Molecular Pathways and Respiratory Involvement in Lysosomal Storage Diseases

 ,
,
Abstract
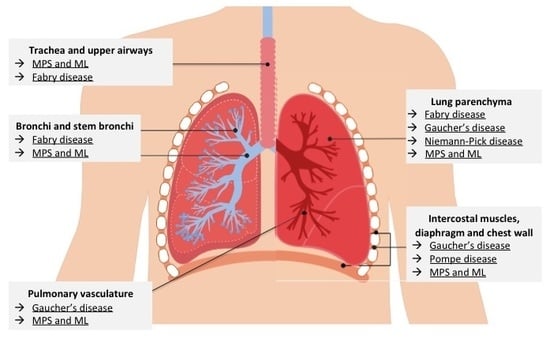
:
1. Introduction
2. Mucopolysaccharidosis and Mucolipidoses
2.1. Type of Respiratory Involvement
2.2. Molecular Pathways Involved
3. Pompe Disease
3.1. Type of Respiratory Involvement
3.2. Molecular Pathways Involved
4. Niemann-Pick Disease
4.1. Type of Respiratory Involvement
4.2. Molecular Pathways Involved
5. Gaucher’s Disease
5.1. Type of Respiratory Involvement
5.2. Molecular Pathways Involved
6. Fabry Disease
6.1. Type of Respiratory Involvement
6.2. Molecular Pathways Involved
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| ABG | Arterial blood gases |
| BAL | Bronchoalveolar lavage |
| BMI | Body mass index |
| CT | Computed tomography |
| DLCO | Diffusion capacity for carbon monoxide |
| EPAP | Expiratory positive airway pressure |
| ERT | Enzyme replacement therapy |
| FEV1 | Forced expiratory volume in the first second |
| FVC | Forced vital capacity |
| GAA | Acid alpha-glucosidase |
| GAG | Glycosaminoglycans |
| Gb3 | Globotriaosylceramide |
| GBA | Acid beta-glucosidase or glucosylceramidase |
| GSD II | Glycogen storage disease type II |
| HRCT | High-resolution computed tomography |
| HSCT | Hematopietic stem-cell transplantation |
| IPAP | Inspiratory positive airway pressure |
| LDL | Low-density lipoprotein |
| LOPD | Late-onset Pompe disease |
| LSD | Lysosomal storage diseases |
| Lyso-Gb3 | Globotriaosylsphingosine |
| MEP | Maximum expiratory pressure |
| MIP | Maximum inspiratory pressure |
| ML | Mucolipidosis |
| MPS | Mucopolysaccharidosis |
| MRI | Magnetic resonance imaging |
| MV | Mechanical ventilation |
| MVV | Maximum voluntary ventilation |
| NIV | Non-invasive ventilation |
| NPC | Niemann-Pick disease type C |
| OSA | Obstructive sleep apnea |
| PaCO2 | Partial pressure of carbon dioxide in arterial blood |
| PCF | Peak cough flow |
| PFTs | Pulmonary function tests |
| PSG | Polysomnography |
| REM | Rapid eye movement |
| SDB | Sleep-disordered breathing |
| SNIP | Sniff nasal inspiratory pressure |
| SRT | Substrate reduction therapy |
| VC | Vital capacity |
References
- Tomatsu, S.; Fujii, T.; Fukushi, M.; Oguma, T.; Shimada, T.; Maeda, M.; Kida, K.; Shibata, Y.; Futatsumori, H.; Montaño, A.M.; et al. Newborn screening and diagnosis of mucopolysaccharidoses. Mol. Genet. Metab. 2013, 110, 42–53. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wraith, J.E. Mucopolysaccharidoses and mucolipidoses. Handb. Clin. Neurol. 2013, 113, 1723–1729. [Google Scholar] [PubMed]
- Lin, S.-P.; Shih, S.-C.; Chuang, C.-K.; Lee, K.-S.; Chen, M.-R.; Niu, D.-M.; Chiu, P.C.; Lin, S.J.; Lin, H.-Y. Characterization of pulmonary function impairments in patients with mucopolysaccharidoses—Changes with age and treatment. Pediatr. Pulmonol. 2014, 49, 277–284. [Google Scholar] [CrossRef] [PubMed]
- Semenza, G.L.; Pyeritz, R.E. Respiratory complications of mucopolysaccharide storage disorders. Medicine 1988, 67, 209–219. [Google Scholar] [CrossRef] [PubMed]
- Muhlebach, M.S.; Wooten, W.; Muenzer, J. Respiratory manifestations in mucopolysaccharidoses. Paediatr. Respir. Rev. 2011, 12, 133–138. [Google Scholar] [CrossRef] [PubMed]
- Santamaria, F.; Andreucci, M.V.; Parenti, G.; Polverino, M.; Viggiano, D.; Montella, S.; Cesaro, A.; Ciccarelli, R.; Capaldo, B.; Andria, G. Upper airway obstructive disease in mucopolysaccharidoses: Polysomnography, computed tomography and nasal endoscopy findings. J. Inherit. Metab. Dis. 2007, 30, 743–749. [Google Scholar] [CrossRef] [PubMed]
- Muenzer, J.; Wraith, J.E.; Clarke, L.A. International Consensus Panel on Management and Treatment of Mucopolysaccharidosis I Mucopolysaccharidosis I: Management and treatment guidelines. Pediatrics 2009, 123, 19–29. [Google Scholar] [CrossRef] [PubMed]
- Keilmann, A.; Bendel, F.; Nospes, S.; Lampe, C.; Läßig, A.K. Alterations of mucosa of the larynx and hypopharynx in patients with mucopolysaccharidoses. J. Laryngol. Otol. 2016, 130, 194–200. [Google Scholar] [CrossRef] [PubMed]
- Simmons, M.A.; Bruce, I.A.; Penney, S.; Wraith, E.; Rothera, M.P. Otorhinolaryngological manifestations of the mucopolysaccharidoses. Int. J. Pediatr. Otorhinolaryngol. 2005, 69, 589–595. [Google Scholar] [CrossRef]
- Kubaski, F.; Tomatsu, S.; Patel, P.; Shimada, T.; Xie, L.; Yasuda, E.; Mason, R.; Mackenzie, W.G.; Theroux, M.; Bober, M.B.; et al. Non-invasive pulmonary function test on Morquio patients. Mol. Genet. Metab. 2015, 115, 186–192. [Google Scholar] [CrossRef] [Green Version]
- Pizarro, C.; Davies, R.R.; Theroux, M.; Spurrier, E.A.; Averill, L.W.; Tomatsu, S. Surgical Reconstruction for Severe Tracheal Obstruction in Morquio A Syndrome. Ann. Thorac. Surg. 2016, 102, e329–e331. [Google Scholar] [CrossRef] [PubMed]
- Doherty, C.; Averill, L.W.; Theroux, M.; Mackenzie, W.G.; Pizarro, C.; Mason, R.W.; Tomatsu, S. Natural history of Morquio A patient with tracheal obstruction from birth to death. Mol. Genet. Metab. Rep. 2018, 14, 59–67. [Google Scholar] [CrossRef] [PubMed]
- Tomatsu, S.; Averill, L.W.; Sawamoto, K.; Mackenzie, W.G.; Bober, M.B.; Pizarro, C.; Goff, C.J.; Xie, L.; Orii, T.; Theroux, M. Obstructive airway in Morquio A syndrome, the past, the present and the future. Mol. Genet. Metab. 2016, 117, 150–156. [Google Scholar] [CrossRef] [Green Version]
- Shinhar, S.Y.; Zablocki, H.; Madgy, D.N. Airway management in mucopolysaccharide storage disorders. Arch. Otolaryngol. Head Neck Surg. 2004, 130, 233–237. [Google Scholar] [CrossRef] [PubMed]
- Rutten, M.; Ciet, P.; van den Biggelaar, R.; Oussoren, E.; Langendonk, J.G.; van der Ploeg, A.T.; Langeveld, M. Severe tracheal and bronchial collapse in adults with type II mucopolysaccharidosis. Orphanet J. Rare Dis. 2016, 11, 50. [Google Scholar] [CrossRef] [PubMed]
- Berger, K.I.; Fagondes, S.C.; Giugliani, R.; Hardy, K.A.; Lee, K.S.; McArdle, C.; Scarpa, M.; Tobin, M.J.; Ward, S.A.; Rapoport, D.M. Respiratory and sleep disorders in mucopolysaccharidosis. J. Inherit. Metab. Dis. 2013, 36, 201–210. [Google Scholar] [CrossRef]
- Moreira, G.A.; Kyosen, S.O.; Patti, C.L.; Martins, A.M.; Tufik, S. Prevalence of obstructive sleep apnea in patients with mucopolysaccharidosis types I, II, and VI in a reference center. Sleep Breath. Schlaf Atm. 2014, 18, 791–797. [Google Scholar] [CrossRef] [PubMed]
- Gönüldaş, B.; Yılmaz, T.; Sivri, H.S.; Güçer, K.Ş.; Kılınç, K.; Genç, G.A.; Kılıç, M.; Coşkun, T. Mucopolysaccharidosis: Otolaryngologic findings, obstructive sleep apnea and accumulation of glucosaminoglycans in lymphatic tissue of the upper airway. Int. J. Pediatr. Otorhinolaryngol. 2014, 78, 944–949. [Google Scholar] [CrossRef] [PubMed]
- Hendriksz, C.J.; Berger, K.I.; Parini, R.; AlSayed, M.D.; Raiman, J.; Giugliani, R.; Mitchell, J.J.; Burton, B.K.; Guelbert, N.; Stewart, F.; et al. Impact of long-term elosulfase alfa treatment on respiratory function in patients with Morquio A syndrome. J. Inherit. Metab. Dis. 2016, 39, 839–847. [Google Scholar] [CrossRef] [Green Version]
- Kamin, W. Diagnosis and management of respiratory involvement in Hunter syndrome. Acta Paediatr. 2008, 97, 57–60. [Google Scholar] [CrossRef]
- Harmatz, P.R.; Mengel, K.E.; Giugliani, R.; Valayannopoulos, V.; Lin, S.-P.; Parini, R.; Guffon, N.; Burton, B.K.; Hendriksz, C.J.; Mitchell, J.J.; et al. Longitudinal analysis of endurance and respiratory function from a natural history study of Morquio A syndrome. Mol. Genet. Metab. 2015, 114, 186–194. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ingelmo, P.M.; Parini, R.; Grimaldi, M.; Mauri, F.; Romagnoli, M.; Tagliabue, G.; Somaini, M.; Sahillioğlu, E.; Frawley, G. Multidetector computed tomography (MDCT) for preoperative airway assessment in children with mucopolysaccharidoses. Minerva Anestesiol. 2011, 77, 774–780. [Google Scholar] [PubMed]
- Dodsworth, C.; Burton, B.K. Increased incidence of neonatal respiratory distress in infants with mucopolysaccharidosis type II (MPS II, Hunter syndrome). Mol. Genet. Metab. 2014, 111, 203–204. [Google Scholar] [CrossRef] [PubMed]
- Wraith, J.E.; Beck, M.; Lane, R.; van der Ploeg, A.; Shapiro, E.; Xue, Y.; Kakkis, E.D.; Guffon, N. Enzyme replacement therapy in patients who have mucopolysaccharidosis I and are younger than 5 years: Results of a multinational study of recombinant human alpha-L-iduronidase (laronidase). Pediatrics 2007, 120, e37–e46. [Google Scholar] [CrossRef] [PubMed]
- Hendriksz, C.J.; Al-Jawad, M.; Berger, K.I.; Hawley, S.M.; Lawrence, R.; Mc Ardle, C.; Summers, C.G.; Wright, E.; Braunlin, E. Clinical overview and treatment options for non-skeletal manifestations of mucopolysaccharidosis type IVA. J. Inherit. Metab. Dis. 2013, 36, 309–322. [Google Scholar] [CrossRef] [PubMed]
- Proia, R.L.; Wu, Y.-P. Blood to brain to the rescue. J. Clin. Investig. 2004, 113, 1108–1110. [Google Scholar] [CrossRef] [Green Version]
- Aldenhoven, M.; Wynn, R.F.; Orchard, P.J.; O’Meara, A.; Veys, P.; Fischer, A.; Valayannopoulos, V.; Neven, B.; Rovelli, A.; Prasad, V.K.; et al. Long-term outcome of Hurler syndrome patients after hematopoietic cell transplantation: An international multicenter study. Blood 2015, 125, 2164–2172. [Google Scholar] [CrossRef]
- Yeung, A.H.; Cowan, M.J.; Horn, B.; Rosbe, K.W. Airway management in children with mucopolysaccharidoses. Arch. Otolaryngol. Head Neck Surg. 2009, 135, 73–79. [Google Scholar] [CrossRef]
- Arn, P.; Bruce, I.A.; Wraith, J.E.; Travers, H.; Fallet, S. Airway-related symptoms and surgeries in patients with mucopolysaccharidosis I. Ann. Otol. Rhinol. Laryngol. 2015, 124, 198–205. [Google Scholar] [CrossRef]
- Ballabio, A.; Gieselmann, V. Lysosomal disorders: From storage to cellular damage. Biochim. Biophys. Acta 2009, 1793, 684–696. [Google Scholar] [CrossRef] [Green Version]
- Fecarotta, S.; Gasperini, S.; Parenti, G. New treatments for the mucopolysaccharidoses: From pathophysiology to therapy. Ital. J. Pediatr. 2018, 44, 124. [Google Scholar] [CrossRef]
- Remiche, G.; Ronchi, D.; Magri, F.; Lamperti, C.; Bordoni, A.; Moggio, M.; Bresolin, N.; Comi, G.P. Extended phenotype description and new molecular findings in late onset glycogen storage disease type II: A northern Italy population study and review of the literature. J. Neurol. 2014, 261, 83–97. [Google Scholar] [CrossRef]
- Chan, J.; Desai, A.K.; Kazi, Z.B.; Corey, K.; Austin, S.; Hobson-Webb, L.D.; Case, L.E.; Jones, H.N.; Kishnani, P.S. The emerging phenotype of late-onset Pompe disease: A systematic literature review. Mol. Genet. Metab. 2017, 120, 163–172. [Google Scholar] [CrossRef]
- Gaeta, M.; Musumeci, O.; Mondello, S.; Ruggeri, P.; Montagnese, F.; Cucinotta, M.; Vinci, S.; Milardi, D.; Toscano, A. Clinical and pathophysiological clues of respiratory dysfunction in late-onset Pompe disease: New insights from a comparative study by MRI and respiratory function assessment. Neuromuscul. Disord. NMD 2015, 25, 852–858. [Google Scholar] [CrossRef] [PubMed]
- Wens, S.C.A.; Ciet, P.; Perez-Rovira, A.; Logie, K.; Salamon, E.; Wielopolski, P.; de Bruijne, M.; Kruijshaar, M.E.; Tiddens, H.A.W.M.; van Doorn, P.A.; et al. Lung MRI and impairment of diaphragmatic function in Pompe disease. BMC Pulm. Med. 2015, 15, 54. [Google Scholar] [CrossRef] [PubMed]
- American Association of Neuromuscular & Electrodiagnostic Medicine Diagnostic criteria for late-onset (childhood and adult) Pompe disease. Muscle Nerve 2009, 40, 149–160.
- Mellies, U.; Ragette, R.; Schwake, C.; Baethmann, M.; Voit, T.; Teschler, H. Sleep-disordered breathing and respiratory failure in acid maltase deficiency. Neurology 2001, 57, 1290–1295. [Google Scholar] [CrossRef] [PubMed]
- Boentert, M.; Prigent, H.; Várdi, K.; Jones, H.N.; Mellies, U.; Simonds, A.K.; Wenninger, S.; Barrot Cortés, E.; Confalonieri, M. Practical Recommendations for Diagnosis and Management of Respiratory Muscle Weakness in Late-Onset Pompe Disease. Int. J. Mol. Sci. 2016, 17. [Google Scholar] [CrossRef] [PubMed]
- Mellies, U.; Stehling, F.; Dohna-Schwake, C.; Ragette, R.; Teschler, H.; Voit, T. Respiratory failure in Pompe disease: Treatment with noninvasive ventilation. Neurology 2005, 64, 1465–1467. [Google Scholar] [CrossRef] [PubMed]
- Boentert, M.; Dräger, B.; Glatz, C.; Young, P. Sleep-Disordered Breathing and Effects of Noninvasive Ventilation in Patients with Late-Onset Pompe Disease. J. Clin. Sleep Med. 2016, 12, 1623–1632. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- American Thoracic Society/European Respiratory Society ATS/ERS Statement on respiratory muscle testing. Am. J. Respir. Crit. Care Med. 2002, 166, 518–624. [CrossRef]
- van der Ploeg, A.T.; Kruijshaar, M.E.; Toscano, A.; Laforêt, P.; Angelini, C.; Lachmann, R.H.; Pascual Pascual, S.I.; Roberts, M.; Rösler, K.; Stulnig, T.; et al. European consensus for starting and stopping enzyme replacement therapy in adult patients with Pompe disease: A 10-year experience. Eur. J. Neurol. 2017, 24, 768–e31. [Google Scholar] [CrossRef] [PubMed]
- Schoser, B.; Stewart, A.; Kanters, S.; Hamed, A.; Jansen, J.; Chan, K.; Karamouzian, M.; Toscano, A. Survival and long-term outcomes in late-onset Pompe disease following alglucosidase alfa treatment: A systematic review and meta-analysis. J. Neurol. 2017, 264, 621–630. [Google Scholar] [CrossRef] [PubMed]
- Filosto, M.; Todeschini, A.; Cotelli, M.S.; Vielmi, V.; Rinaldi, F.; Rota, S.; Scarpelli, M.; Padovani, A. Non-muscle involvement in late-onset glycogenosis II. Acta Myol. Myopathies Cardiomyopathies 2013, 32, 91–94. [Google Scholar]
- Llerena Junior, J.C.; Nascimento, O.J.; Oliveira, A.S.B.; Dourado Junior, M.E.T.; Marrone, C.D.; Siqueira, H.H.; Sobreira, C.F.R.; Dias-Tosta, E.; Werneck, L.C. Guidelines for the diagnosis, treatment and clinical monitoring of patients with juvenile and adult Pompe disease. Arq. Neuropsiquiatr. 2016, 74, 166–176. [Google Scholar] [CrossRef] [PubMed]
- Shea, L.; Raben, N. Autophagy in skeletal muscle: Implications for Pompe disease. Int. J. Clin. Pharmacol. Ther. 2009, 47 (Suppl 1), S42–S47. [Google Scholar] [CrossRef]
- Von Ranke, F.M.; Pereira Freitas, H.M.; Mançano, A.D.; Rodrigues, R.S.; Hochhegger, B.; Escuissato, D.; Araujo Neto, C.A.; da Silva, T.K.B.; Marchiori, E. Pulmonary Involvement in Niemann-Pick Disease: A State-of-the-Art Review. Lung 2016, 194, 511–518. [Google Scholar] [CrossRef]
- Mendelson, D.S.; Wasserstein, M.P.; Desnick, R.J.; Glass, R.; Simpson, W.; Skloot, G.; Vanier, M.; Bembi, B.; Giugliani, R.; Mengel, E.; et al. Type B Niemann-Pick disease: Findings at chest radiography, thin-section CT, and pulmonary function testing. Radiology 2006, 238, 339–345. [Google Scholar] [CrossRef]
- Minai, O.A.; Sullivan, E.J.; Stoller, J.K. Pulmonary involvement in Niemann-Pick disease: Case report and literature review. Respir. Med. 2000, 94, 1241–1251. [Google Scholar] [CrossRef]
- Chung, M.J.; Lee, K.S.; Franquet, T.; Müller, N.L.; Han, J.; Kwon, O.J. Metabolic lung disease: Imaging and histopathologic findings. Eur. J. Radiol. 2005, 54, 233–245. [Google Scholar] [CrossRef]
- Rossi, G.; Cavazza, A.; Spagnolo, P.; Bellafiore, S.; Kuhn, E.; Carassai, P.; Caramanico, L.; Montanari, G.; Cappiello, G.; Andreani, A.; et al. The role of macrophages in interstitial lung diseases: Number 3 in the Series “Pathology for the clinician” Edited by Peter Dorfmüller and Alberto Cavazza. Eur. Respir. Rev. 2017, 26, 170009. [Google Scholar] [CrossRef] [PubMed]
- Staretz-Chacham, O.; Aviram, M.; Morag, I.; Goldbart, A.; Hershkovitz, E. Pulmonary involvement in Niemann-Pick C type 1. Eur. J. Pediatr. 2018, 177, 1609–1615. [Google Scholar] [CrossRef] [PubMed]
- Stirnemann, J.; Belmatoug, N.; Camou, F.; Serratrice, C.; Froissart, R.; Caillaud, C.; Levade, T.; Astudillo, L.; Serratrice, J.; Brassier, A.; et al. A Review of Gaucher Disease Pathophysiology, Clinical Presentation and Treatments. Int. J. Mol. Sci. 2017, 18, 441. [Google Scholar] [CrossRef] [PubMed]
- Galiè, N.; Humbert, M.; Vachiery, J.-L.; Gibbs, S.; Lang, I.; Torbicki, A.; Simonneau, G.; Peacock, A.; Vonk Noordegraaf, A.; Beghetti, M.; et al. 2015 ESC/ERS Guidelines for the diagnosis and treatment of pulmonary hypertension: The Joint Task Force for the Diagnosis and Treatment of Pulmonary Hypertension of the European Society of Cardiology (ESC) and the European Respiratory Society (ERS): Endorsed by: Association for European Paediatric and Congenital Cardiology (AEPC), International Society for Heart and Lung Transplantation (ISHLT). Eur. Respir. J. 2015, 46, 903–975. [Google Scholar]
- Gülhan, B.; Ozçelik, U.; Gürakan, F.; Güçer, S.; Orhan, D.; Cinel, G.; Yalçin, E.; Ersöz, D.D.; Kiper, N.; Yüce, A.; et al. Different features of lung involvement in Niemann-Pick disease and Gaucher disease. Respir. Med. 2012, 106, 1278–1285. [Google Scholar] [CrossRef] [Green Version]
- Ross, D.J.; Spira, S.; Buchbinder, N.A. Gaucher cells in pulmonary-capillary blood in association with pulmonary hypertension. N. Engl. J. Med. 1997, 336, 379–381. [Google Scholar] [CrossRef]
- Lachmann, R.H.; Wight, D.G.; Lomas, D.J.; Fisher, N.C.; Schofield, J.P.; Elias, E.; Cox, T.M. Massive hepatic fibrosis in Gaucher’s disease: Clinico-pathological and radiological features. Mon. J. Assoc. Physicians 2000, 93, 237–244. [Google Scholar] [CrossRef]
- Goitein, O.; Elstein, D.; Abrahamov, A.; Hadas-Halpern, I.; Melzer, E.; Kerem, E.; Zimran, A. Lung involvement and enzyme replacement therapy in Gaucher’s disease. Mon. J. Assoc. Physicians 2001, 94, 407–415. [Google Scholar] [CrossRef]
- Fuller, M.; Meikle, P.J.; Hopwood, J.J. Epidemiology of lysosomal storage diseases: An overview. In Fabry Disease: Perspectives from 5 Years of FOS; Mehta, A., Beck, M., Sunder-Plassmann, G., Eds.; Oxford PharmaGenesis: Oxford, UK, 2006; ISBN 978-1-903539-03-3. [Google Scholar]
- Svensson, C.K.; Feldt-Rasmussen, U.; Backer, V. Fabry disease, respiratory symptoms, and airway limitation—A systematic review. Eur. Clin. Respir. J. 2015, 2. [Google Scholar] [CrossRef]
- Bagdade, J.D.; Parker, F.; Ways, P.O.; Morgan, T.E.; Lagunoff, D.; Eidelman, S. Fabry’s disease. A correlative clinical, morphologic, and biochemical study. Lab. Investig. J. Tech. Methods Pathol. 1968, 18, 681–688. [Google Scholar]
- Kelly, M.M.; Leigh, R.; McKenzie, R.; Kamada, D.; Ramsdale, E.H.; Hargreave, F.E. Induced sputum examination: Diagnosis of pulmonary involvement in Fabry’s disease. Thorax 2000, 55, 720–721. [Google Scholar] [CrossRef] [PubMed]
- Smith, P.; Heath, D.; Rodgers, B.; Helliwell, T. Pulmonary vasculature in Fabry’s disease. Histopathology 1991, 19, 567–569. [Google Scholar] [CrossRef]
- Kariman, K.; Singletary, W.V.; Sieker, H.O. Pulmonary involvement in Fabry’s disease. Am. J. Med. 1978, 64, 911–912. [Google Scholar] [CrossRef]
- Rosenberg, D.M.; Ferrans, V.J.; Fulmer, J.D.; Line, B.R.; Barranger, J.A.; Brady, R.O.; Crystal, R.G. Chronic airflow obstruction in Fabry’s disease. Am. J. Med. 1980, 68, 898–905. [Google Scholar] [CrossRef]
- Magage, S.; Lubanda, J.-C.; Susa, Z.; Bultas, J.; Karetová, D.; Dobrovolný, R.; Hrebícek, M.; Germain, D.P.; Linhart, A. Natural history of the respiratory involvement in Anderson-Fabry disease. J. Inherit. Metab. Dis. 2007, 30, 790–799. [Google Scholar] [CrossRef] [PubMed]
- Brown, L.K.; Miller, A.; Bhuptani, A.; Sloane, M.F.; Zimmerman, M.I.; Schilero, G.; Eng, C.M.; Desnick, R.J. Pulmonary involvement in Fabry disease. Am. J. Respir. Crit. Care Med. 1997, 155, 1004–1010. [Google Scholar] [CrossRef] [PubMed]
- Barbey, F.; Widmer, U.; Brack, T.; Vogt, B.; Aubert, J. Spirometric abnormalities in patients with Fabry disease and effect of enzyme replacement therapy. In Proceedings of the 4th International Symposium on Lysosomal Storage, Seville, Spain, 23–24 April 2004; p. 105. [Google Scholar]
- Aubert, J.D.; Barbey, F. Pulmonary involvement in Fabry disease. In Fabry Disease: Perspectives from 5 Years of FOS; Mehta, A., Beck, M., Sunder-Plassmann, G., Eds.; Oxford PharmaGenesis: Oxford, UK, 2006; ISBN 1-903539-03-X. [Google Scholar]
- Faverio, P.; Mantero, M.; Pieruzzi, F.; Torti, G.; Di Giacomo, A.; Pesci, A. Early recognition of airway obstruction In Fabry disease and correlation with dyspnea: A case series. Minerva Pneumol. 2016, 55, 1–6. [Google Scholar]
- Pauwels, R.A.; Buist, A.S.; Calverley, P.M.; Jenkins, C.R.; Hurd, S.S. GOLD Scientific Committee Global strategy for the diagnosis, management, and prevention of chronic obstructive pulmonary disease. NHLBI/WHO Global Initiative for Chronic Obstructive Lung Disease (GOLD) Workshop summary. Am. J. Respir. Crit. Care Med. 2001, 163, 1256–1276. [Google Scholar] [CrossRef] [PubMed]
- Brown, C.J.; Friedkin, R.J.; Inouye, S.K. Prevalence and outcomes of low mobility in hospitalized older patients. J. Am. Geriatr. Soc. 2004, 52, 1263–1270. [Google Scholar] [CrossRef]
- Franzen, D.; Krayenbuehl, P.A.; Lidove, O.; Aubert, J.-D.; Barbey, F. Pulmonary involvement in Fabry disease: Overview and perspectives. Eur. J. Intern. Med. 2013, 24, 707–713. [Google Scholar] [CrossRef] [Green Version]
- Kendrick, K.R.; Baxi, S.C.; Smith, R.M. Usefulness of the modified 0-10 Borg scale in assessing the degree of dyspnea in patients with COPD and asthma. J. Emerg. Nurs. 2000, 26, 216–222. [Google Scholar] [CrossRef]
- Bierer, G.; Kamangar, N.; Balfe, D.; Wilcox, W.R.; Mosenifar, Z. Cardiopulmonary Exercise Testing in Fabry Disease. Respiration 2005, 72, 504–511. [Google Scholar] [CrossRef] [PubMed]
- Odler, B.; Cseh, Á.; Constantin, T.; Fekete, G.; Losonczy, G.; Tamási, L.; Benke, K.; Szilveszter, B.; Müller, V. Long time enzyme replacement therapy stabilizes obstructive lung disease and alters peripheral immune cell subsets in Fabry patients. Clin. Respir. J. 2016, 11, 942–950. [Google Scholar] [CrossRef] [PubMed]
- Faverio, P.; Binaggia, A.; Pieruzzi, F.; Torti, G.; Pesci, A. Progression of obstructive ventilatory disorder in Fabry disease: Only a matter of time? Clin. Respir. J. 2018, 12, 832–834. [Google Scholar] [CrossRef] [PubMed]
- Franzen, D.P.; Nowak, A.; Haile, S.R.; Mottet, D.; Bonani, M.; Dormond, O.; Kohler, M.; Krayenbuehl, P.A.; Barbey, F. Long-term follow-up of pulmonary function in Fabry disease: A bi-center observational study. PLoS ONE 2017, 12, e0180437. [Google Scholar] [CrossRef]
- Kim, W.; Pyeritz, R.E.; Bernhardt, B.A.; Casey, M.; Litt, H.I. Pulmonary manifestations of Fabry disease and positive response to enzyme replacement therapy. Am. J. Med. Genet. A 2007, 143, 377–381. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.Y.; Abe, J.T.; Cohen, A.H.; Wilcox, W.R. Enzyme replacement therapy stabilizes obstructive pulmonary Fabry disease associated with respiratory globotriaosylceramide storage. J. Inherit. Metab. Dis. 2008, 31 (Suppl 2), S369–S374. [Google Scholar] [CrossRef]
- Ahuja, J.; Kanne, J.P.; Meyer, C.A.; Pipavath, S.N.J.; Schmidt, R.A.; Swanson, J.O.; Godwin, J.D. Histiocytic disorders of the chest: Imaging findings. Radiographics 2015, 35, 357–370. [Google Scholar] [CrossRef]
- Duning, T.; Deppe, M.; Keller, S.; Schiffbauer, H.; Stypmann, J.; Böntert, M.; Schaefer, R.; Young, P. Excessive Daytime Sleepiness Is a Common Symptom in Fabry Disease. Case Rep. Neurol. 2009, 1, 33–40. [Google Scholar] [CrossRef] [Green Version]
- Desnick, R.J.; Brady, R.; Barranger, J.; Collins, A.J.; Germain, D.P.; Goldman, M.; Grabowski, G.; Packman, S.; Wilcox, W.R. Fabry disease, an under-recognized multisystemic disorder: Expert recommendations for diagnosis, management, and enzyme replacement therapy. Ann. Intern. Med. 2003, 138, 338–346. [Google Scholar] [CrossRef]
- Franzen, D.; Gerard, N.; Bratton, D.J.; Wons, A.; Gaisl, T.; Sievi, N.A.; Clarenbach, C.F.; Kohler, M.; Krayenbühl, P.A. Prevalence and Risk Factors of Sleep Disordered Breathing in Fabry disease: A Prospective Cohort Study. Medicine 2015, 94, e2413. [Google Scholar] [CrossRef] [PubMed]
- Talbot, A.; Hammerschlag, G.; Goldin, J.; Nicholls, K. Sleep Disturbance, Obstructive Sleep Apnoea and Abnormal Periodic Leg Movements: Very Common Problems in Fabry Disease. In JIMD Reports; Springer: Berlin/Heidelberg, Germany, 2016; Volume 31, pp. 37–41. [Google Scholar]
- Mehta, A.; Widmer, U. Natural history of Fabry disease. In Fabry Disease: Perspectives from 5 Years of FOS; Mehta, A., Beck, M., Sunder-Plassmann, G., Eds.; Oxford PharmaGenesis: Oxford, UK, 2006; ISBN 978-1-903539-03-3. [Google Scholar]
- Rozenfeld, P.; Feriozzi, S. Contribution of inflammatory pathways to Fabry disease pathogenesis. Mol. Genet. Metab. 2017, 122, 19–27. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
Lysosomal Storage Diseases (OR Mucopolysaccharidosis OR Mucolipidoses OR Pompe disease OR Niemann-Pick disease OR Gaucher’s disease OR Fabry disease) AND Lung involvement (OR Lung manifestations OR Respiratory involvement OR Respiratory manifestations OR pulmonary involvement OR pulmonary manifestations OR Pulmonary function tests OR restrictive lung disease OR obstructive lung disease OR airway obstruction OR interstitial lung disease OR pulmonary hypertension OR obstructive sleep apnea OR sleep-disordered breathing OR hypoventilation OR muscle weakness OR diaphragmatic weakness OR respiratory failure) |
| Current Treatments | Mucopolysaccharidosis | Mucolipidoses | Pompe Disease | Niemann-Pick Disease | Gaucher’s Disease | Fabry Disease |
|---|---|---|---|---|---|---|
| Symptomatic treatments for rhinitis and otitis (nasal wash and decongestions, pressure equalization tubes) | X | X | ||||
| Inhaled Beta2-agonists and anticholinergic agents ± inhaled corticosteroids to optimize bronchodilation | X | X | X | |||
| Airway clearance techniques, including cough assistance when appropriate | X | X | X | |||
| Prompt chest infections’ management | X | X | X | X | X | X |
| Non-Invasive Ventilation with positive airway pressure treatment (continuous or bilevel) with or without oxygen supplementation (for obstructive sleep apneas and/or muscle weakness) | X | X | X | |||
| Tracheostomy for tracheal obstruction and/or invasive mechanical ventilation | X | X | X | |||
| Tonsillectomy and adenoidectomy | X | X | ||||
| Tracheal (-vascular) reconstructive surgery | X | X | ||||
| Stents or laser excision of tracheal lesions | X | X | ||||
| Whole lung lavage | X | |||||
| Enzyme Replacement Therapy | X | X | X | X | X | |
| Substrate Reduction Therapy | X | |||||
| Hematopoietic Stem Cell transplantation | X | X | ||||
| Smoking cessation | X | X | X | X | X | X |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Faverio, P.; Stainer, A.; De Giacomi, F.; Gasperini, S.; Motta, S.; Canonico, F.; Pieruzzi, F.; Monzani, A.; Pesci, A.; Biondi, A. Molecular Pathways and Respiratory Involvement in Lysosomal Storage Diseases. Int. J. Mol. Sci. 2019, 20, 327. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20020327
Faverio P, Stainer A, De Giacomi F, Gasperini S, Motta S, Canonico F, Pieruzzi F, Monzani A, Pesci A, Biondi A. Molecular Pathways and Respiratory Involvement in Lysosomal Storage Diseases. International Journal of Molecular Sciences. 2019; 20(2):327. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20020327
Chicago/Turabian StyleFaverio, Paola, Anna Stainer, Federica De Giacomi, Serena Gasperini, Serena Motta, Francesco Canonico, Federico Pieruzzi, Anna Monzani, Alberto Pesci, and Andrea Biondi. 2019. "Molecular Pathways and Respiratory Involvement in Lysosomal Storage Diseases" International Journal of Molecular Sciences 20, no. 2: 327. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20020327