Current Structural Knowledge on the CNNM Family of Magnesium Transport Mediators

 ,
,
Abstract
:1. Introduction
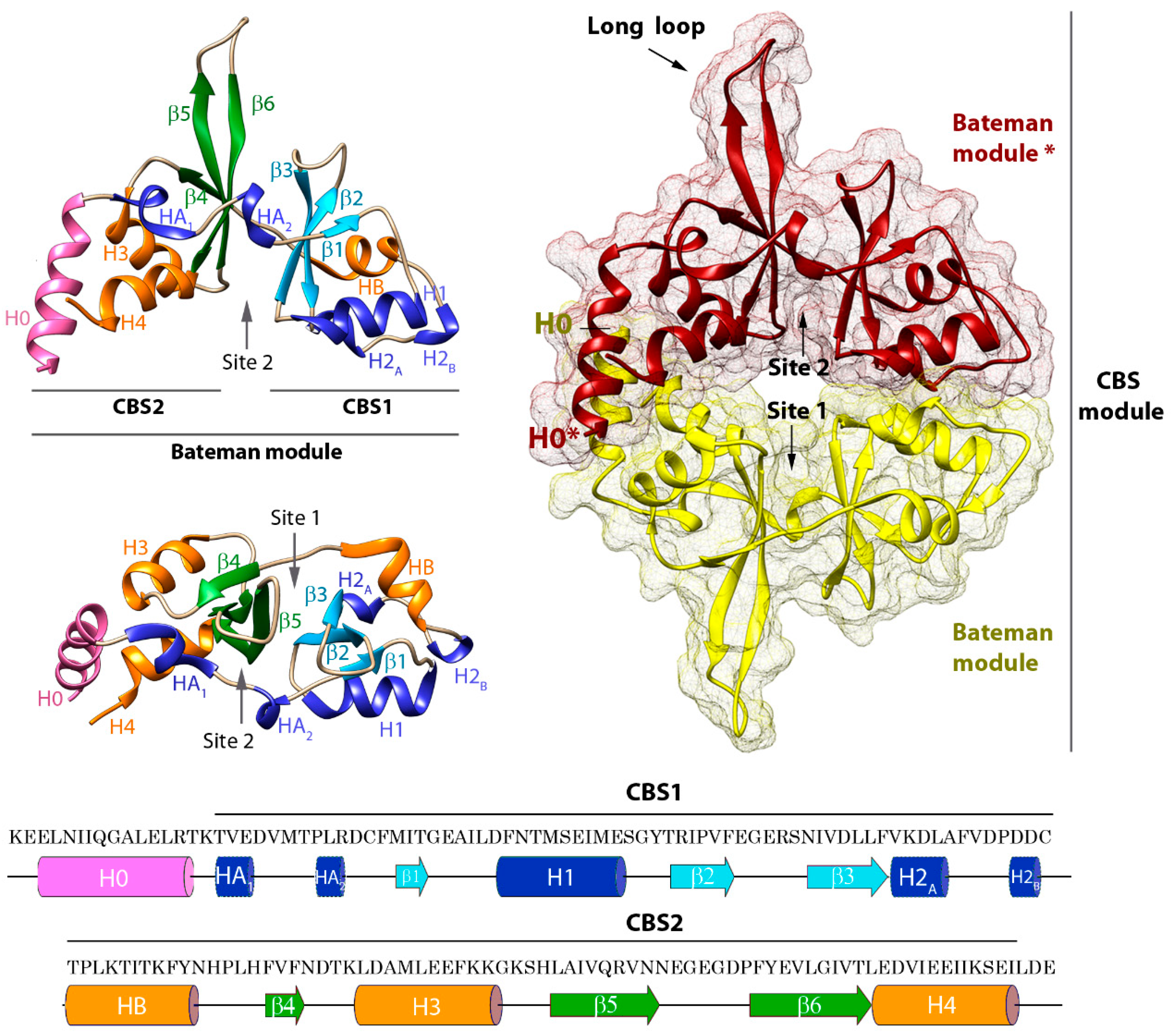
2. Structure of CNNMs
3. CNNM Ligands and Interacting Partners
3.1. Modulators of the CNNMs’ Cellular Location
3.2. Small Molecules That Modulate CNNMs’ Activity
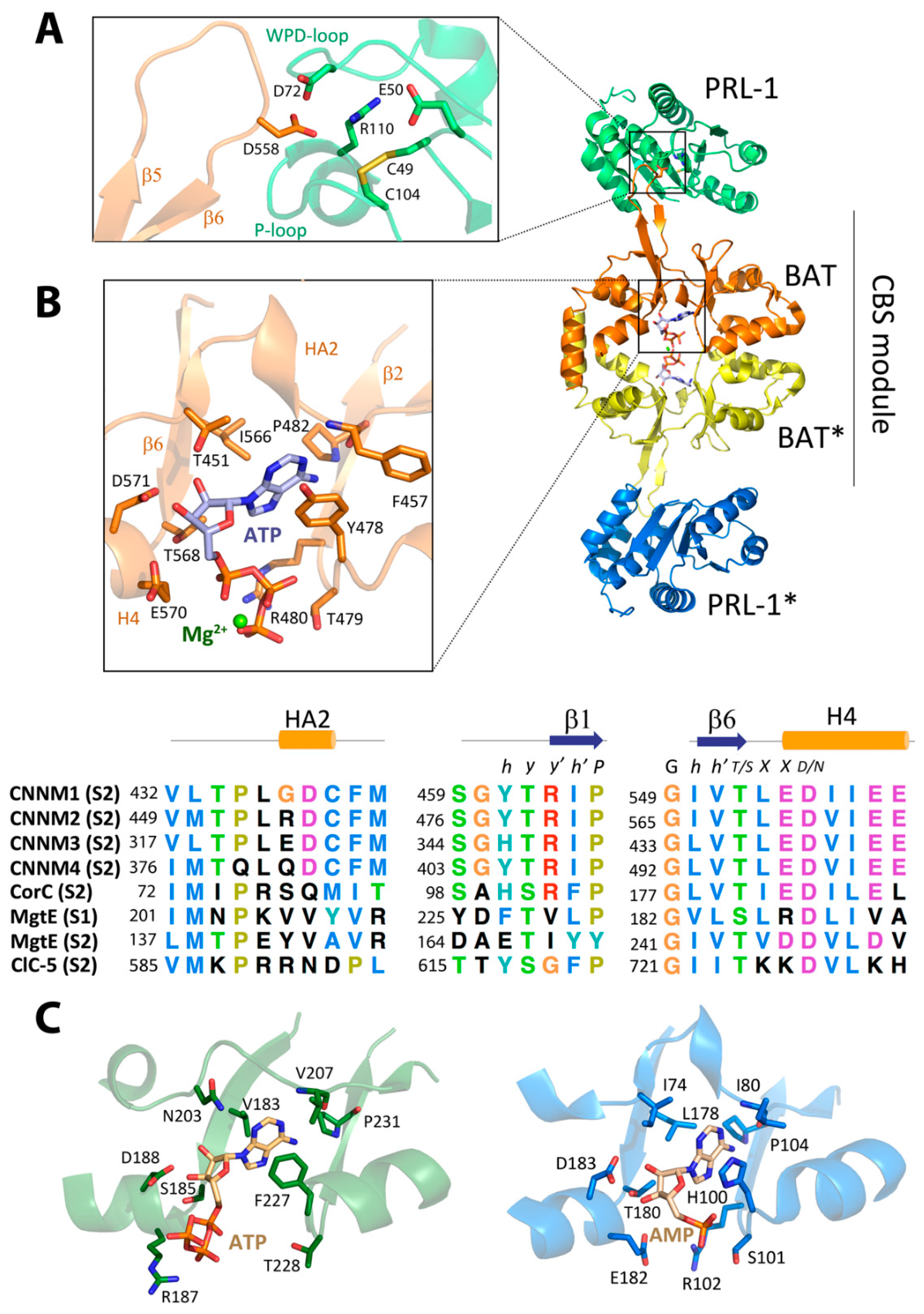
3.3. ATP Binding Site of CNNMs Differs from That of Its Homologs
3.4. Large Modulators of CNNMs’ Activity
4. Ligand-Induced Conformational Changes
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Maguire, M.E.; Cowan, J.A. Magnesium chemistry and biochemistry. Biometals 2002, 15, 203–210. [Google Scholar] [CrossRef] [PubMed]
- Gwanyanya, A.; Amuzescu, B.; Zakharov, S.I.; Macianskiene, R.; Sipido, K.R.; Bolotina, V.M.; Vereecke, J.; Mubagwa, K. Magnesium-inhibited, TRPM6/7-like channel in cardiac myocytes: Permeation of divalent cations and pH-mediated regulation. J. Physiol. 2004, 559, 761–776. [Google Scholar] [CrossRef] [PubMed]
- Saris, N.E.; Mervaala, E.; Karppanen, H.; Khawaja, J.A.; Lewenstam, A. Magnesium. An update on physiological, clinical and analytical aspects. Clin. Chim. Acta 2000, 294, 1–26. [Google Scholar] [CrossRef]
- Volpe, S.L. Magnesium in disease prevention and overall health. Adv. Nutr. 2013, 4, 378S–383S. [Google Scholar] [CrossRef] [PubMed]
- Iseri, L.T. Magnesium in coronary artery disease. Drugs 1984, 28, 151–160. [Google Scholar] [CrossRef] [PubMed]
- Li, F.Y.; Chaigne-Delalande, B.; Kanellopoulou, C.; Davis, J.C.; Matthews, H.F.; Douek, D.C.; Cohen, J.I.; Uzel, G.; Su, H.C.; Lenardo, M.J. Second messenger role for Mg2+ revealed by human T-cell immunodeficiency. Nature 2011, 475, 471–476. [Google Scholar] [CrossRef] [PubMed]
- Alfrey, A.C.; Miller, N.L. Bone Magnesium Pools in Uremia. J. Clin. Investig. 1973, 52, 3019–3027. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Baaij, J.H.; Hoenderop, J.G.; Bindels, R.J. Magnesium in man: Implications for health and disease. Physiol. Rev. 2015, 95, 1–46. [Google Scholar] [CrossRef] [PubMed]
- Ferré, S.; Hoenderop, J.G.; Bindels, R.J. Insight into renal Mg2+ transporters. Curr. Opin. Nephrol. Hypertens. 2011, 20, 169–176. [Google Scholar] [CrossRef] [PubMed]
- Giménez-Mascarell, P.; Schirrmacher, C.E.; Martínez-Cruz, L.A.; Müller, D. Novel Aspects of Renal Magnesium Homeostasis. Front. Pediatr. 2018, 6, 77. [Google Scholar] [CrossRef] [PubMed]
- Schäffers, O.J.M.; Hoenderop, J.G.J.; Bindels, R.J.M.; de Baaij, J.H.F. The rise and fall of novel renal magnesium transporters. Am. J. Physiol. Ren. Physiol. 2018, 314, 1027–1033. [Google Scholar] [CrossRef] [PubMed]
- Walder, R.Y.; Landau, D.; Meyer, P.; Shalev, H.; Tsolia, M.; Borochowitz, Z.; Boettger, M.B.; Beck, G.E.; Englehardt, R.K.; Carmi, R.; et al. Mutation of TRPM6 causes familial hypomagnesemia with secondary hypocalcemia. Nat. Genet. 2002, 31, 171–174. [Google Scholar] [CrossRef] [PubMed]
- Rude, R.K.; Gruber, H.E. Magnesium deficiency and osteoporosis: Animal and human observations. J. Nutr. Biochem. 2004, 15, 710–716. [Google Scholar] [CrossRef] [PubMed]
- Polok, B.; Escher, P.; Ambresin, A.; Chouery, E.; Bolay, S.; Meunier, I.; Nan, F.; Hamel, C.; Munier, F.L.; Thilo, B.; et al. Mutations in CNNM4 cause recessive cone-rod dystrophy with amelogenesis imperfecta. Am. J. Hum. Genet. 2009, 84, 259–265. [Google Scholar] [CrossRef] [PubMed]
- Wolf, F.I.; Trapani, V. Magnesium and its transporters in cancer: A novel paradigm in tumour development. Clin. Sci. 2012, 123, 417–427. [Google Scholar] [CrossRef] [PubMed]
- Luder, H.U.; Gerth-Kahlert, C.; Ostertag-Benzinger, S.; Schorderet, D.F. Dental phenotype in Jalili syndrome due to a c.1312 dupC homozygous mutation in the CNNM4 gene. PLoS ONE 2013, 8, e78529. [Google Scholar] [CrossRef] [PubMed]
- Arjona, F.J.; de Baaij, J.H.; Schlingmann, K.P.; Lameris, A.L.; van Wijk, E.; Flik, G.; Regele, S.; Korenke, G.C.; Neophytou, B.; Rust, S.; et al. CNNM2 mutations cause impaired brain development and seizures in patients with hypomagnesemia. PLoS Genet. 2014, 10, e1004267. [Google Scholar] [CrossRef] [PubMed]
- Funato, Y.; Yamazaki, D.; Mizukami, S.; Du, L.; Kikuchi, K.; Miki, H. Membrane protein CNNM4-dependent Mg2+ efflux suppresses tumor progression. J. Clin. Investig. 2014, 124, 5398–5410. [Google Scholar] [CrossRef] [PubMed]
- Yamazaki, D.; Miyata, H.; Funato, Y.; Fujihara, Y.; Ikawa, M.; Miki, H. The Mg2+ transporter CNNM4 regulates sperm Ca2+ homeostasis and is essential for reproduction. J. Cell Sci. 2016, 129, 1940–1949. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.Y.; Shi, J.D.; Yang, P.; Kumar, P.G.; Li, Q.Z.; Run, Q.G.; Su, Y.C.; Scott, H.S.; Kao, K.J.; She, J.X. Molecular cloning and characterization of a novel gene family of four ancient conserved domain proteins (ACDP). Gene 2003, 306, 37–44. [Google Scholar] [CrossRef]
- Ereño-Orbea, J.; Oyenarte, I.; Martínez-Cruz, L.A. CBS domains: Ligand binding sites and conformational variability. Arch. Biochem. Biophys. 2013, 540, 70–81. [Google Scholar] [CrossRef] [PubMed]
- Stuiver, M.; Lainez, S.; Will, C.; Terryn, S.; Günzel, D.; Debaix, H.; Sommer, K.; Kopplin, K.; Thumfart, J.; Kampik, N.B.; et al. CNNM2, encoding a basolateral protein required for renal Mg2+ handling, is mutated in dominant hypomagnesemia. Am. J. Hum. Genet. 2011, 88, 333–343. [Google Scholar] [CrossRef] [PubMed]
- Yamazaki, D.; Funato, Y.; Miura, J.; Sato, S.; Toyosawa, S.; Furutani, K.; Kurachi, Y.; Omori, Y.; Furukawa, T.; Tsuda, T.; et al. Basolateral Mg2+ extrusion via CNNM4 mediates transcellular Mg2+ transport across epithelia: A mouse model. PLoS Genet. 2013, 9, e1003983. [Google Scholar] [CrossRef] [PubMed]
- Hardy, S.; Uetani, N.; Wong, N.; Kostantin, E.; Labbé, D.P.; Bégin, L.R.; Mes-Masson, A.; Miranda-Saavedra, D.; Tremblay, M.L. The protein tyrosine phosphatase PRL-2 interacts with the magnesium transporter CNNM3 to promote oncogenesis. Oncogene 2015, 34, 986–995. [Google Scholar] [CrossRef] [PubMed]
- Sponder, G.; Mastrototaro, L.; Kurth, K.; Merolle, L.; Zhang, Z.; Abdulhanan, N.; Smorodchenko, A.; Wolf, K.; Fleig, A.; Penner, R.; et al. Human CNNM2 is not a Mg2+ transporter per se. Pflugers Arch. 2016, 468, 1223–1240. [Google Scholar] [CrossRef] [PubMed]
- Kolisek, M.; Sponder, G.; Pilchova, I.; Cibulka, M.; Tatarkova, Z.; Werner, T.; Racay, P. Magnesium Extravaganza: A critical compendium of current research into cellular Mg2+ transporters other than TRPM6/7. Rev. Physiol. Biochem. Pharmacol. 2018. [Google Scholar] [CrossRef]
- De Baaij, J.H.; Stuiver, M.; Meij, I.C.; Lainez, S.; Kopplin, K.; Venselaar, H.; Müller, D.; Bindels, R.J.; Hoenderop, J.G. Membrane topology and intracellular processing of cyclin M2 (CNNM2). J. Biol. Chem. 2012, 287, 13644–13655. [Google Scholar] [CrossRef] [PubMed]
- Alderton, A.; Davies, P.; Illman, K.; Brown, D.R. Ancient conserved domain protein-1 binds copper and modifies its retention in cells. J. Neurochem. 2007, 103, 312–321. [Google Scholar] [CrossRef] [PubMed]
- Chandran, U.; Indu, S.; Kumar, A.T.; Devi, A.N.; Khan, I.; Srivastava, D.; Kumar, P.G. Expression of Cnnm1 and Its Association with Stemness, Cell Cycle, and Differentiation in Spermatogenic Cells in Mouse Testis. Biol. Reprod. 2016, 95, 7. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.Y.; Yang, P.; Shi, J.D.; Purohit, S.; Guo, D.; An, H.; Gu, J.G.; Ling, J.; Dong, Z.; She, J.X. Molecular cloning and characterization of the mouse Acdp gene family. BMC Genom. 2004, 5, 7. [Google Scholar] [CrossRef] [PubMed]
- Chandran, U.; Laloraya, M.; Pradeep Kumar, G. Identification of Testis-Expressed Cell Cycle Regulating Proteins with Special Reference to Meiosis. J. Endocrinol. Reprod. 2007, 11, 45–48. [Google Scholar]
- Voets, T.; Janssens, A.; Droogmans, G.; Nilius, B. Outer pore architecture of a Ca2+-selective TRP channel. J. Biol. Chem. 2004, 279, 15223–15230. [Google Scholar] [CrossRef] [PubMed]
- Glaudemans, B.; Knoers, N.V.; Hoenderop, J.G.; Bindels, R.J. New molecular players facilitating Mg2+ reabsorption in the distal convoluted tubule. Kidney Int. 2010, 77, 17–22. [Google Scholar] [CrossRef] [PubMed]
- Goytain, A.; Quamme, G.A. Functional characterization of ACDP2 (ancient conserved domain protein) a divalent metal transporter. Physiol. Genom. 2005, 22, 382–389. [Google Scholar] [CrossRef] [PubMed]
- Quamme, G.A. Molecular identification of ancient and modern mammalian magnesium transporters. Am. J. Physiol. Cell Physiol. 2010, 298, 407–429. [Google Scholar] [CrossRef] [PubMed]
- Won, J.; Kim, J.H.; Oh, S.B. Molecular expression of Mg2+ regulator TRPM7 and CNNM4 in rat odontoblasts. Arch. Oral Biol. 2018, 96, 182–188. [Google Scholar] [CrossRef] [PubMed]
- Quamme, G.; Biber, J.; Murer, H. Sodium-phosphate cotransport in OK cells: Inhibition by PTH and “adaptation” to low phosphate. Am. J. Physiol. 1989, 257, 967–973. [Google Scholar] [CrossRef] [PubMed]
- Ohi, K.; Hashimoto, R.; Ikeda, M.; Yamamori, H.; Yasuda, Y.; Fujimoto, M.; Umeda-Yano, S.; Fukunaga, M.; Fujino, H.; Watanabe, Y.; et al. Glutamate Networks Implicate Cognitive Impairments in Schizophrenia: Genome-Wide Association Studies of 52 Cognitive Phenotypes. Schizophr Bull. 2015, 41, 909–918. [Google Scholar] [CrossRef] [PubMed]
- Funato, Y.; Yamazaki, D.; Miki, H. Renal function of cyclin M2 Mg2+ transporter maintains blood pressure. J. Hypertens. 2017, 35, 585–592. [Google Scholar] [CrossRef] [PubMed]
- Kieboom, B.C.T.; Ligthart, S.; Dehghan, A.; Kurstjens, S.; de Baaij, J.H.F.; Franco, O.H.; Hofman, A.; Zietse, R.; Stricker, B.H.; Hoorn, E.J. Serum magnesium and the risk of prediabetes: A population-based cohort study. Diabetologia 2017, 60, 843–853. [Google Scholar] [CrossRef] [PubMed]
- Lv, W.Q.; Zhang, X.; Zhang, Q.; He, J.Y.; Liu, H.M.; Xia, X.; Fan, K.; Zhao, Q.; Shi, X.Z.; Zhang, W.D.; et al. Novel common variants associated with body mass index and coronary artery disease detected using a pleiotropic cFDR method. J. Mol. Cell Cardiol. 2017, 112, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Hirata, Y.; Funato, Y.; Takano, Y.; Miki, H. Mg2+-dependent interactions of ATP with the cystathionine-β-synthase (CBS) domains of a magnesium transporter. J. Biol. Chem. 2014, 289, 14731–14739. [Google Scholar] [CrossRef] [PubMed]
- Gulerez, I.; Funato, Y.; Wu, H.; Yang, M.; Kozlov, G.; Miki, H.; Gehring, K. Phosphocysteine in the PRL-CNNM pathway mediates magnesium homeostasis. EMBO Rep. 2016, 12, 1890–1900. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Kozlov, G.; Li, X.; Wu, H.; Gulerez, I.; Gehring, K. PRL3 phosphatase active site is required for binding the putative magnesium transporter CNNM3. Sci. Rep. 2017, 7, 48. [Google Scholar] [CrossRef] [PubMed]
- Giménez-Mascarell, P.; Oyenarte, I.; Hardy, S.; Breiderhoff, T.; Stuiver, M.; Kostantin, E.; Diercks, T.; Pey, A.L.; Ereño-Orbea, J.; Martínez-Chantar, M.L.; et al. Structural Basis of the Oncogenic Interaction of Phosphatase PRL-1 with the Magnesium Transporter CNNM2. J. Biol. Chem. 2017, 292, 786–801. [Google Scholar] [CrossRef] [PubMed]
- Kostantin, E.; Hardy, S.; Valinsky, W.C.; Kompatscher, A.; de Baaij, J.H.; Zolotarov, Y.; Landry, M.; Uetani, N.; Martínez-Cruz, L.A.; Hoenderop, J.G.; et al. Inhibition of PRL-2·CNNM3 Protein Complex Formation Decreases Breast Cancer Proliferation and Tumor Growth. J. Biol. Chem. 2016, 291, 10716–10725. [Google Scholar] [CrossRef] [PubMed]
- Parry, D.A.; Mighell, A.J.; El-Sayed, W.; Shore, R.C.; Jalili, I.K.; Dollfus, H.; Bloch-Zupan, A.; Carlos, R.; Carr, I.M.; Downey, L.M.; et al. Mutations in CNNM4 cause Jalili syndrome, consisting of autosomal-recessive cone-rod dystrophy and amelogenesis imperfecta. Am. J. Hum. Genet. 2009, 84, 266–273. [Google Scholar] [CrossRef] [PubMed]
- Cherkaoui Jaouad, I.; Lyahyai, J.; Guaoua, S.; El Alloussi, M.; Zrhidri, A.; Doubaj, Y.; Boulanouar, A.; Sefiani, A. Novel splice site mutation in CNNM4 gene in a family with Jalili syndrome. Eur. J. Med. Genet. 2017, 60, 239–244. [Google Scholar] [CrossRef] [PubMed]
- Coppieters, F.; Van Schil, K.; Bauwens, M.; Verdin, H.; De Jaegher, A.; Syx, D.; Sante, T.; Lefever, S.; Abdelmoula, N.B.; Depasse, F.; et al. Identity-by descent -guided mutation analysis and exome sequencing in consanguineous families reveals unusual clinical and molecular findings in retinal dystrophy. Genet. Med. 2014, 16, 671–680. [Google Scholar] [CrossRef] [PubMed]
- Rahimi-Aliabadi, S.; Daftarian, N.; Ahmadieh, H.; Emamalizadeh, B.; Jamshidi, J.; Tafakhori, A.; Ghaedi, H.; Noroozi, R.; Taghav, S.; et al. A novel mutation and variable phenotypic expression in a large consanguineous pedigree with Jalili. Eye 2016, 30, 1424–1432. [Google Scholar] [CrossRef] [PubMed]
- Lopez Torres, L.T.; Schorderet, D.; Valmaggia, C.; Todorova, M. A novel mutation in CNNM4 (G492C) associated with Jalili Syndrome. Acta Ophthalmol. 2015, 93, S255. [Google Scholar] [CrossRef]
- Abu-Safieh, L.; Alrashed, M.; Anazi, S.; Alkuraya, H.; Khan, A.O.; Al-Owain, M.; Al-Zahrani, J.; Al-Abdi, L.; Hashem, M.; Al-Tarimi, S.; et al. Autozygomeguided exome sequencing in retinal dystrophy patients reveals pathogenetic mutations and novel candidate disease genes. Genome Res. 2013, 23, 236–247. [Google Scholar] [CrossRef] [PubMed]
- Prasad, K.; Geoffroy, V.; Vicaire, S.; Jost, B.; Dumas, M.; Le Gras, S.; Switala, M.; Gasse, B.; Laugel-Haushalter, V.; Paschaki, M.; et al. A targeted next-generation sequencing assay for the molecular diagnosis of genetic disorders with orodental involvement. J. Med. Genet. 2016, 53, 98–110. [Google Scholar] [CrossRef] [PubMed]
- Doucette, L.; Green, J.; Black, C.; Schwartzentruber, J.; Johnson, G.J.; Galutira, D.; Young, T.L. Molecular genetics of achromatopsia in Newfoundland reveal genetic heterogeneity, founder effects and the first cases of Jalili syndrome in North America. Ophthalmic Genet. 2013, 34, 119–129. [Google Scholar] [CrossRef] [PubMed]
- Topçu, V.; Alp, M.Y.; Alp, C.K.; Bakır, A.; Geylan, D.; Yılmazoğlu, M.Ö. A new familial case of Jalili syndrome caused by a novel mutation in CNNM4. Ophthalmic Genet. 2016, 12, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Maia, C.M.F.; Machado, R.A.; Gil-da-Silva-Lopes, V.L.; Lustosa-Mendes, E.; Rim, P.H.H.; Dias, V.O.; Martelli, D.R.B.; Nasser, L.S.; Coletta, R.D.; Martelli-Júnior, H. Report of two unrelated families with Jalili syndrome and a novel nonsense heterozygous mutation in CNNM4 gene. Eur. J. Med. Genet. 2018, 61, 384–387. [Google Scholar] [CrossRef] [PubMed]
- Armitano, J.; Redder, P.; Guimarães, V.A.; Linder, P. An Essential Factor for High Mg2+ Tolerance of Staphylococcus aureus. Front. Microbiol. 2016, 7, 1888. [Google Scholar] [CrossRef] [PubMed]
- Corral-Rodríguez, M.A.; Stuiver, M.; Abascal-Palacios, G.; Diercks, T.; Oyenarte, I.; Ereño-Orbea, J.; Ibáñez de Opakua, A.; Blanco, F.J.; Encinar, J.A.; Spiwok, V.; et al. Nucleotide binding triggers a conformational change of the CBS module of the magnesium transporter CNNM2 from a twisted towards a flat structure. Biochem. J. 2014, 464, 23–34. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.S.; Kozlov, G.; Fakih, R.; Funato, Y.; Miki, H.; Gehring, K. The cyclic nucleotide-binding homology domain of the integral membrane protein CNNM mediates dimerization and is required for Mg2+ efflux activity. J. Biol. Chem. 2018, 293, 19998–20007. [Google Scholar] [CrossRef] [PubMed]
- Kushwaha, H.R.; Singh, A.K.; Sopory, S.K.; Singla-Pareek, S.L.; Pareek, A. Genome wide expression analysis of CBS domain containing proteins in Arabidopsis thaliana (L.) Heynh and Oryza sativa L. reveals their developmental and stress regulation. BMC Genom. 2009, 10, 200. [Google Scholar] [CrossRef] [PubMed]
- Sinharoy, S.; Liu, C.; Breakspear, A.; Guan, D.; Shailes, S.; Nakashima, J.; Zhang, S.; Wen, J.; Torres-Jerez, I.; Oldroyd, G.; et al. A Medicago truncatula Cystathionine Beta Synthase like domain-containing protein is required for rhizobial infection and symbiotic nitrogen fixation. Plant Physiol. 2016, 170, 2204–2217. [Google Scholar] [CrossRef] [PubMed]
- Baykov, A.A.; Tuominen, H.K.; Lahti, R. The CBS domain: A protein module with an emerging prominent role in regulation. ACS Chem. Biol. 2011, 6, 1156–1163. [Google Scholar] [CrossRef] [PubMed]
- Bateman, A. The structure of a domain common to archaebacteria and the homocystinuria disease protein. Tresarnds Biochem. Sci. 1997, 22, 12–13. [Google Scholar] [CrossRef]
- Mahmood, N.A.B.N.; Biemans-Oldehinkel, E.; Poolman, B. Engineering of Ion Sensing by the Cystathionine β-Synthase Module of the ABC Transporter OpuA. J. Biol. Chem. 2009, 284, 21. [Google Scholar] [CrossRef] [PubMed]
- Zeng, Q.; Hong, W.; Tan, Y.H. Mouse PRL-2 and PRL-3, two potentially prenylated protein tyrosine phosphatases homologous to PRL-1. Biochem. Biophys. Res. Commun. 1998, 244, 421–427. [Google Scholar] [CrossRef] [PubMed]
- Lolicato, M.; Nardini, M.; Gazzarrini, S.; Möller, S.; Bertinetti, D.; Herberg, F.W.; Bolognesi, M.; Martin, H.; Fasolini, M.; Bertrand, J.A.; et al. Tetramerization dynamics of C-terminal domain underlies isoform-specific cAMP gating in hyperpolarization-activated cyclic nucleotide-gated channels. J. Biol. Chem. 2011, 286, 44811–44820. [Google Scholar] [CrossRef] [PubMed]
- Pessoa, J.; Fonseca, F.; Furini, S.; Morais-Cabral, J.H. Determinants of ligand selectivity in a cyclic nucleotide-regulated potassium channel. J. Gen. Physiol. 2014, 144, 41–54. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brelidze, T.I.; Carlson, A.E.; Sankaran, B.; Zagotta, W.N. Structure of the carboxy-terminal region of a KCNH channel. Nature 2012, 481, 530–533. [Google Scholar] [CrossRef] [PubMed]
- Feng, N.; Qi, C.; Hou, Y.J.; Zhang, Y.; Wang, D.C.; Li, D.F. The C2′- and C3′-endo equilibrium for AMP molecules bound in the cystathionine-beta-synthase domain. Biochem. Biophys. Res. Commun. 2018, 497, 646–651. [Google Scholar] [CrossRef] [PubMed]
- Bonifacino, J.S. Adaptor proteins involved in polarized sorting. J. Cell Biol. 2014, 204, 7–17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- González, A.; Rodriguez-Boulan, E.; Clathrin, A.P.B. Key roles in basolateral trafficking through trans-endosomal routes. FEBS Lett. 2009, 583, 3784–3795. [Google Scholar]
- Traub, L.M.; Bonifacino, J.S. Cargo recognition in clathrin-mediated endocytosis. Cold Spring Harb. Perspect. Biol. 2013, 5, a016790. [Google Scholar] [CrossRef] [PubMed]
- Scott, J.W.; Hawley, S.A.; Green, K.A.; Anis, M.; Stewart, G.; Scullion, G.A.; Norman, D.G.; Hardie, D.G. CBS domains form energy sensing modules whose binding of adenosine ligands is disrupted by disease mutations. J. Clin. Investig. 2004, 113, 274–284. [Google Scholar] [CrossRef] [PubMed]
- Shabb, J.B.; Corbin, J.D. Cyclic nucleotide-binding domains in proteins having diverse functions. J. Biol. Chem. 1992, 267, 5723–5726. [Google Scholar] [PubMed]
- Tomita, A.; Zhang, M.; Jin, F.; Zhuang, W.; Takeda, H.; Maruyama, T.; Osawa, M.; Hashimoto, K.I.; Kawasaki, H.; Ito, K.; et al. ATP-dependent modulation of MgtE in Mg2+ homeostasis. Nat. Commun. 2017, 8, 148. [Google Scholar] [CrossRef] [PubMed]
- Meyer, S.; Savaresi, S.; Forster, I.C.; Dutzler, R. Nucleotide recognition by the cytoplasmic domain of the human chloride transporter ClC-5. Nat. Struct. Mol. Biol. 2007, 14, 60–67. [Google Scholar] [CrossRef] [PubMed]
- Julien, S.G.; Dubé, N.; Hardy, S.; Tremblay, M.L. Inside the human cancer tyrosine phosphatome. Nat. Rev. Cancer 2011, 11, 35–49. [Google Scholar] [CrossRef] [PubMed]
- Hardy, S.; Wong, N.N.; Muller, W.J.; Park, M.; Tremblay, M.L. Overexpression of the protein tyrosine phosphatase PRL-2 correlates with breast tumor formation and progression. Cancer Res. 2010, 70, 8959–8967. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, A.; Mugford, J.W.; Krautzberger, A.M.; Naiman, N.; Liao, J.; McMahon, A.P. Identification of a multipotent self-renewing stromal progenitor population during mammalian kidney organogenesis. Stem Cell Rep. 2014, 3, 650–662. [Google Scholar] [CrossRef] [PubMed]
- Hattori, M.; Tanaka, Y.; Fukai, S.; Ishitani, R.; Nureki, O. Crystal structure of the MgtE Mg2+ transporter. Nature 2007, 448, 1072–1075. [Google Scholar] [CrossRef] [PubMed]
- Hattori, M.; Iwase, N.; Furuya, N.; Tanaka, Y.; Tsukazaki, T.; Ishitani, R.; Maguire, M.E.; Ito, K.; Maturana, A.; Nureki, O. Mg(2+)-dependent gating of bacterial MgtE channel underlies Mg(2+) homeostasis. EMBO J. 2009, 28, 3602–3612. [Google Scholar] [CrossRef] [PubMed]
- Sponder, G.; Svidova, S.; Schweigel, M.; Vormann, J.; Kolisek, M. Splice-variant 1 of the ancient domain protein 2 (ACDP2) complements the magnesium-deficient growth phenotype of Salmonella enterica sv. typhimurium strain MM281. Magnes Res. 2010, 23, 105–114. [Google Scholar] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Protein | Localization | References |
|---|---|---|
| CNNM1 | stomach, kidney, skeletal muscles, heart, lungs, liver, colon, spleen, small intestine, brain, testis | [11,20,24,29,43,44] |
| CNNM2 | odontoblasts, small intestine, colon, kidney, lung, spleen, testis, brain, liver, heart | [20,24,27,34,35,36,43,44,45] |
| CNNM3 | odontoblasts, skeletal muscles, kidney, brain, lung, spleen, heart, liver | [20,24,27,36,43,44,46] |
| CNNM4 | odontoblasts, colon, mature ameloblasts, sperm gastrointestinal tract | [18,23,24,27,36,43,44] |
| Protein | Mutation | Domain | Pathology | References |
|---|---|---|---|---|
| CNNM2 | R38Q, E122K | Extracellular | Hypomagnesemia | [17,22] |
| S269W, L330F, E357K | DUF21 | Hypomagnesemia | [17] | |
| T568I | Bateman module | Hypomagnesemia | [46] | |
| CNNM4 | D63E | Extracellular | Jalili Syndrome | [49] |
| S196P, S200Y, I232P, R236Q, L324P | DUF21 | Jalili Syndrome | [14,47,50] | |
| G364V, G492C, L438P, T495I, V499M | Bateman Module | Jalili Syndrome | [14,50,51,52,53] | |
| R519X | Linker Bateman-CNBH | Jalili Syndrome | [54] | |
| N594S, Q564X, R605X, T581 * | CNBH | Jalili Syndrome | [47,55,56] |
| Protein | Domain | Partner | PDB Code | Reference |
|---|---|---|---|---|
| CNNM2 | Bateman module | -/AMP/ADP/ATP/Mg2+ | 4IYS,4P1O, 4P1G, 4IY0, 4IY4 | [58] |
| Bateman module | PRL-1/ATP, Zn2+ | 5LXQ, 5MMZ | [45] | |
| CNBH | -- | 6DJ3 | [59] | |
| CNNM3 | Bateman module | PRL-2 | 5K22, 5K23, 5K24, 5K25 | [43,44] |
| Bateman module | PRL-3 | 5TSR | [44] | |
| CNBH | -- | 6DFD | [59] | |
| CNNM4 | Bateman module | -- | 4IY3 | [58] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Giménez-Mascarell, P.; González-Recio, I.; Fernández-Rodríguez, C.; Oyenarte, I.; Müller, D.; Martínez-Chantar, M.L.; Martínez-Cruz, L.A. Current Structural Knowledge on the CNNM Family of Magnesium Transport Mediators. Int. J. Mol. Sci. 2019, 20, 1135. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20051135
Giménez-Mascarell P, González-Recio I, Fernández-Rodríguez C, Oyenarte I, Müller D, Martínez-Chantar ML, Martínez-Cruz LA. Current Structural Knowledge on the CNNM Family of Magnesium Transport Mediators. International Journal of Molecular Sciences. 2019; 20(5):1135. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20051135
Chicago/Turabian StyleGiménez-Mascarell, Paula, Irene González-Recio, Cármen Fernández-Rodríguez, Iker Oyenarte, Dominik Müller, María Luz Martínez-Chantar, and Luis Alfonso Martínez-Cruz. 2019. "Current Structural Knowledge on the CNNM Family of Magnesium Transport Mediators" International Journal of Molecular Sciences 20, no. 5: 1135. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20051135