A Novel Rare Missense Variation of the NOD2 Gene: Evidences of Implication in Crohn’s Disease

 , ,
, ,
Abstract
:1. Introduction
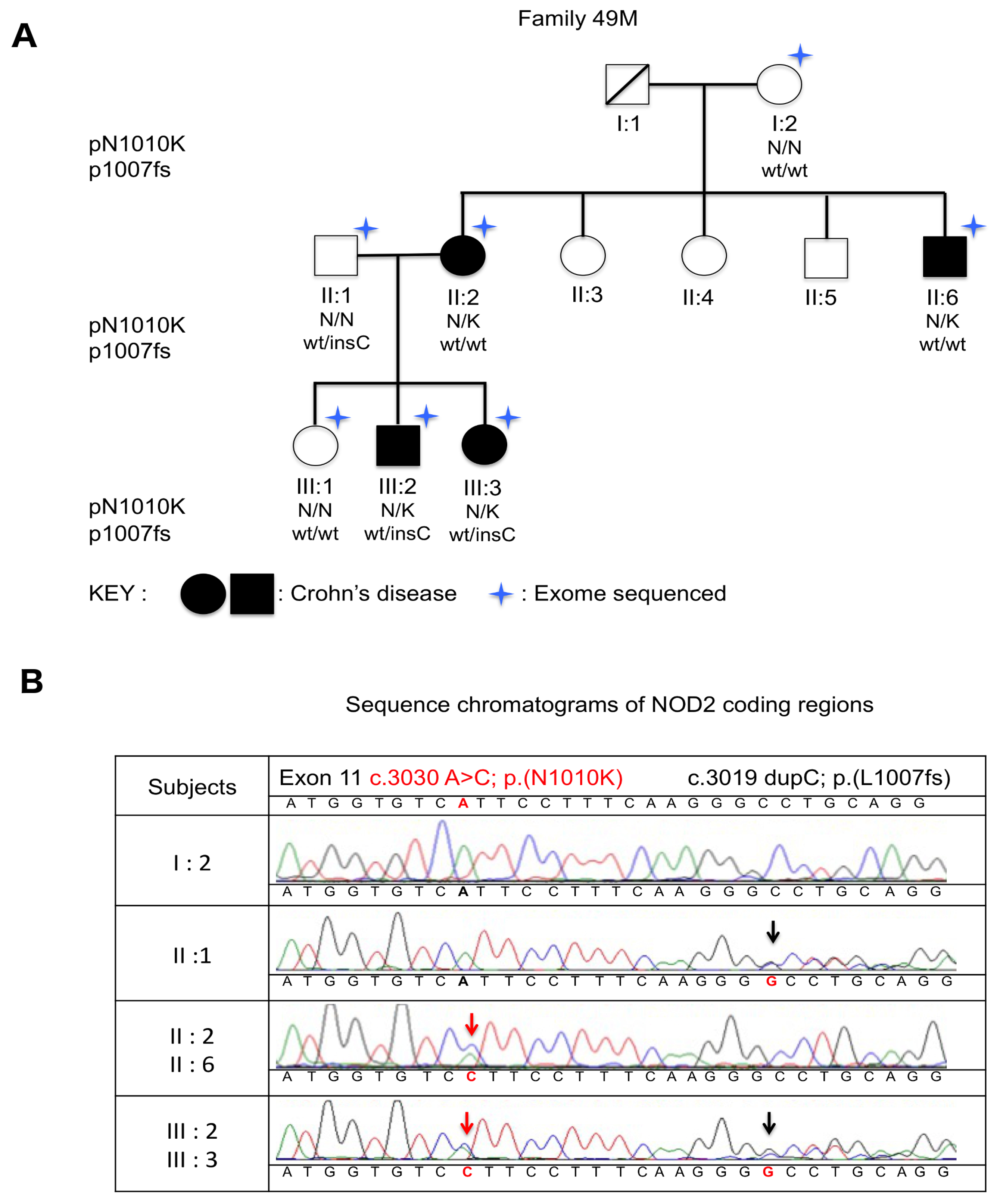
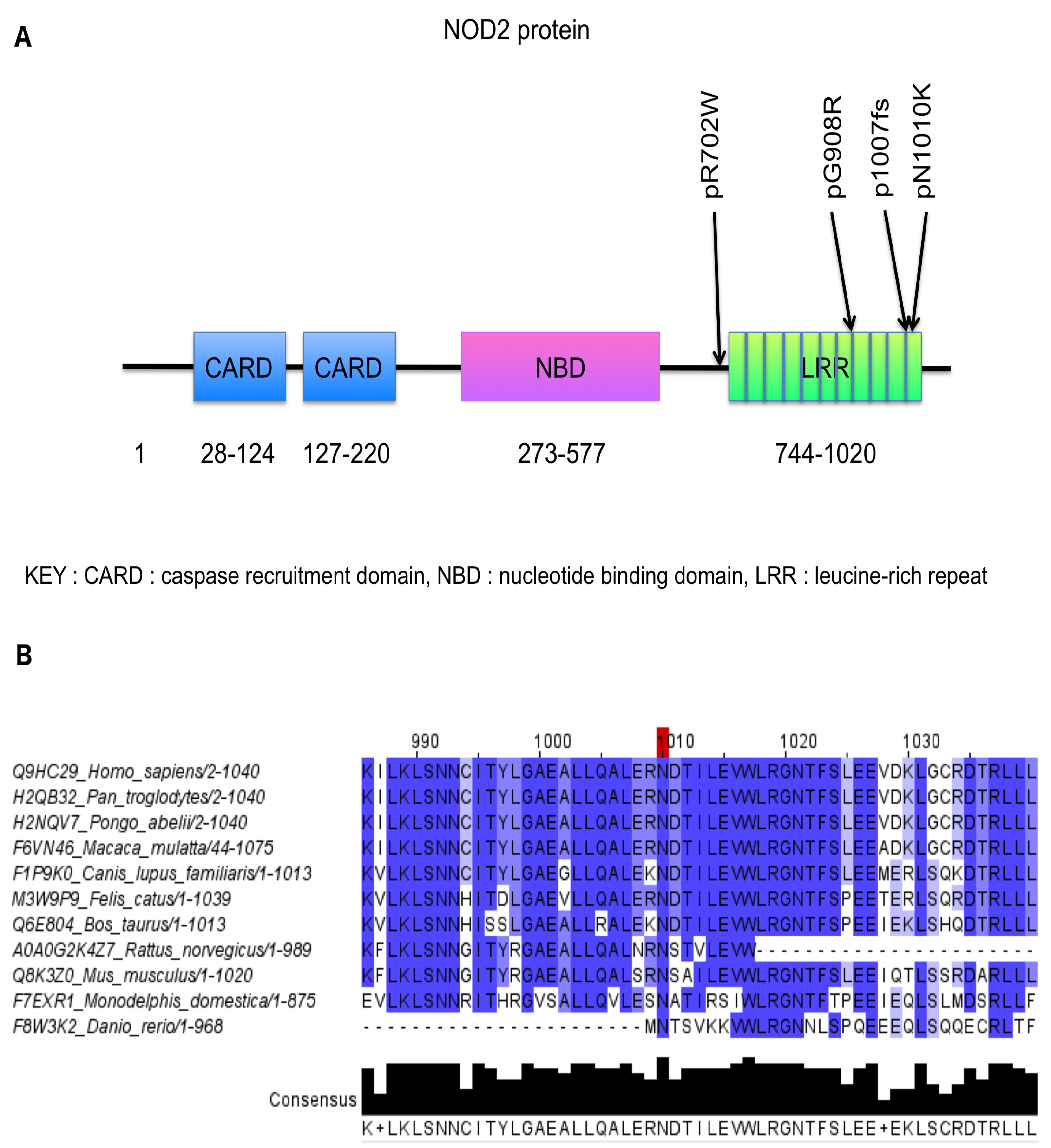
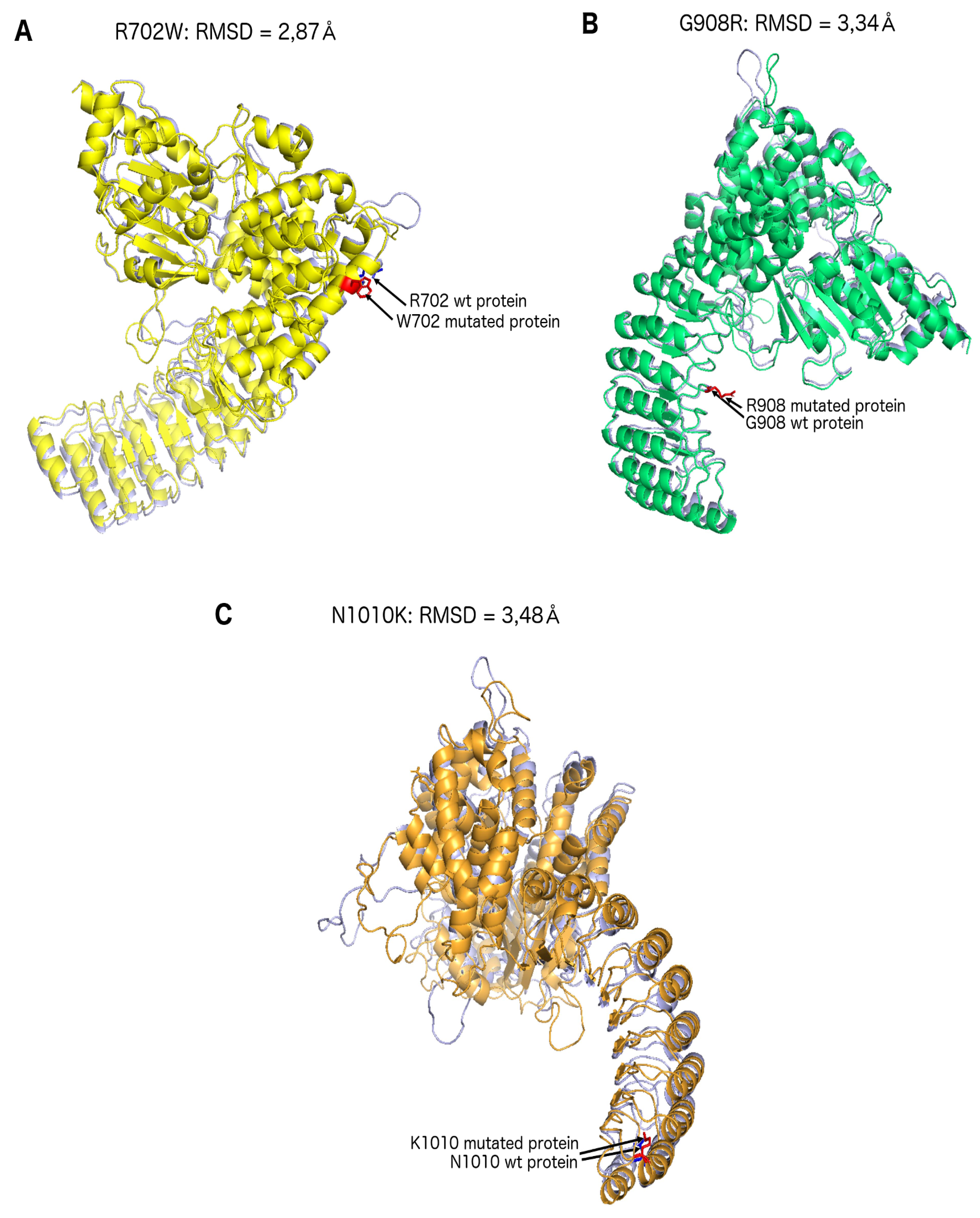
2. Results
3. Discussion
4. Materials and Methods
4.1. Subjects
4.2. Exome Sequencing Analysis and Computer Analyses
4.3. Sanger Sequencing Confirmation of NOD2 Variants
4.4. Structural Predictions: NOD2
4.5. In Silico Predictions and Annotations
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| CD | Crohn’s disease |
| DOAJ | Directory of open access journals |
| GWAS | Genome wide association study |
| IBD | Inflammatory Bowel Disease |
| LD | Linear Dichroism |
| LRR | Leucine-Rich Repeat |
| MAF | Minor Allele Frequency |
| MDPI | Multidisciplinary Digital Publishing Institute |
| NLR | NOD-like receptor |
| PCR | Polymerase Chain Reaction |
| RMSD | Root-Mean-Square-Deviation |
| TLA | Three letter acronym |
| WES | Whole Exome Sequencing |
References
- Qin, X. Etiology of inflammatory bowel disease: A unified hypothesis. World J. Gastroenterol. 2012, 18, 1708–1722. [Google Scholar] [CrossRef] [PubMed]
- Hugot, J.P.; Chamaillard, M.; Zouali, H.; Lesage, S.; Cézard, J.P.; Belaiche, J.; Almer, S.; Tysk, C.; O’Morain, C.A.; Gassull, M.; et al. Association of NOD2 leucine-rich repeat variants with susceptibility to Crohn’s disease. Nature 2001, 411, 599–603. [Google Scholar] [CrossRef] [PubMed]
- Lesage, S.; Zouali, H.; Cézard, J.-P.; Colombel, J.-F.; Belaiche, J.; Almer, S.; Tysk, C.; O’Morain, C.; Gassull, M.; Binder, V.; et al. CARD15/NOD2 Mutational Analysis and Genotype-Phenotype Correlation in 612 Patients with Inflammatory Bowel Disease. Am. J. Hum. Genet. 2002, 70, 845–857. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rivas, M.A.; Beaudoin, M.; Gardet, A.; Stevens, C.; Sharma, Y.; Zhang, C.K.; Boucher, G.; Ripke, S.; Ellinghaus, D.; Burtt, N.; et al. Deep resequencing of GWAS loci identifies independent rare variants associated with inflammatory bowel disease. Nat. Genet. 2011, 43, 1066–1073. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Franke, A.; McGovern, D.P.B.; Barrett, J.C.; Wang, K.; Radford-Smith, G.L.; Ahmad, T.; Lees, C.W.; Balschun, T.; Lee, J.; Roberts, R.; et al. Genome-wide meta-analysis increases to 71 the number of confirmed Crohn’s disease susceptibility loci. Nat. Genet. 2010, 43, 1118–1125. [Google Scholar] [CrossRef] [PubMed]
- Gower-Rousseau, C.; Salomez, J.L.; Dupas, J.L.; Marti, R.; Nuttens, M.C.; Votte, A.; Lemahieu, M.; Lemaire, B.; Colombel, J.F.; Cortot, A. Incidence of inflammatory bowel disease in northern France (1988–1990). Gut 1994, 35, 1433–1438. [Google Scholar] [CrossRef] [PubMed]
- Gower-Rousseau, C.; Vasseur, F.; Fumery, M.; Savoye, G.; Salleron, J.; Dauchet, L.; Turck, D.; Cortot, A.; Peyrin-Biroulet, L.; Colombel, J.-F. Epidemiology of inflammatory bowel diseases: New insights from a French population-based registry (EPIMAD). Dig. Liver Dis. 2013, 45, 89–94. [Google Scholar] [CrossRef] [PubMed]
- Vasseur, F.; Sendid, B.; Jouault, T.; Standaert-Vitse, A.; Dubuquoy, L.; Francois, N.; Gower-Rousseau, C.; Desreumaux, P.; Broly, F.; Vermeire, S.; et al. Variants of NOD1 and NOD2 genes display opposite associations with familial risk of Crohn’s disease and anti-saccharomyces cerevisiae antibody levels. Inflamm. Bowel Dis. 2012, 18, 430–438. [Google Scholar] [CrossRef]
- Ogura, Y.; Bonen, D.K.; Inohara, N.; Nicolae, D.L.; Chen, F.F.; Ramos, R.; Britton, H.; Moran, T.; Karaliuskas, R.; Duerr, R.H.; et al. A frameshift mutation in NOD2 associated with susceptibility to Crohn’s disease. Nature 2001, 411, 603–606. [Google Scholar] [CrossRef]
- Philpott, D.J.; Sorbara, M.T.; Robertson, S.J.; Croitoru, K.; Girardin, S.E. NOD proteins: Regulators of inflammation in health and disease. Nat. Rev. Immunol. 2014, 14, 9–23. [Google Scholar] [CrossRef]
- Inohara; Chamaillard; McDonald, C.; Nuñez, G. NOD-LRR proteins: Role in host-microbial interactions and inflammatory disease. Annu. Rev. Biochem. 2005, 74, 355–383. [Google Scholar] [CrossRef] [PubMed]
- Inohara, N.; Ogura, Y.; Fontalba, A.; Gutierrez, O.; Pons, F.; Crespo, J.; Fukase, K.; Inamura, S.; Kusumoto, S.; Hashimoto, M.; et al. Host recognition of bacterial muramyl dipeptide mediated through NOD2. Implications for Crohn’s disease. J. Biol. Chem. 2003, 278, 5509–5512. [Google Scholar] [CrossRef] [PubMed]
- Cho, J.H. The genetics and immunopathogenesis of inflammatory bowel disease. Nat. Rev. Immunol. 2008, 8, 458–466. [Google Scholar] [CrossRef] [PubMed]
- Gavel, Y.; von Heijne, G. Sequence differences between glycosylated and non-glycosylated Asn-X-Thr/Ser acceptor sites: Implications for protein engineering. Protein Eng. 1990, 3, 433–442. [Google Scholar] [CrossRef]
- Maekawa, S.; Ohto, U.; Shibata, T.; Miyake, K.; Shimizu, T. Crystal structure of NOD2 and its implications in human disease. Nat. Commun. 2016, 7, 11813. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barnich, N.; Hisamatsu, T.; Aguirre, J.E.; Xavier, R.; Reinecker, H.-C.; Podolsky, D.K. GRIM-19 interacts with nucleotide oligomerization domain 2 and serves as downstream effector of anti-bacterial function in intestinal epithelial cells. J. Biol. Chem. 2005, 280, 19021–19026. [Google Scholar] [CrossRef] [PubMed]
- Girardelli, M.; Vuch, J.; Tommasini, A.; Crovella, S.; Bianco, A.M. Novel missense mutation in the NOD2 gene in a patient with early onset ulcerative colitis: Causal or chance association? Int. J. Mol. Sci. 2014, 15, 3834–3841. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Salem, M.; Boyd, M.; Bornholdt, J.; Li, Y.; Coskun, M.; Seidelin, J.B.; Sandelin, A.; Nielsen, O.H. Relation between NOD2 genotype and changes in innate signaling in Crohn’s disease on mRNA and miRNA levels. NPJ Genom. Med. 2017, 2, 3. [Google Scholar] [CrossRef] [PubMed]
- Molinié, F.; Gower-Rousseau, C.; Yzet, T.; Merle, V.; Grandbastien, B.; Marti, R.; Lerebours, E.; Dupas, J.L.; Colombel, J.F.; Salomez, J.L.; et al. Opposite evolution in incidence of Crohn’s disease and ulcerative colitis in Northern France (1988–1999). Gut 2004, 53, 843–848. [Google Scholar] [CrossRef] [PubMed]
- Maiti, R.; Van Domselaar, G.H.; Zhang, H.; Wishart, D.S. SuperPose: A simple server for sophisticated structural superposition. Nucleic Acids Res. 2004, 117, 761–771. [Google Scholar] [CrossRef] [PubMed]
- Kircher, M.; Witten, D.M.; Jain, P.; O’Roak, B.J.; Cooper, G.M.; Shendure, J. A general framework for estimating the relative pathogenicity of human genetic variants. Nat. Genet. 2014, 46, 310–315. [Google Scholar] [CrossRef] [PubMed]
- Ng, P.C.; Henikoff, S. Predicting deleterious amino acid substitutions. Genome Res. 2001, 11, 863–874. [Google Scholar] [CrossRef] [PubMed]
- Adzhubei, I.A.; Schmidt, S.; Peshkin, L.; Ramensky, V.E.; Gerasimova, A.; Bork, P.; Kondrashov, A.S.; Sunyaev, S.R. A method and server for predicting damaging missense mutations. Nat. Methods 2010, 7, 248–249. [Google Scholar] [CrossRef] [PubMed] [Green Version]




{kind=link}
{kind=link}
{kind=link}
| R702W | G908R | L1007fs | N1010K | |
|---|---|---|---|---|
| ExAC MAF in Non-Finnish CEU | 0.04307 | 0.01187 | 0.02319 | 0 |
| GnomAD MAF | 0.02355 | 0.007589 | 0.01520 | 0 |
| Kaviar MAF | 0.2409 | 0.009595 | 0.01279 | 0 |
| CADD Phred | 24.6 | 29.8 | 35.0 | 22.6 |
| SIFT | 0.01 | 0.01 | N/A | 0 |
| PolyPhen2 | 0.72 | 0.986 | N/A | 0.996 |
| Grantham Score | 101 | 125 | N/A | 94 |
| Modelisation gap | 2.87 Å | 3.34 Å | N/A | 3.48 Å |
| Patient Identification | Age at Diagnosis (y) | Location at Diagnosis | Behaviour at Diagnosis |
|---|---|---|---|
| II:2 N1010K | 30/A2 | L3 | B2 |
| II:6 N1010K | 24/A2 | L1 | B3 |
| III:2 1007fs + N1010K | 8/A1 | L3 | B1 |
| III:3 1007fs + N1010K | 15/A1 | L3 | B1 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Frade-Proud’Hon-Clerc, S.; Smol, T.; Frenois, F.; Sand, O.; Vaillant, E.; Dhennin, V.; Bonnefond, A.; Froguel, P.; Fumery, M.; Guillon-Dellac, N.; et al. A Novel Rare Missense Variation of the NOD2 Gene: Evidences of Implication in Crohn’s Disease. Int. J. Mol. Sci. 2019, 20, 835. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20040835
Frade-Proud’Hon-Clerc S, Smol T, Frenois F, Sand O, Vaillant E, Dhennin V, Bonnefond A, Froguel P, Fumery M, Guillon-Dellac N, et al. A Novel Rare Missense Variation of the NOD2 Gene: Evidences of Implication in Crohn’s Disease. International Journal of Molecular Sciences. 2019; 20(4):835. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20040835
Chicago/Turabian StyleFrade-Proud’Hon-Clerc, Sara, Thomas Smol, Frédéric Frenois, Olivier Sand, Emmanuel Vaillant, Véronique Dhennin, Amélie Bonnefond, Philippe Froguel, Mathurin Fumery, Nathalie Guillon-Dellac, and et al. 2019. "A Novel Rare Missense Variation of the NOD2 Gene: Evidences of Implication in Crohn’s Disease" International Journal of Molecular Sciences 20, no. 4: 835. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20040835