev. Immunol. 2009, 9, 93–407. [Google Scholar] [CrossRef] [PubMed]
- de Rosbo, N.K.; Kaye, J.F.; Eisenstein, M.; Mendel, I.; Hoeftberger, R.; Lassmann, H.; Milo, R.; Ben-Nun, A. The myelin-associated oligodendrocytic basic protein regionMOBP15-36 encompasses the immunodominant major encephalitogenic epitope(s) for SJL/J mice and predicted epitope(s) for multiple sclerosis-associated HLA-DRB1*1501. J. Immunol. 2004, 173, 1426–1435. [Google Scholar] [CrossRef] [PubMed]
- Laatsch, R.; Kies Mw Gordon, S.; Alvord, E., Jr. The encephalomyelitic activity of myelin isolated by ultracentrifugation. J. Exp. Med. 1962, 115, 777–788. [Google Scholar] [CrossRef] [PubMed]
- Eaton, V.L.; Vasquez, K.O.; Goings, G.E.; Hunter, Z.N.; Peterson, J.D.; Miller, S.D. Optical tomographic imaging of near infrared imaging agents quantifies disease severity and immunomodulation of experimental autoimmune encephalomyelitis in vivo. J. Neuroinflamm. 2013, 10, 138. [Google Scholar] [CrossRef] [Green Version]
- Firestein, G.S. Evolving concepts of rheumatoid arthritis. Nature 2003, 423, 356–361. [Google Scholar] [CrossRef] [PubMed]
- Kim, W.U.; Cho, M.L.; Jung, Y.O.; Min, S.Y.; Park, S.W.; Min, D.J.; Yoon, J.H.; Kim, H.Y. Type II collagen autoimmunity in rheumatoid arthritis. Am. J. Med. Sci. 2004, 327, 202–211. [Google Scholar] [CrossRef] [PubMed]
- Staines, N.A.; Wooley, P.H. Collagen arthritis; what can it teach us? Br. J. Rheumatol. 1994, 33, 798–807. [Google Scholar] [CrossRef]
- Kiyono, H.; Fukuyama, S. NALT- versus Peyer’s-patch-mediated mucosal immunity. Nat. Rev. Immunol. 2004, 4, 699–710. [Google Scholar] [CrossRef]
- Barker, J.M. Clinical review: Type 1 diabetes-associated autoimmunity: Natural history, genetic associations, and screening. J. Clin. Endocrinol. MeTable 2006, 91, 1210–1217. [Google Scholar] [CrossRef]
- Pan, Z.; Ding, J. Poly(lactide-co-glycolide) porous scaffolds for tissue engineering and regenerative medicine. Interface Focus 2012, 2, 366–377. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Mooney, D.J. Designing hydrogels for controlled drug delivery. Nat. Rev. Mater. 2016, 1, 16071. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Zhao, M.; Yu, C.; Zhang, X.; Liu, J.; Cheng, X.; Lee, R.J.; Sun, F.; Teng, L.; Li, Y. Multifunctional folate receptor-targeting and pH-responsive nanocarriers loaded with methotrexate for treatment of rheumatoid arthritis. Int. J. Nanomed. 2017, 12, 6735–6746. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.J.; Lee, S.M.; Park, K.H.; Mun, C.H.; Park, Y.B.; Yoo, K.H. Drug-loaded gold/iron/gold plasmonic nanoparticles for magnetic targeted chemo-photothermal treatment of rheumatoid arthritis. Biomaterials 2015, 61, 95–102. [Google Scholar] [CrossRef]
- Scheinman, R.I.; Trivedi, R.; Vermillion, S.; Kompella, U.B. Functionalized STAT1 siRNA nanoparticles regress rheumatoid arthritis in a mouse model. Nanomedicine (Lond.) 2011, 6, 1669–16682. [Google Scholar] [CrossRef] [PubMed]
- Langert, K.A.; Goshu, B.; Stubbs, E.B. Attenuation of experimental autoimmune neuritis with locally administered lovastatin-encapsulating poly(lactic-co-glycolic) acid nanoparticles. J. Neurochem. 2016, 140, 334–346. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y. FDA’s regulatory science program for generic PLA/PLGA-based drug products. Am. Pharm. Rev. 2016, 19, 5–9. [Google Scholar]


© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cappellano, G.; Comi, C.; Chiocchetti, A.; Dianzani, U. Exploiting PLGA-Based Biocompatible Nanoparticles for Next-Generation Tolerogenic Vaccines against Autoimmune Disease. Int. J. Mol. Sci. 2019, 20, 204. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20010204
Cappellano G, Comi C, Chiocchetti A, Dianzani U. Exploiting PLGA-Based Biocompatible Nanoparticles for Next-Generation Tolerogenic Vaccines against Autoimmune Disease. International Journal of Molecular Sciences. 2019; 20(1):204. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20010204
Chicago/Turabian StyleCappellano, Giuseppe, Cristoforo Comi, Annalisa Chiocchetti, and Umberto Dianzani. 2019. "Exploiting PLGA-Based Biocompatible Nanoparticles for Next-Generation Tolerogenic Vaccines against Autoimmune Disease" International Journal of Molecular Sciences 20, no. 1: 204. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20010204