3. Discussion
In the present investigation, we wanted to elucidate which chromosomal regions and annotated genes are involved in the genesis and progression of astrocytic brain tumors. Cancer genomes suffer many structural changes [
5] and CNAs have been commonly found in glioma [
19]. However, CNAs differ in their frequency of recurrence even among the patients suffering from the same type of malady. Which specific CNAs are attributed as early events and which are responsible for progression still remains to be fully understood.
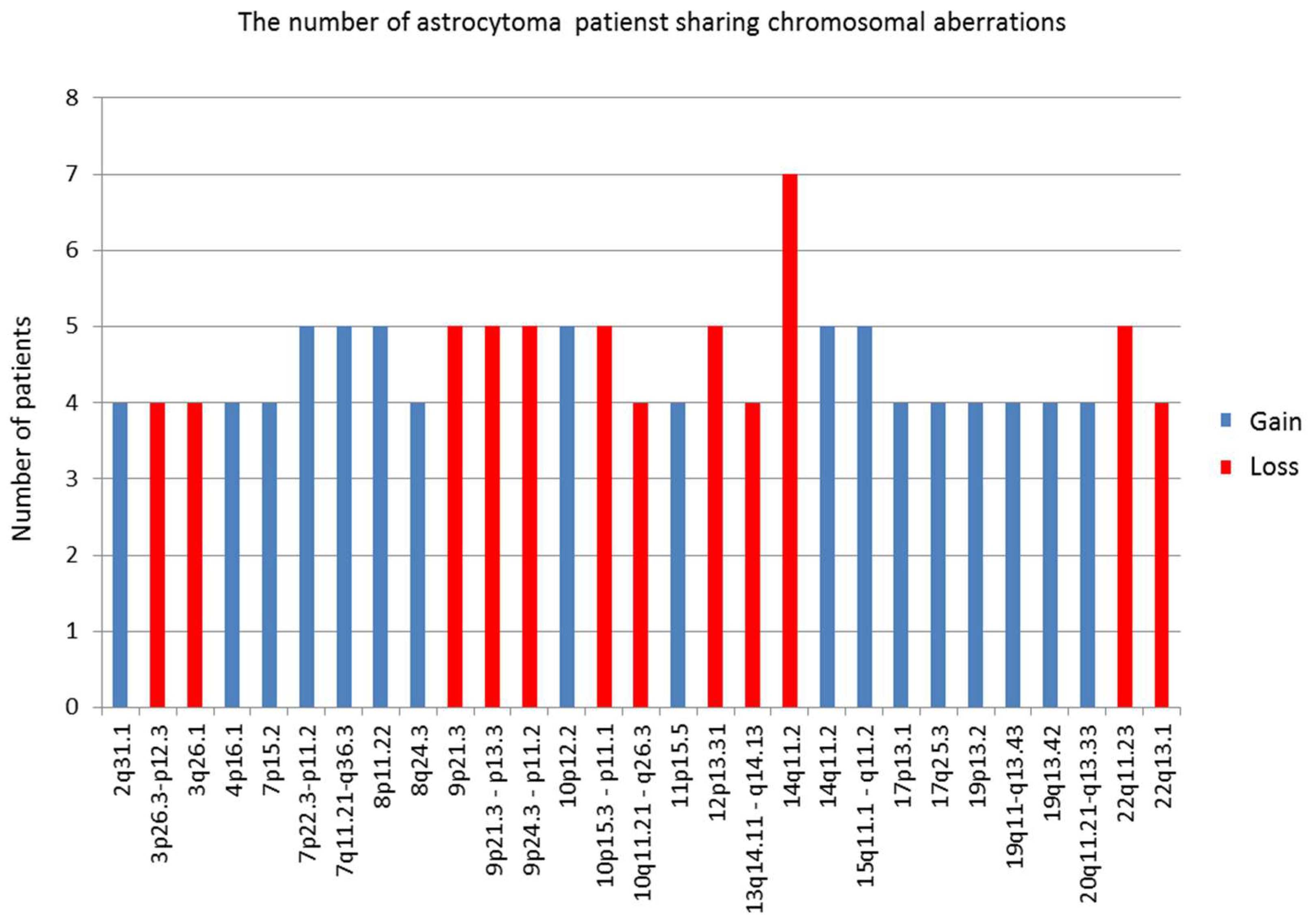
In our total sample, we found that the number of losses significantly exceeded the number of observed gains and amplifications. This finding is not unusual since it has been reported as a general pattern in cancer [
27] that losses are more frequent than amplifications. Furthermore, we have found that the mean number of CNA is much higher in malignancy grades III and IV when compared to lower grades. In addition, a great number of aberrant regions were recurring in grades III and IV.
Our study also revealed similarities and differences in the aberrations across astrocytoma grades. The CNA that were found to be shared among grade I benign pilocytic astrocytomas indicated relatively different patterns than observed in the malignant group. It has been postulated that pilocytic astrocytomas differ from other histopathological types as they are slow-growing and non-infiltrative. Although they usually exhibit a normal karyotype, ~32% display chromosomal abnormalities. Chromosomal regions that have been reported to hold abnormalities include 1p, 2p, 4q–9q and 13q and losses on 1p, 9q, 12q and 19–22 [
28,
29,
30,
31,
32]. The situation found in our study is compatible to some of the aberrations reported previously, but also differed from the literature. We found losses in pilocytic astrocytomas of which: 3q; 10q; 11p; 12p; 14q; 15q and 18p have not previously been reported, while there were fewer gains found in our study, only on 7p15.2 and 15q11.1–q11.2.
Grade II astrocytomas harbored very few recurrent aberrations, only losses on 1p36.33–p11.2 and 1q21.1 and gains on 1q21.1–q25.1. None of them recurred in grade I tumors. However, regions with recurrent losses in grade II astrocytomas were also repeatedly affected in higher grade tumors.
Malignant high grades tumors, III and IV, on the other hand, harbored numerous recurrent changes, which indicates the augmentation of aberrations as the disease progresses.
The majority of CNA that have been reported in the literature were also discovered and confirmed with our experiments [
24,
25]. However, the frequencies differed as well as their previous assignments to specific grade. Seifert et al. [
33] in their computational study revealed similarities and differences in gene expression levels between astrocytomas of all four WHO grades. The authors report that transcriptional alterations of individual signaling pathways typically increase with WHO grade of astrocytoma. The high number of copy number changes found to be increasing with the grade can also be indicative of the acquisition of genomic instability in glioblastoma, especially since deleted regions may harbor genes involved in mismatch DNA repair.
The most common amplification—the one on chromosome 7 [
8,
22], was also frequently found in our investigated sample with 77.8% of tumors displaying this type of change. Another frequent event—deletions of chromosome 10 [
22]—has been discovered in 88.9% of our patients. This finding, which is in accordance with literature and included loss of heterozygosity on chromosomal arm 10p, is commonly reported for high-grade gliomas, usually concentrated in the region 10p14–p15 [
4,
34]. Our study found losses of 10p11.1-p15.3 region in 77.8% of glioblastomas.
Brennan et al. [
8] found higher frequencies of the common amplification events reported for astrocytoma than other investigators. However, such frequencies were not confirmed in our study. Only 22.2% of our cases had gains on chromosome 12 (CDK4 and MDM2), and 28.6% on chromosome 4 (PDGFRA).
Several of our results corroborate the findings of previous studies regarding relevant genes [
8,
35]. We found
EGFR amplification to be targeted in 89% of glioblastomas and one astrocytoma grade II, with a total of 90% of cases with amplified
EGFR genes.
In order to comprehend CNA events in astrocytomas of different pathohistological types and identify alterations that are biologically and functionally significant, we used the GISTIC algorithm. We were interested in differentiating founder events and subclonal drivers from passenger mutations [
2,
36]. The software was utilized previously in numerous cancer studies, including lung [
37], colorectal carcinoma [
38] and melanoma [
39], and has facilitated the identification of new significant cancer associated targets.
By exploring a range of cutoff
q-values, we identified additional segments of significance. Thus, significant deletions affecting 14 chromosomal regions were found, out of which deletions of 17p13.2, 9p21.3, 13q12.11 and 22q12.3 remained significant even at 0.05
q-value. Of importance is that locus 9p22.1–p21.3 (p16INK4a/p14ARF/p15INK4b) has been known to encompass the
CDKN2A gene frequently deleted in gliomas [
23]. Furthermore, other regions also harbored important genes,
RB gene in the region 13q and
TP53 gene in 17p13. In accordance with our findings is the study by Yin et al. [
23], who found that the long arm of chromosome 13 was lost in nearly 50% of cases.
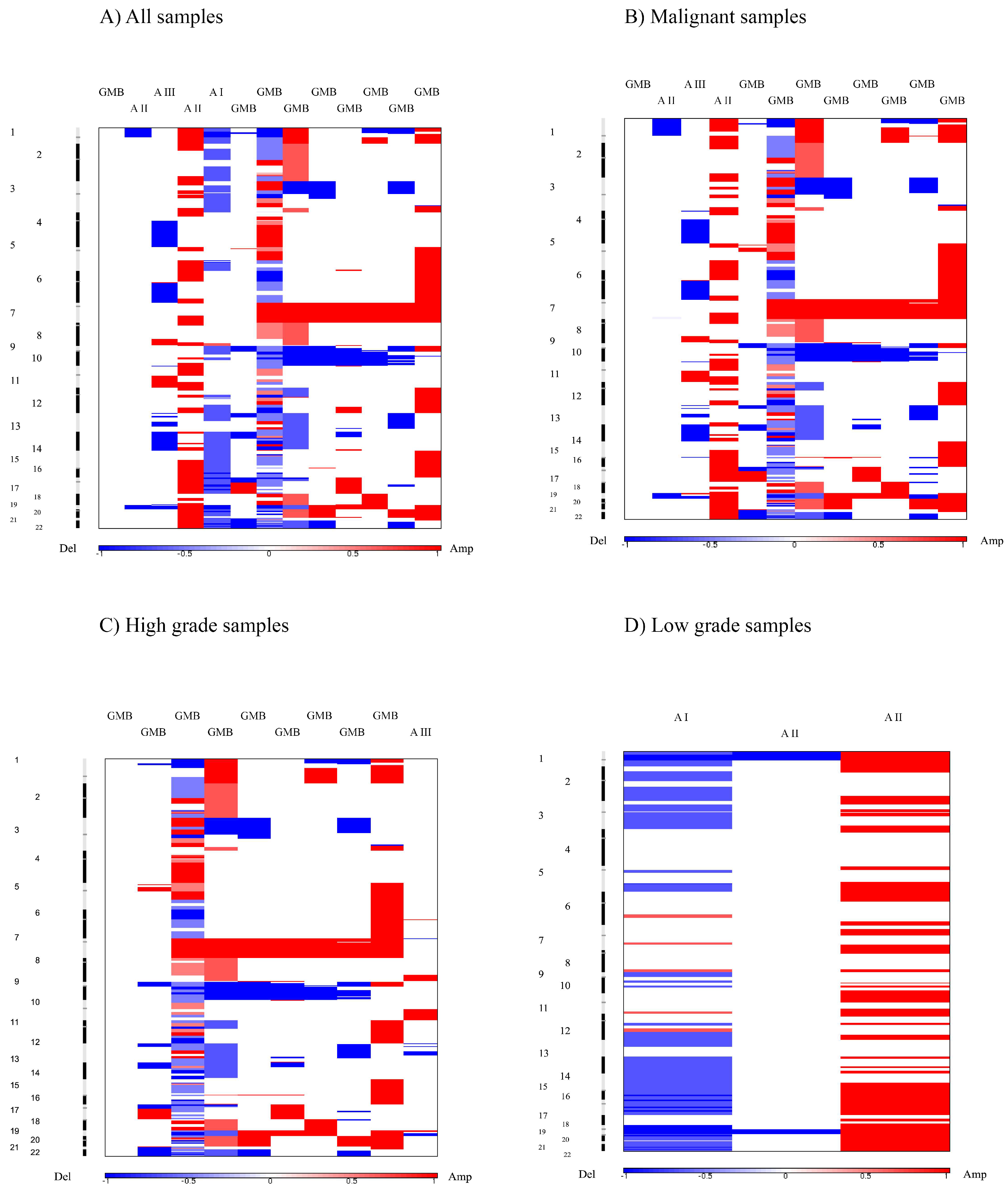
Low grade astrocytomas demonstrated the lack of both significant deletions and amplifications, suggesting a general pattern associated with these grades. Common genetic changes and tumor associated mutations found in higher grade gliomas, p53, PDGF, p16 (CDKN2A), IDH1 and IDH2 are rarely reported in pilocytic astrocytomas, which is consistent with our results that also indicate a lack of focal abnormalities in loci where those genes reside.
When excluding pilocytic cases, the GISTIC reanalysis resulted in three novel, previously disregarded regions to be identified as significantly amplified, 3p28, 14q32.33 and 18q12.2. Since the number of significantly deleted regions decreased by more than a half, it seems that deletions are characteristic of benign cases. Two of the deleted regions, 17p13.2 and 9p21.3, still remained significant at a threshold level of q-value 0.05. Out of six remaining regions, four overlapped with those in the previous analysis (17p13.2, 13q12.11; 10q24.2, 9p21.3). We can assume that these regions could represent the early events in the consecutive steps of gliomagenesis. Of note is that the region 14q32.33 found to be amplified on a total sample at q-0.45 was in malignant cases amplified at q-0.25.
In the analysis performed only on high grade astrocytomas (III and IV), none of the previously identified amplified regions, classified as significant at a 0.25 q-threshold, were observed. On the other hand, all previously identified deletions found in malignant groups were present, constituting a stable result. When the cutoff value was raised to 0.35, significant amplifications became evident and, in line with three previously identified ones of which 3q28 was significant at a 0.25 threshold for a malignant astrocytoma group and at p-0.45 on our total sample. This is in line with the identified stable cross sample amplification region. Thus, the significantly deleted regions in high grade astrocytoma groups were: 9p21.3; 17p13.2; 10q24.2; 14q21.3; 1p36.11 and 13q12.11, while significantly amplified were 3q28; 12q13.3 and 21q22.3. None of these aberrations were significant for low grade astrocytoma tumors, and we believe they might be associated with progression events.
Although we cannot be sure if our findings represent genetic “malignancy switch”, the majority of regions and genes within were previously reported for the process of progression towards malignancy.
Regions 17q25.3 and 8q24.3 that were found to be amplified on our total sample did not emerge in subsequent analyses and therefore may be characteristic for lower grades. Of note is that deletions 3p14.3, 11p15.4, 15q15.1, 16q22.1, 20q11.22 and 22q12.3 were all found in low grade samples at a threshold level of q-0.45 and also on our total sample at q-0.25, but were not repeatedly found in high grades. This may indicate that these regions and genes within may be involved as early events as well. Although many observed changes were similar to the literary reports, some were identified for the first time in our patients and associated with progression or as an early event.
We could not establish any differences between IDH1 mutant and WT tumors in regard to the presence of listed CNAs.
Even though drawing conclusions is complicated perhaps because of the inherent heterogeneity of astrocytomas [
40] and complexity of cancer genomes per se, our bioinformatics results indicate compatibility with the previously reported regions. At first, cancer-related aCGH studies have showed a high level of discordance in the reported genomic aberrations [
41], leading to conclusions that random mutations and CNA are prevalent. However, a newly developed tool GISTIC can distinguish which CNA are more functionally relevant to the cancer evolution. The accordance rate among different studies improved and a concordant picture of biologically significant CNAs in the glioma genome emerged [
15].
Genes known to be the most frequently amplified in glioblastoma,
EGFR, CDK4, PDGFRA, MDM2, MDM4 [
8,
19] are all found to be involved in tumorigenesis of a variety of cancers and are members of several signaling pathways notoriously involved in cancer. Although these genes are highly involved in glioblastoma evolution, they cannot be considered as solely astrocytoma-specific since they are malfunctioning in a great number of different cancers. EGFR (7p11.2) is one of the most renowned members of the protein kinase superfamily and a member of Ras-Raf-MEK-ERK pathway, but can also activate PI3 kinase-AKT-mTOR signaling. The gene is amplified in 40% of glioblastomas and was associated with the so-called classical subtype. Nevertheless,
EGFR amplification and mutations have been shown to be responsible for many other cancer types.
Another common amplification is of the chromosome 12 on which genes
CDK4 and
MDM2 reside.
CDK4 (cyclin dependent kinase 4), yet another candidate gene for glioblastoma, is responsible for the cell cycle’s G1 to S transition but is also involved in a variety of cancers. MDM2 is an E3 ubiquitin ligase localized in the nucleus that mediates ubiquitination of p53, leading to its degradation by the proteasome and inhibits p53- and p73-mediated cell cycle arrest and apoptosis. Similar involvement in glioblastoma displays gene
MDM4 [
42].
The region 10q23.31 where tumor suppressor PTEN resides is also known to be frequently lost in glioblastoma, but also mutated or lost in a large number of other human tumors (prostate cancer, glioblastoma, endometrial, lung and breast cancer). The gene encodes a phosphatidylinositol-3,4,5-trisphosphate 3-phosphatase that contains a tensin like domain. It negatively regulates the AKT/PKB signaling pathway. We have observed significantly deleted region 10q24.2 distant 5814367bp from the PTEN region.
Another well-known amplification event is the one on chromosome 4 [
8] where a gene for receptor tyrosine-protein kinase
PDGFRA (4q12) resides. PDGFRA acts as a receptor for PDGFA, PDGFB and PDGFC growth factors necessary among other things for the growth of glial cells, too [
43]. It has been shown that kinase PDGFRA mediates the activation of both PI3K/Akt/mTOR and Ras/Raf/MEK/ERK signaling.
Of note is our result on the deletions of loci at 9p21.3, where genes
CDKN2A/CDKN2B reside, that have been identified with GISTIC as significantly deleted regions both on our total sample as well as on malignant cases only. It has been shown that the region is significant for glioblastomas and highly recurrent homozygous deletions of
CDKN2A/B genes were established [
8]. CDKN2A functions as inhibitors of CDK4 kinase, which denotes this gene as a tumor suppressor. Its protein can also stabilize the p53 protein. Adjacent to
CDKN2A lays
CDKN2B gene which encodes a cyclin-dependent kinase inhibitor that disables the activation of CDK4 or CDK6. Both genes are involved in the G1 cell-cycle control.
Beroukhim et al. [
15] report on amplifications of 4q12 and 7p11.2 (18–26% of samples) and deletions of 1p36.31 and 9p21.3 (35–49%). Their paper argues that, in some cases, a high degree of amplification renders amplifications highly significant even though they occur in only 6–7% of samples. Because the background rate of deletions across the genome is higher, deletions usually must occur at higher frequencies than amplifications to attain similar levels of significance.
Roerig et al. [
13] found novel sites of losses such as 15q14–q26 in anaplastic astrocytomas, supporting our GISTIC results where region 15q15.1 was significantly deleted (even at 0.15 cutoff value). Another reported region 18q11.2–qter for secondary glioblastomas was significantly amplified in our group of pooled malignant cases (18q12.2;
q=0.25), but was missing from glioblastomas.
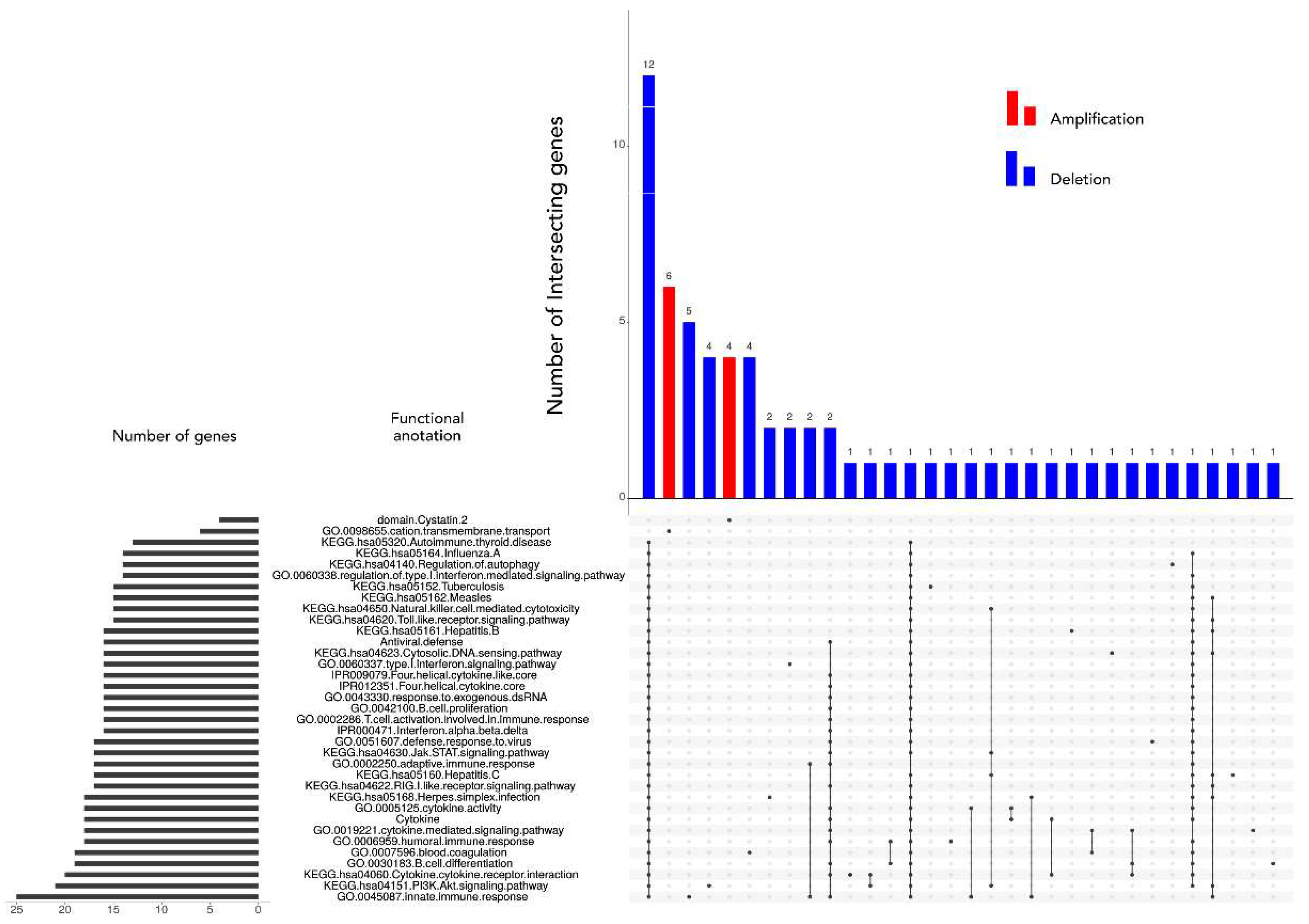
Several significant aberrant regions and genes within were further investigated using functional enrichment strategies [
44]. According to DAVID, 65 genes were assigned to a pathway or a significantly over-represented functional category. Our results on annotated genes possibly involved in astrocytoma tumors brought many candidates which we allocated to the regions identified by GISTIC. In such a manner, potentially important genes in high grade samples were:
SOS2, FCN3, ZNF683, FGF9, IL17D, TNFRSF19, FLT3, POLR1D, FLT1, HMGB1, genes for several interferon molecules,
C1QBP, CXCL16, DHX33, GP1BA, NLRP1, P2RX1, P2RX5, CLDN7, CLEC10A, GABARAP, XAF1, DVL2, RTN4RL1, YWHAE BLNK, CHUK, ENTPD1, FGF8, HPS6, NFKB2, PIK3AP1, TAF5, TRIM8 WNT8B. Only one significantly amplified region in high grades harbored functionally relevant annotated gene—
CLDN1.
A significantly deleted region suspected as an early event harbored just one functionally annotated gene—MAP1LC3A (LC3).
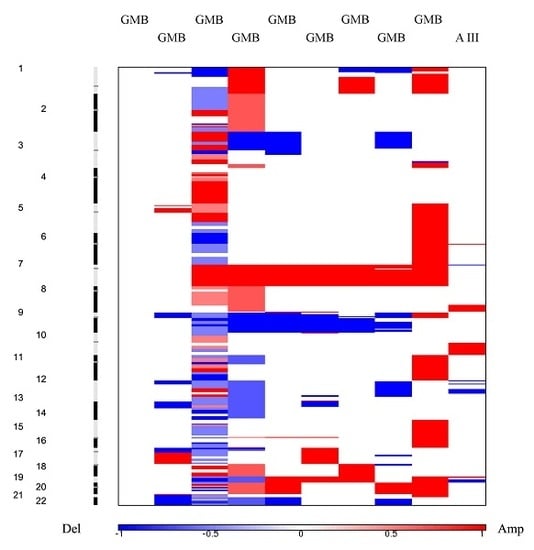
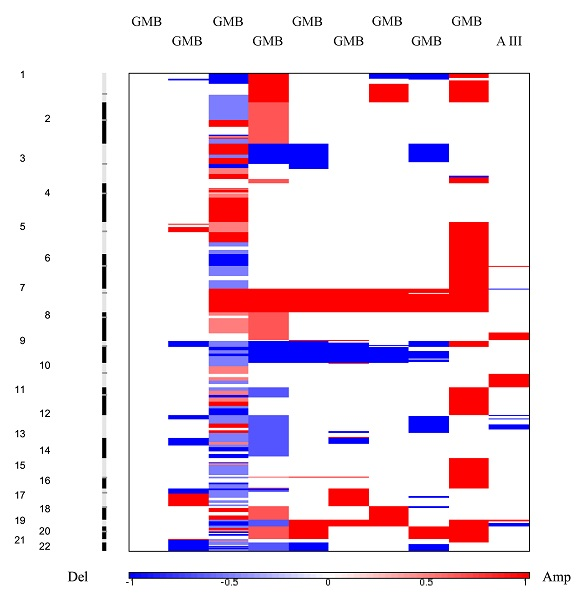
Heat maps revealed a clear distinction between low and high grade samples showing that high grades were reflecting the general heat map images of the entire batch. Furthermore, in the majority of malignant samples, systematic amplification of segments in chromosome 7 and respective deletion in chromosome 10 were evident, a pattern previously reported for glioblastoma patients [
26].
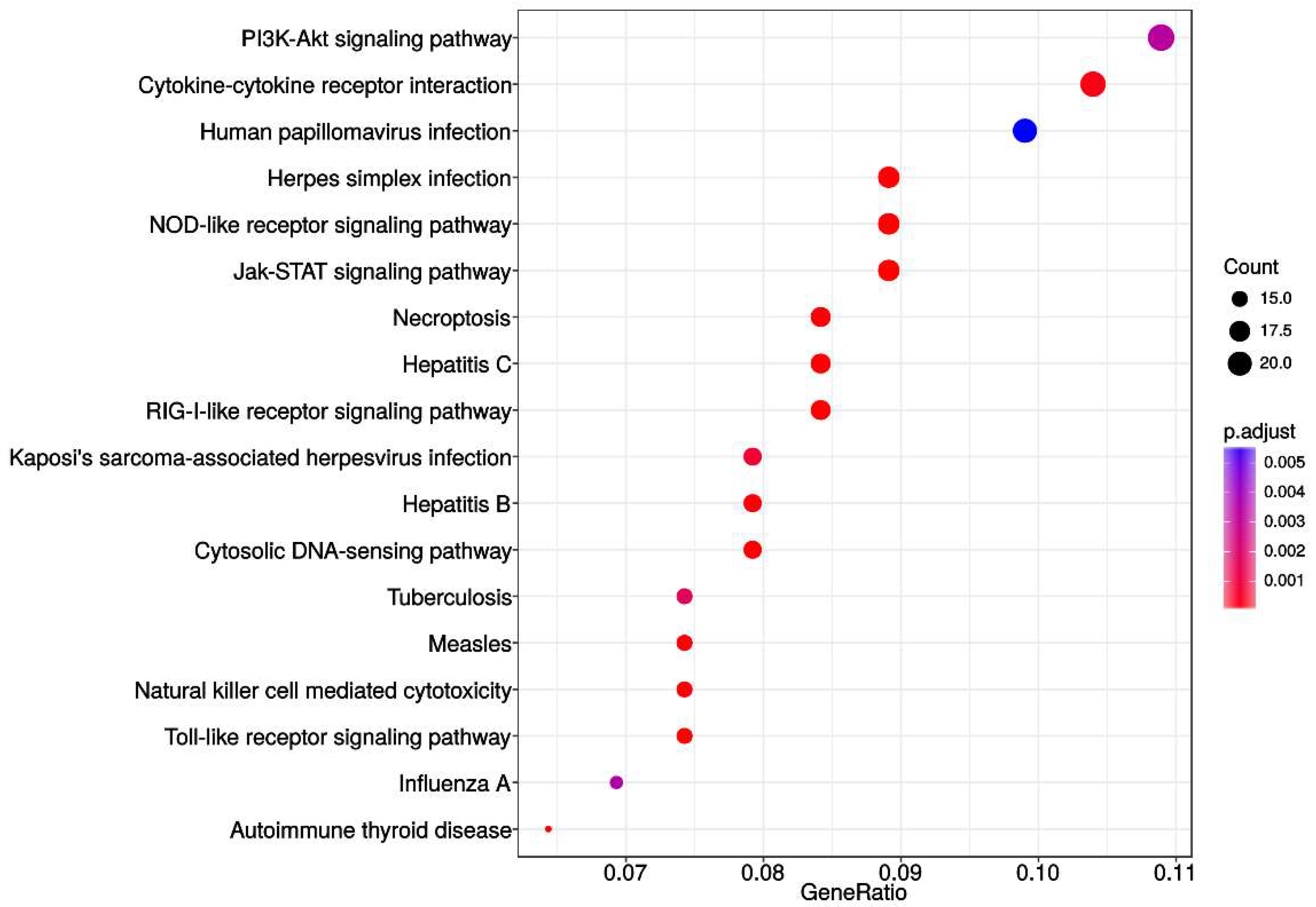
Next, we restricted our analysis to KEGG database and evidenced that only genes associated with deleted segments were significantly enriched in 18 out of 325 total
Homo sapiens associated KEGG pathways. The most significantly represented pathways were PI3K-Akt, Cytokine-cytokine receptor interaction, NOD-like receptor, Jak-STAT, RIG-I-like receptor and Toll-like receptor. In addition, pathways involved in viral infections and inflammation were all significantly enriched too. The enrichment map revealed roughly equal systematic involvement of all genes across the identified pathways. Probably the most intriguing of those are enrichments within HPV and Herpes simplex infection pathways as several studies indicated that the infectious agents have previously been associated with the carcinogenesis of brain and head and neck cancers [
45,
46,
47,
48,
49]. There is evidence for a viral etiology for glioblastoma. It has been shown that many viruses can drive glioma formation in vitro and in xenograft models [
50]. The most evident association is with the human Cytomegalovirus. Nevertheless, Hashida et al. [
47] demonstrated the presence of the HPV viral genome and protein as well in a subset of patients with glioblastoma. However, the majority of literary findings are still contradictory.
The involvement of cytokine and pathways connected to inflammation that emerged as significantly represented in our study is not unusual. It has long been known that numerous cytokines are strongly implicated in the development and progression of cancer [
51,
52], but the mechanisms behind their complex involvement are not completely elucidated. Tumor cells communicate with various types of cells in the tumor microenvironment and this interaction can both promote and inhibit cancer progression depending on the context. Besides being involved in inflammation, cytokines and their receptors also mediating the host response to cancer, the relationship between cancer and inflammation is an important novel topic that needs to be explored further.
Interferons are small signaling proteins released by host cells with the aim to eradicate pathogens or tumors. Interferon gene cluster region on 9p21.3 has long been shown to be deleted in glioblastoma [
53,
54]. The same region was deleted in our study and 16 interferon genes (INF) emerged as significantly annotated by DAVID. This is in accordance with the study by Olopade et al. [
55], who showed that loss of DNA sequences on 9p, particularly the IFN genes, occurred at a significant frequency in gliomas, and is important for the progression of these tumors. The cBioPortal for Cancer Genomics website (
http://www.cbioportal.org/, accessed on 23 February 2019) data mining validated this finding since all of the genes within 9p21.3 region were also reported to be substantially deleted in high grade gliomas. Our results on many significantly implicated interferon genes are consistent with a model of tumorigenesis in which the development or progression of cancer involves the loss or inactivation of genes that normally act to fight tumorigenesis. This may suggest involvement of immunological impairment in gliomas.
It is relevant to discuss potential pharmaceuticals employed against the signaling pathways and genes described above. Recent therapeutic approaches target many levels of glioblastoma biology. One approach is the inhibition of cell cycle molecules. A great number of compounds have been tested as cyclin-dependent kinase (CDK) inhibitors in many malignancies including glioblastoma, yet the majority of them are in pre-clinical or phase I/II trials. Another strategy is immunotherapy that is also being tested in glioblastoma in pre-clinical or phase I/II trials. Furthermore, oligodeoxynucleotides that act on Toll signaling by binding to intracellular Toll-like receptor 9 (TLR9) and thus activate innate and adaptive immunity at first showed no improvement of overall survival of glioblastoma patients, but are being further investigated. Another interesting therapeutic target, and in line with our findings, is the targeting of EGFR. For example, the use of Rindopepimut1—the EGFRvIII mutation vaccine—shows great promise. EGFRvIII is a glioblastoma-specific EGFR mutation consisting of a deletion that causes constitutive activity of tyrosine kinase contributing to glioblastoma aggressiveness. It is important to mention that STAT signaling emerged as another potential therapeutic target in glioblastoma, since siRNAs or pharmacological inhibitors of STAT 3 and its activator, IL-6, showed promising results for several other malignancies including multiple myeloma, head and neck cancer and prostate cancer. Employing miRNAs and siRNA are trialed for suppression of Akt signaling, too [
56]. Attempts to target the PI3K-Akt-mTOR pathway with PI3K, AKT, or mTORC1 inhibitors failed to improve survival, but switching to the inhibition of another player of this pathway, mTORC2, shows promise [
57]. However, effective crossing of drugs and cells through the blood–brain barrier still represents a big problem; therefore, nanobodies and micelles are being investigated to bypass this obstacle.
The major limitation of our study is the small number of patients in our cohort. Nevertheless, the minute CNA investigation brings important findings. We are also aware that the roles of involved genes within lost or gained regions need to be further explored by measuring their differential expression, but we must leave these experiments for future studies.

 ,
, 





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}