Protein Interaction with Charged Macromolecules: From Model Polymers to Unfolded Proteins and Post-Translational Modifications


Abstract
: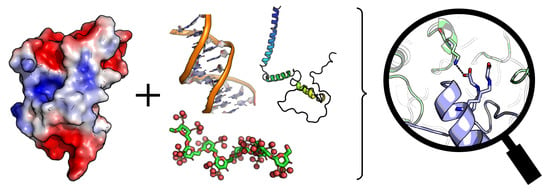
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
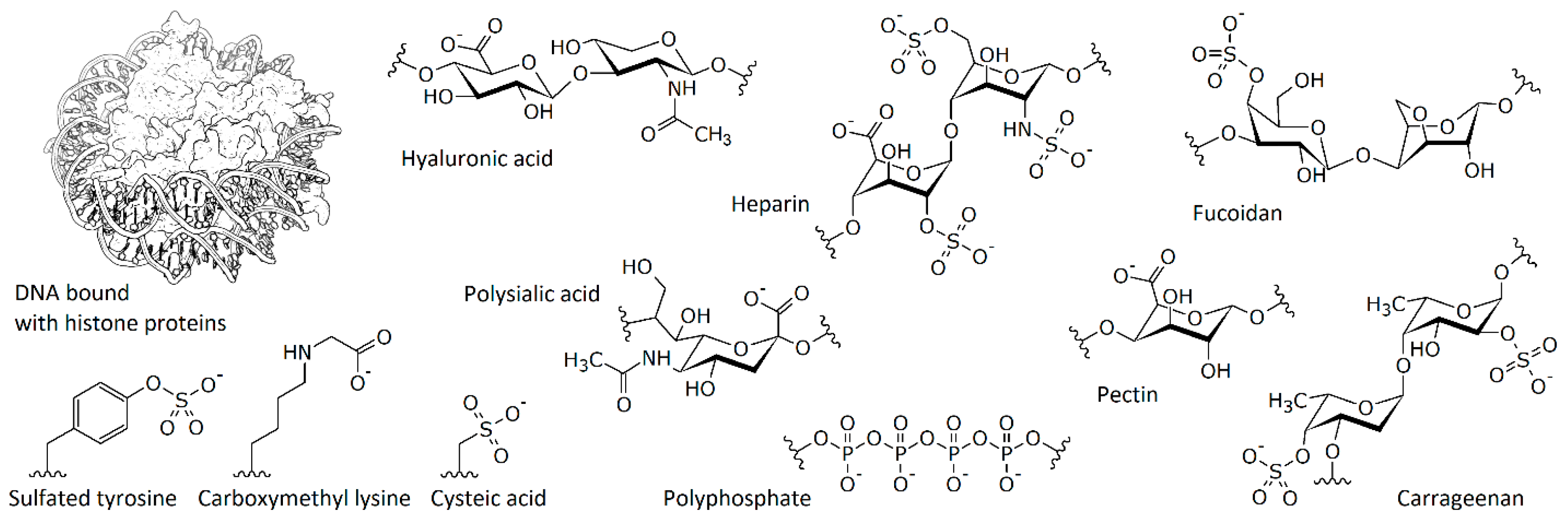
2. Protein Interaction with Model Polymers and Nucleic Acids
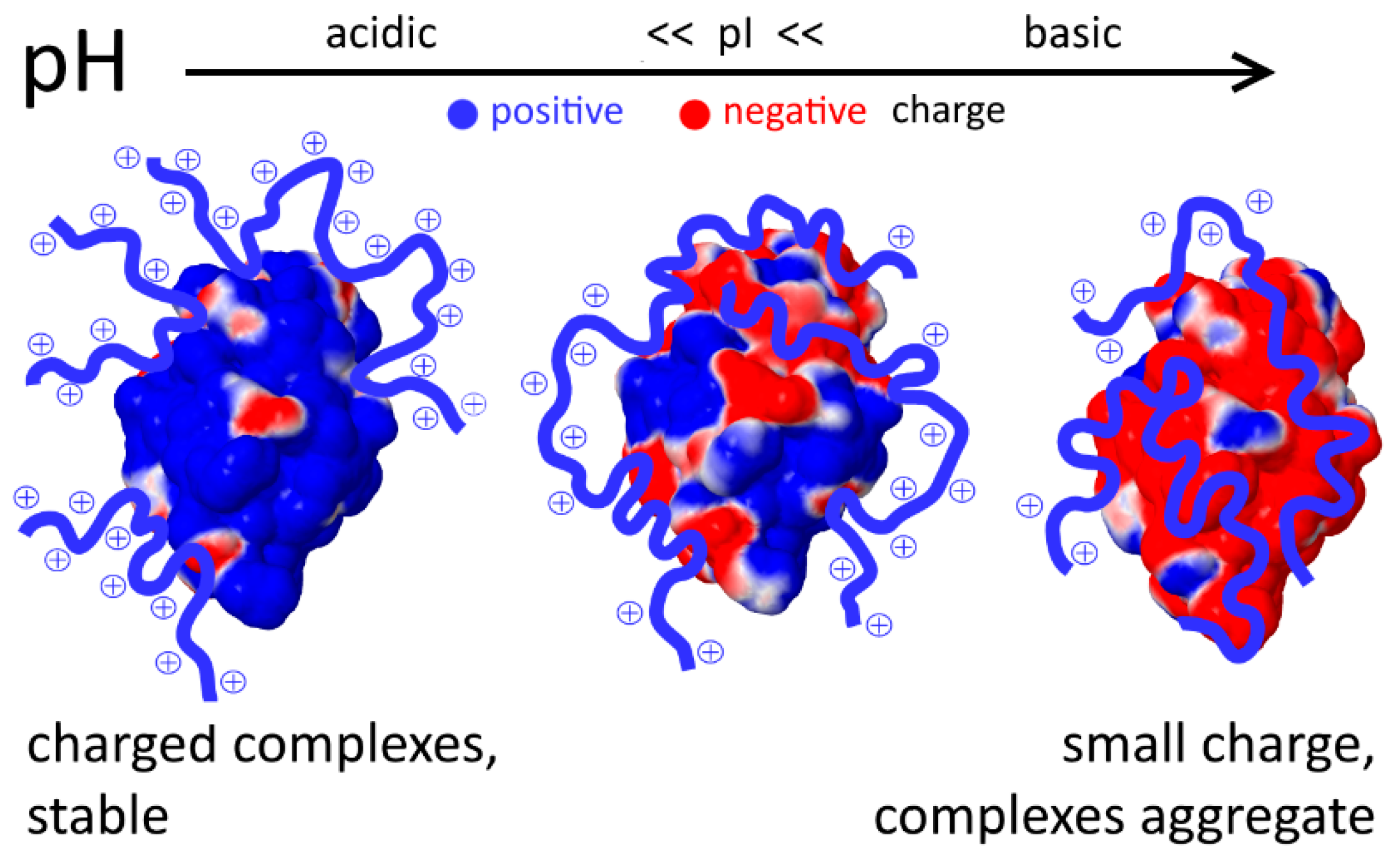
3. Interaction with Other Charged Proteins
4. Unfolded Proteins
5. Effect of Post-Translational Modifications
5.1. Phosphorylation
5.2. Sulfation
5.3. Glycation
5.4. Cysteine Oxidation
6. Concluding Remarks
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| ATP | Adenosine triphosphate |
| ADP | Adenosine diphosphate |
| DNA | Deoxyribonucleic acid |
| GAPDH | Glyceraldehyde-3-phosphate dehydrogenase |
| MD | Molecular dynamics |
| QM/MM | Quantum mechanics/Molecular mechanics |
| RNA | Ribonucleic acid |
References
- Nelson, D.L.; Cox, M.M. Lehninger Principles of Biochemistry; W.H. Freeman: New York, NY, USA, 2005; ISBN 978-0-7167-4339-2. [Google Scholar]
- Kozlowski, L.P. Proteome-pI: Proteome isoelectric point database. Nucleic Acids Res. 2017, 45, D1112–D1116. [Google Scholar] [CrossRef] [PubMed]
- Azevedo, C.; Livermore, T.; Saiardi, A. Protein Polyphosphorylation of Lysine Residues by Inorganic Polyphosphate. Mol. Cell 2015, 58, 71–82. [Google Scholar] [CrossRef] [PubMed]
- Niehrs, C.; Beisswanger, R.; Huttner, W.B. Protein tyrosine sulfation, 1993–an update. Chem. Biol. Interact. 1994, 92, 257–271. [Google Scholar] [CrossRef]
- Moore, K.L. The Biology and Enzymology of Protein Tyrosine O-Sulfation. J. Biol. Chem. 2003, 278, 24243–24246. [Google Scholar] [CrossRef] [PubMed]
- Salahuddin, P.; Rabbani, G.; Khan, R. The role of advanced glycation end products in various types of neurodegenerative disease: A therapeutic approach. Cell. Mol. Biol. Lett. 2014, 19. [Google Scholar] [CrossRef]
- Sadowska-Bartosz, I.; Bartosz, G. Effect of glycation inhibitors on aging and age-related diseases. Mech. Ageing Dev. 2016, 160, 1–18. [Google Scholar] [CrossRef]
- Muronetz, V.I.; Melnikova, A.K.; Saso, L.; Schmalhausen, E.V. Influence of Oxidative Stress on Catalytic and Non-glycolytic Functions of Glyceraldehyde-3-phosphate dehydrogenase. Curr. Med. Chem. 2018. [Google Scholar] [CrossRef]
- Mühlenhoff, M.; Rollenhagen, M.; Werneburg, S.; Gerardy-Schahn, R.; Hildebrandt, H. Polysialic Acid: Versatile Modification of NCAM, SynCAM 1 and Neuropilin-2. Neurochem. Res. 2013, 38, 1134–1143. [Google Scholar] [CrossRef]
- Cameron, I.L.; Jeter, J.R. Acidic Proteins of the Nucleus; Cell Biology; Academic Press: New York, NY, USA, 1974; ISBN 978-0-12-156930-3. [Google Scholar]
- Klein, D.J.; Moore, P.B.; Steitz, T.A. The Roles of Ribosomal Proteins in the Structure Assembly, and Evolution of the Large Ribosomal Subunit. J. Mol. Biol. 2004, 340, 141–177. [Google Scholar] [CrossRef]
- Rohs, R.; Jin, X.; West, S.M.; Joshi, R.; Honig, B.; Mann, R.S. Origins of Specificity in Protein-DNA Recognition. Annu. Rev. Biochem. 2010, 79, 233–269. [Google Scholar] [CrossRef]
- Jones, S. Protein–RNA interactions: Structural biology and computational modeling techniques. Biophys. Rev. 2016, 8, 359–367. [Google Scholar] [CrossRef]
- Seyrek, E.; Dubin, P. Glycosaminoglycans as polyelectrolytes. Adv. Colloid Interface Sci. 2010, 158, 119–129. [Google Scholar] [CrossRef]
- de Kruif, C.G.; Tuinier, R. Polysaccharide protein interactions. Food Hydrocoll. 2001, 15, 555–563. [Google Scholar] [CrossRef]
- Van Haver, L.; Nayar, S. Polyelectrolyte flocculants in harvesting microalgal biomass for food and feed applications. Algal Res. 2017, 24, 167–180. [Google Scholar] [CrossRef]
- Kusaykin, M.; Bakunina, I.; Sova, V.; Ermakova, S.; Kuznetsova, T.; Besednova, N.; Zaporozhets, T.; Zvyagintseva, T. Structure, biological activity, and enzymatic transformation of fucoidans from the brown seaweeds. Biotechnol. J. 2008, 3, 904–915. [Google Scholar] [CrossRef]
- de Kruif, C.G.; Weinbreck, F.; de Vries, R. Complex coacervation of proteins and anionic polysaccharides. Curr. Opin. Colloid Interface Sci. 2004, 9, 340–349. [Google Scholar] [CrossRef]
- Kulaev, I.S.; Vagabov, V.; Kulakovskaya, T. The Biochemistry of Inorganic Polyphosphates; John Wiley & Sons: Hoboken, NJ, USA, 2005; ISBN 978-0-470-85818-9. [Google Scholar]
- Schröder, H.C.; Müller, W.E.G. Inorganic Polyphosphates: Biochemistry, Biology, Biotechnology; Springer Science & Business Media: Berlin/Heidelberg, Germany, 2012; ISBN 978-3-642-58444-2. [Google Scholar]
- Bentley-DeSousa, A.; Downey, M. From underlying chemistry to therapeutic potential: Open questions in the new field of lysine polyphosphorylation. Curr. Genet. 2018. [Google Scholar] [CrossRef]
- Karplus, M.; McCammon, J.A. Molecular dynamics simulations of biomolecules. Nat. Struct. Mol. Biol. 2002, 9, 646–652. [Google Scholar] [CrossRef]
- Warshel, A. Multiscale Modeling of Biological Functions: From Enzymes to Molecular Machines (Nobel Lecture). Angew. Chem. Int. Ed. 2014, 53, 10020–10031. [Google Scholar] [CrossRef]
- Gao, J.; Truhlar, D.G. Quantum mechanical methods for enzyme kinetics. Annu. Rev. Phys. Chem. 2002, 53, 467–505. [Google Scholar] [CrossRef]
- Dror, R.O.; Dirks, R.M.; Grossman, J.P.; Xu, H.; Shaw, D.E. Biomolecular Simulation: A Computational Microscope for Molecular Biology. Annu. Rev. Biophys. 2012, 41, 429–452. [Google Scholar] [CrossRef]
- Takada, S. Coarse-grained molecular simulations of large biomolecules. Curr. Opin. Struct. Biol. 2012, 22, 130–137. [Google Scholar] [CrossRef]
- Wallin, T.; Linse, P. Monte Carlo Simulations of Polyelectrolytes at Charged Micelles. 1. Effects of Chain Flexibility. Langmuir 1996, 12, 305–314. [Google Scholar] [CrossRef]
- Carlsson, F.; Linse, P.; Malmsten, M. Monte Carlo Simulations of Polyelectrolyte−Protein Complexation. J. Phys. Chem. B 2001, 105, 9040–9049. [Google Scholar] [CrossRef]
- de Vries, R. Monte Carlo simulations of flexible polyanions complexing with whey proteins at their isoelectric point. J. Chem. Phys. 2004, 120, 3475–3481. [Google Scholar] [CrossRef]
- Kayitmazer, A.B.; Quinn, B.; Kimura, K.; Ryan, G.L.; Tate, A.J.; Pink, D.A.; Dubin, P.L. Protein specificity of charged sequences in polyanions and heparins. Biomacromolecules 2010, 11, 3325–3331. [Google Scholar] [CrossRef]
- Hofzumahaus, C.; Hebbeker, P.; Schneider, S. Monte Carlo simulations of weak polyelectrolyte microgels: pH-dependence of conformation and ionization. Soft Matter 2018. [Google Scholar] [CrossRef]
- Harrison, R.E.S.; Morikis, D. Molecular Mechanisms of Macular Degeneration Associated with the Complement Factor H Y402H Mutation. Biophys. J. 2018. [Google Scholar] [CrossRef]
- Yu, S.; Xu, X.; Yigit, C.; van der Giet, M.; Zidek, W.; Jankowski, J.; Dzubiella, J.; Ballauff, M. Interaction of human serum albumin with short polyelectrolytes: A study by calorimetry and computer simulations. Soft Matter 2015, 11, 4630–4639. [Google Scholar] [CrossRef]
- Yigit, C.; Heyda, J.; Ballauff, M.; Dzubiella, J. Like-charged protein-polyelectrolyte complexation driven by charge patches. J. Chem. Phys. 2015, 143, 064905. [Google Scholar] [CrossRef]
- Perlmutter, J.D.; Drasler, W.J.; Xie, W.; Gao, J.; Popot, J.-L.; Sachs, J.N. All-Atom and Coarse-Grained Molecular Dynamics Simulations of a Membrane Protein Stabilizing Polymer. Langmuir 2011, 27, 10523–10537. [Google Scholar] [CrossRef]
- Schneider, C.P.; Shukla, D.; Trout, B.L. Effects of Solute-Solute Interactions on Protein Stability Studied Using Various Counterions and Dendrimers. PLoS ONE 2011, 6. [Google Scholar] [CrossRef]
- Sofronova, A.A.; Evstafyeva, D.B.; Izumrudov, V.A.; Muronetz, V.I.; Semenyuk, P.I. Protein-polyelectrolyte complexes: Molecular dynamics simulations and experimental study. Polymer 2017, 113, 39–45. [Google Scholar] [CrossRef]
- Tian, W.; Ma, Y. Theoretical and computational studies of dendrimers as delivery vectors. Chem. Soc. Rev. 2012, 42, 705–727. [Google Scholar] [CrossRef]
- Kayitmazer, A.B.; Seeman, D.; Minsky, B.B.; Dubin, P.L.; Xu, Y. Protein-polyelectrolyte interactions. Soft Matter 2013, 9, 2553–2583. [Google Scholar] [CrossRef]
- Xu, X.; Angioletti-Uberti, S.; Lu, Y.; Dzubiella, J.; Ballauff, M. Interaction of Proteins with Polyelectrolytes: Comparison of Theory to Experiment. Langmuir 2018. [Google Scholar] [CrossRef]
- Capila, I.; Linhardt, R.J. Heparin–Protein Interactions. Angew. Chem. Int. Ed. 2002, 41, 390–412. [Google Scholar] [CrossRef]
- Gandhi, N.S.; Mancera, R.L. Molecular Dynamics Simulations of CXCL-8 and Its Interactions with a Receptor Peptide, Heparin Fragments, and Sulfated Linked Cyclitols. J. Chem. Inf. Model. 2011, 51, 335–358. [Google Scholar] [CrossRef]
- Pichert, A.; Samsonov, S.A.; Theisgen, S.; Thomas, L.; Baumann, L.; Schiller, J.; Beck-Sickinger, A.G.; Huster, D.; Pisabarro, M.T. Characterization of the interaction of interleukin-8 with hyaluronan, chondroitin sulfate, dermatan sulfate and their sulfated derivatives by spectroscopy and molecular modeling. Glycobiology 2012, 22, 134–145. [Google Scholar] [CrossRef]
- Sapay, N.; Cabannes, E.; Petitou, M.; Imberty, A. Molecular modeling of the interaction between heparan sulfate and cellular growth factors: Bringing pieces together. Glycobiology 2011, 21, 1181–1193. [Google Scholar] [CrossRef]
- Salbach-Hirsch, J.; Samsonov, S.A.; Hintze, V.; Hofbauer, C.; Picke, A.-K.; Rauner, M.; Gehrcke, J.-P.; Moeller, S.; Schnabelrauch, M.; Scharnweber, D.; et al. Structural and functional insights into sclerostin-glycosaminoglycan interactions in bone. Biomaterials 2015, 67, 335–345. [Google Scholar] [CrossRef]
- Singh, A.; Kett, W.C.; Severin, I.C.; Agyekum, I.; Duan, J.; Amster, I.J.; Proudfoot, A.E.I.; Coombe, D.R.; Woods, R.J. The Interaction of Heparin Tetrasaccharides with Chemokine CCL5 Is Modulated by Sulfation Pattern and pH. J. Biol. Chem. 2015, 290, 15421–15436. [Google Scholar] [CrossRef]
- Hintze, V.; Samsonov, S.A.; Anselmi, M.; Moeller, S.; Becher, J.; Schnabelrauch, M.; Scharnweber, D.; Pisabarro, M.T. Sulfated Glycosaminoglycans Exploit the Conformational Plasticity of Bone Morphogenetic Protein-2 (BMP-2) and Alter the Interaction Profile with Its Receptor. Biomacromolecules 2014, 15, 3083–3092. [Google Scholar] [CrossRef]
- Valle-Delgado, J.J.; Alfonso-Prieto, M.; de Groot, N.S.; Ventura, S.; Samitier, J.; Rovira, C.; Fernàndez-Busquets, X. Modulation of Aβ42 fibrillogenesis by glycosaminoglycan structure. FASEB J. 2010, 24, 4250–4261. [Google Scholar] [CrossRef]
- Björk, I.; Lindahl, U. Mechanism of the anticoagulant action of heparin. Mol. Cell. Biochem. 1982, 48, 161–182. [Google Scholar] [CrossRef]
- Jones, L.S.; Yazzie, B.; Middaugh, C.R. Polyanions and the proteome. Mol. Cell Proteom. 2004, 3, 746–769. [Google Scholar] [CrossRef]
- Pol-Fachin, L.; Verli, H. Structural glycobiology of heparin dynamics on the exosite 2 of coagulation cascade proteases: Implications for glycosaminoglycans antithrombotic activity. Glycobiology 2014, 24, 97–105. [Google Scholar] [CrossRef]
- Singh, A.; Tessier, M.B.; Pederson, K.; Wang, X.; Venot, A.P.; Boons, G.-J.; Prestegard, J.H.; Woods, R.J. Extension and validation of the GLYCAM force field parameters for modeling glycosaminoglycans. Can. J. Chem. 2016, 94, 927–935. [Google Scholar] [CrossRef]
- Carter, W.J.; Cama, E.; Huntington, J.A. Crystal Structure of Thrombin Bound to Heparin. J. Biol. Chem. 2005, 280, 2745–2749. [Google Scholar] [CrossRef]
- Boetsch, C.; Aguayo-Villegas, D.R.; Gonzalez-Nilo, F.D.; Lisa, Á.T.; Beassoni, P.R. Putative binding mode of Escherichia coli exopolyphosphatase and polyphosphates based on a hybrid in silico/biochemical approach. Arch. Biochem. Biophys. 2016, 606, 64–72. [Google Scholar] [CrossRef]
- Sánchez-Moreno, I.; Bordes, I.; Castillo, R.; Ruiz-Pernía, J.J.; Moliner, V.; García-Junceda, E. Tuning the Phosphoryl Donor Specificity of Dihydroxyacetone Kinase from ATP to Inorganic Polyphosphate. An Insight from Computational Studies. Int. J. Mol. Sci. 2015, 16, 27835–27849. [Google Scholar] [CrossRef]
- Svenson, S.; Tomalia, D.A. Dendrimers in biomedical applications—Reflections on the field. Adv. Drug Deliv. Rev. 2005, 57, 2106–2129. [Google Scholar] [CrossRef]
- Martinho, N.; Florindo, H.; Silva, L.; Brocchini, S.; Zloh, M.; Barata, T. Molecular Modeling to Study Dendrimers for Biomedical Applications. Molecules 2014, 19, 20424–20467. [Google Scholar] [CrossRef]
- Moiani, D.; Salvalaglio, M.; Cavallotti, C.; Bujacz, A.; Redzynia, I.; Bujacz, G.; Dinon, F.; Pengo, P.; Fassina, G. Structural Characterization of a Protein A Mimetic Peptide Dendrimer Bound to Human IgG. J. Phys. Chem. B 2009, 113, 16268–16275. [Google Scholar] [CrossRef]
- Sorokina, S.; Semenyuk, P.; Stroylova, Y.; Muronetz, V.; Shifrina, Z. Complexes between cationic pyridylphenylene dendrimers and ovine prion protein: Do hydrophobic interactions matter? RSC Adv. 2017, 7, 16565–16574. [Google Scholar] [CrossRef]
- Shen, Z.; Tian, W.; Chen, K.; Ma, Y. Molecular dynamics simulation of G-actin interacting with PAMAM dendrimers. J. Mol. Graph. Model. 2018, 84, 145–151. [Google Scholar] [CrossRef]
- Giri, J.; Diallo, M.S.; Simpson, A.J.; Liu, Y.; Goddard, W.A.; Kumar, R.; Woods, G.C. Interactions of Poly(amidoamine) Dendrimers with Human Serum Albumin: Binding Constants and Mechanisms. ACS Nano 2011, 5, 3456–3468. [Google Scholar] [CrossRef]
- Nandy, B.; Saurabh, S.; Sahoo, A.K.; Dixit, N.M.; Maiti, P.K. The SPL7013 dendrimer destabilizes the HIV-1 gp120–CD4 complex. Nanoscale 2015, 7, 18628–18641. [Google Scholar] [CrossRef]
- Sepúlveda-Crespo, D.; Vacas-Córdoba, E.; Márquez-Miranda, V.; Araya-Durán, I.; Gómez, R.; Mata, F.J.D.L.; González-Nilo, F.D.; Muñoz-Fernández, M.Á. Effect of Several HIV Antigens Simultaneously Loaded with G2-NN16 Carbosilane Dendrimer in the Cell Uptake and Functionality of Human Dendritic Cells. Bioconjugate Chem. 2016, 27, 2844–2849. [Google Scholar] [CrossRef]
- Camarada, M.B.; Márquez-Miranda, V.; Araya-Durán, I.; Yévenes, A.; González-Nilo, F. PAMAM G4 dendrimers as inhibitors of the iron storage properties of human L-chain ferritin. Phys. Chem. Chem. Phys. 2015, 17, 19001–19011. [Google Scholar] [CrossRef]
- Stroylova, Y.; Sorokina, S.; Stroylov, V.; Melnikova, A.; Gaillard, C.; Shifrina, Z.; Haertlé, T.; Muronetz, V.I. Spontaneous formation of nanofilms under interaction of 4th generation pyrydylphenylene dendrimer with proteins. Polymer 2018, 137, 186–194. [Google Scholar] [CrossRef]
- Mandal, T.; Kanchi, S.; Ayappa, K.G.; Maiti, P.K. pH controlled gating of toxic protein pores by dendrimers. Nanoscale 2016, 8, 13045–13058. [Google Scholar] [CrossRef] [PubMed]
- Sofronova, A.A.; Izumrudov, V.A.; Muronetz, V.I.; Semenyuk, P.I. Similarly charged polyelectrolyte can be the most efficient suppressor of the protein aggregation. Polymer 2017, 108, 281–287. [Google Scholar] [CrossRef]
- Shalova, I.N.; Asryants, R.A.; Sholukh, M.V.; Saso, L.; Kurganov, B.I.; Muronetz, V.I.; Izumrudov, V.A. Interaction of polyanions with basic proteins, 2(a): Influence of complexing polyanions on the thermo-aggregation of oligomeric enzymes. Macromolar Biosci. 2005, 5, 1184–1192. [Google Scholar] [CrossRef]
- Shalova, I.N.; Naletova, I.N.; Saso, L.; Muronetz, V.I.; Izumrudov, V.A. Interaction of polyelectrolytes with proteins, 3. Influence of complexing polycations on the thermoaggregation of oligomeric enzymes. Macromol. Biosci. 2007, 7, 929–939. [Google Scholar] [CrossRef] [PubMed]
- Semenyuk, P.I.; Moiseeva, E.V.; Stroylova, Y.Y.; Lotti, M.; Izumrudov, V.A.; Muronetz, V.I. Sulfated and sulfonated polymers are able to solubilize efficiently the protein aggregates of different nature. Arch. Biochem. Biophys. 2015, 567, 22–29. [Google Scholar] [CrossRef]
- Semenyuk, P.I.; Kurochkina, L.P.; Gusev, N.B.; Izumrudov, V.A.; Muronetz, V.I. Chaperone-like activity of synthetic polyanions can be higher than the activity of natural chaperones at elevated temperature. Biochem. Biophys. Res. Commun. 2017, 489, 200–205. [Google Scholar] [CrossRef] [PubMed]
- Sagui, C.; Darden, T.A. MOLECULAR DYNAMICS SIMULATIONS OF BIOMOLECULES: Long-Range Electrostatic Effects. Annu. Rev. Biophys. Biomol. Struct. 1999, 28, 155–179. [Google Scholar] [CrossRef] [PubMed]
- Šponer, J.; Bussi, G.; Krepl, M.; Banáš, P.; Bottaro, S.; Cunha, R.A.; Gil-Ley, A.; Pinamonti, G.; Poblete, S.; Jurečka, P.; et al. RNA Structural Dynamics As Captured by Molecular Simulations: A Comprehensive Overview. Chem. Rev. 2018. [Google Scholar] [CrossRef] [PubMed]
- Cheatham, T.E., III; Brooks, B.R. Recent advances in molecular dynamics simulation towards the realistic representation of biomolecules in solution. Theor. Chem. Acc. 1998, 99, 279–288. [Google Scholar] [CrossRef]
- Margreitter, C.; Petrov, D.; Zagrovic, B. Vienna-PTM web server: A toolkit for MD simulations of protein post-translational modifications. Nucleic Acids Res. 2013, 41, W422–W426. [Google Scholar] [CrossRef] [PubMed]
- Khoury, G.A.; Thompson, J.P.; Smadbeck, J.; Kieslich, C.A.; Floudas, C.A. Forcefield_PTM: Ab Initio Charge and AMBER Forcefield Parameters for Frequently Occurring Post-Translational Modifications. J. Chem. Theory Comput. 2013, 9, 5653–5674. [Google Scholar] [CrossRef]
- Homeyer, N.; Horn, A.H.C.; Lanig, H.; Sticht, H. AMBER force-field parameters for phosphorylated amino acids in different protonation states: Phosphoserine, phosphothreonine, phosphotyrosine, and phosphohistidine. J. Mol. Model. 2006, 12, 281–289. [Google Scholar] [CrossRef] [PubMed]
- Dupradeau, F.-Y.; Pigache, A.; Zaffran, T.; Savineau, C.; Lelong, R.; Grivel, N.; Lelong, D.; Rosanski, W.; Cieplak, P. The R.E.D. Tools: Advances in RESP and ESP charge derivation and force field library building. Phys. Chem. Chem. Phys. 2010, 12, 7821–7839. [Google Scholar] [CrossRef] [PubMed]
- Schüttelkopf, A.W.; van Aalten, D.M.F. PRODRG: A tool for high-throughput crystallography of protein–ligand complexes. Acta Crystallogr. Sect. D Biol. Crystallogr. 2004, 60, 1355–1363. [Google Scholar] [CrossRef] [PubMed]
- Malde, A.K.; Zuo, L.; Breeze, M.; Stroet, M.; Poger, D.; Nair, P.C.; Oostenbrink, C.; Mark, A.E. An Automated Force Field Topology Builder (ATB) and Repository: Version 1.0. J. Chem. Theory Comput. 2011, 7, 4026–4037. [Google Scholar] [CrossRef]
- Record, M.T.; Anderson, C.F.; Lohman, T.M. Thermodynamic analysis of ion effects on the binding and conformational equilibria of proteins and nucleic acids: The roles of ion association or release, screening, and ion effects on water activity. Q. Rev. Biophys. 1978, 11, 103–178. [Google Scholar] [CrossRef] [PubMed]
- Kabanov, V.A. Polyelectrolyte complexes in solution and in bulk. Russ. Chem. Rev. 2005, 74, 3. [Google Scholar] [CrossRef]
- Henzler, K.; Haupt, B.; Lauterbach, K.; Wittemann, A.; Borisov, O.; Ballauff, M. Adsorption of β-Lactoglobulin on Spherical Polyelectrolyte Brushes: Direct Proof of Counterion Release by Isothermal Titration Calorimetry. J. Am. Chem. Soc. 2010, 132, 3159–3163. [Google Scholar] [CrossRef]
- Wittemann, A.; Ballauff, M. Interaction of proteins with linear polyelectrolytes and spherical polyelectrolyte brushes in aqueous solution. Phys. Chem. Chem. Phys. 2006, 8, 5269–5275. [Google Scholar] [CrossRef]
- Xu, X.; Ran, Q.; Dey, P.; Nikam, R.; Haag, R.; Ballauff, M.; Dzubiella, J. Counterion-Release Entropy Governs the Inhibition of Serum Proteins by Polyelectrolyte Drugs. Biomacromolecules 2018, 19, 409–416. [Google Scholar] [CrossRef] [PubMed]
- de Vries, R.; Cohen Stuart, M. Theory and simulations of macroion complexation. Curr. Opin. Colloid Interface Sci. 2006, 11, 295–301. [Google Scholar] [CrossRef]
- Kudlay, A.; Ermoshkin, A.V.; Olvera de la Cruz, M. Complexation of Oppositely Charged Polyelectrolytes: Effect of Ion Pair Formation. Macromolecules 2004, 37, 9231–9241. [Google Scholar] [CrossRef]
- Bharadwaj, S.; Montazeri, R.; Haynie, D.T. Direct Determination of the Thermodynamics of Polyelectrolyte Complexation and Implications Thereof for Electrostatic Layer-by-Layer Assembly of Multilayer Films. Langmuir 2006, 22, 6093–6101. [Google Scholar] [CrossRef]
- Andreev, M.; Prabhu, V.M.; Douglas, J.F.; Tirrell, M.; de Pablo, J.J. Complex Coacervation in Polyelectrolytes from a Coarse-Grained Model. Macromolecules 2018, 51, 6717–6723. [Google Scholar] [CrossRef]
- Becker, A.L.; Henzler, K.; Welsch, N.; Ballauff, M.; Borisov, O. Proteins and polyelectrolytes: A charged relationship. Curr. Opin. Colloid Interface Sci. 2012, 17, 90–96. [Google Scholar] [CrossRef]
- Heyda, J.; Dzubiella, J. Ion-specific counterion condensation on charged peptides: Poisson–Boltzmann vs. atomistic simulations. Soft Matter 2012, 8, 9338–9344. [Google Scholar] [CrossRef]
- Ahmed, M.C.; Papaleo, E.; Lindorff-Larsen, K. How well do force fields capture the strength of salt bridges in proteins? PeerJ 2018, 6, e4967. [Google Scholar] [CrossRef] [PubMed]
- Grosberg, A.Y.; Nguyen, T.T.; Shklovskii, B.I. Colloquium: The physics of charge inversion in chemical and biological systems. Rev. Mod. Phys. 2002, 74, 329–345. [Google Scholar] [CrossRef]
- Joung, I.S.; Cheatham, T.E. Determination of Alkali and Halide Monovalent Ion Parameters for Use in Explicitly Solvated Biomolecular Simulations. J. Phys. Chem. B 2008, 112, 9020–9041. [Google Scholar] [CrossRef] [PubMed]
- Semenyuk, P.I.; Zhiryakova, M.V.; Izumrudov, V.A. Supercharged Polyplexes: Full-Atom Molecular Dynamics Simulations and Experimental Study. Macromolecules 2018, 51, 5450–5459. [Google Scholar] [CrossRef]
- Faller, C.E.; Guvench, O. Sulfation and Cation Effects on the Conformational Properties of the Glycan Backbone of Chondroitin Sulfate Disaccharides. J. Phys. Chem. B 2015, 119, 6063–6073. [Google Scholar] [CrossRef] [PubMed]
- Antila, H.S.; Sammalkorpi, M. Polyelectrolyte Decomplexation via Addition of Salt: Charge Correlation Driven Zipper. J. Phys. Chem. B 2014, 118, 3226–3234. [Google Scholar] [CrossRef] [PubMed]
- Antila, H.S.; Härkönen, M.; Sammalkorpi, M. Chemistry specificity of DNA–polycation complex salt response: A simulation study of DNA, polylysine and polyethyleneimine. Phys. Chem. Chem. Phys. 2015, 17, 5279–5289. [Google Scholar] [CrossRef]
- Jones, S.; Shanahan, H.P.; Berman, H.M.; Thornton, J.M. Using electrostatic potentials to predict DNA-binding sites on DNA-binding proteins. Nucleic Acids Res. 2003, 31, 7189–7198. [Google Scholar] [CrossRef] [PubMed]
- Jayaram, B.; Sharp, K.A.; Honig, B. The electrostatic potential of B-DNA. Biopolymers 1989, 28, 975–993. [Google Scholar] [CrossRef]
- Cherstvy, A.G. Positively Charged Residues in DNA-Binding Domains of Structural Proteins Follow Sequence-specific Positions of DNA Phosphate Groups. J. Phys. Chem. B 2009, 113, 4242–4247. [Google Scholar] [CrossRef]
- Savelyev, A.; Materese, C.K.; Papoian, G.A. Is DNA’s Rigidity Dominated by Electrostatic or Nonelectrostatic Interactions? J. Am. Chem. Soc. 2011, 133, 19290–19293. [Google Scholar] [CrossRef]
- McDowell, S.E.; Špačková, N.; Šponer, J.; Walter, N.G. Molecular dynamics simulations of RNA: An in silico single molecule approach. Biopolymers 2007, 85, 169–184. [Google Scholar] [CrossRef]
- MacKerell, A.D.; Nilsson, L. Molecular dynamics simulations of nucleic acid–protein complexes. Curr. Opin. Struct. Biol. 2008, 18, 194–199. [Google Scholar] [CrossRef] [PubMed]
- Honig, B.; Nicholls, A. Classical electrostatics in biology and chemistry. Science 1995, 268, 1144–1149. [Google Scholar] [CrossRef] [PubMed]
- Sheinerman, F.B.; Norel, R.; Honig, B. Electrostatic aspects of protein–protein interactions. Curr. Opin. Struct. Biol. 2000, 10, 153–159. [Google Scholar] [CrossRef]
- Muronetz, V.I.; Wang, Z.X.; Keith, T.J.; Knull, H.R.; Srivastava, D.K. Binding constants and stoichiometries of glyceraldehyde 3-phosphate dehydrogenase-tubulin complexes. Arch. Biochem. Biophys. 1994, 313, 253–260. [Google Scholar] [CrossRef] [PubMed]
- Barinova, K.; Khomyakova, E.; Semenyuk, P.; Schmalhausen, E.; Muronetz, V. Binding of alpha-synuclein to partially oxidized glyceraldehyde-3-phosphate dehydrogenase induces subsequent inactivation of the enzyme. Arch. Biochem. Biophys. 2018, 642, 10–22. [Google Scholar] [CrossRef]
- Shcherbatova, N.A.; Nagradova, N.K.; Muronets, V.I. Effect of erythrocyte membranes and tubulin on the activity of NAD-dependent dehydrogenases. Biokhimiia 1996, 61, 1512–1525. [Google Scholar]
- Chu, H.; Low, P.S. Mapping of glycolytic enzyme-binding sites on human erythrocyte band 3. Biochem. J. 2006, 400, 143–151. [Google Scholar] [CrossRef]
- Yi, H.; Qiu, S.; Cao, Z.; Wu, Y.; Li, W. Molecular basis of inhibitory peptide maurotoxin recognizing Kv1.2 channel explored by ZDOCK and molecular dynamic simulations. Proteins: Struct. Funct. Bioinform. 2008, 70, 844–854. [Google Scholar] [CrossRef]
- Han, S.; Yin, S.; Yi, H.; Mouhat, S.; Qiu, S.; Cao, Z.; Sabatier, J.-M.; Wu, Y.; Li, W. Protein−Protein Recognition Control by Modulating Electrostatic Interactions. J. Proteome Res. 2010, 9, 3118–3125. [Google Scholar] [CrossRef]
- Buckle, A.M.; Schreiber, G.; Fersht, A.R. Protein-protein recognition: Crystal structural analysis of a barnase-barstar complex at 2.0-.ANG. resolution. Biochemistry 1994, 33, 8878–8889. [Google Scholar] [CrossRef]
- Schreiber, G.; Fersht, A.R. Rapid, electrostatically assisted association of proteins. Nat. Struct. Biol. 1996, 3, 427–431. [Google Scholar] [CrossRef]
- Demchenko, A.P. Recognition between flexible protein molecules: Induced and assisted folding. J. Mol. Recognit. 2001, 14, 42–61. [Google Scholar] [CrossRef]
- Fiorucci, S.; Zacharias, M. Prediction of Protein-Protein Interaction Sites Using Electrostatic Desolvation Profiles. Biophys. J. 2010, 98, 1921–1930. [Google Scholar] [CrossRef]
- Tsuchiya, Y.; Kinoshita, K.; Nakamura, H. Structure-based prediction of DNA-binding sites on proteins Using the empirical preference of electrostatic potential and the shape of molecular surfaces. Proteins Struct. Funct. Bioinform. 2004, 55, 885–894. [Google Scholar] [CrossRef]
- Lee, L.-P.; Tidor, B. Optimization of binding electrostatics: Charge complementarity in the barnase-barstar protein complex. Protein Sci. 2001, 10, 362–377. [Google Scholar] [CrossRef]
- Sheinerman, F.B.; Honig, B. On the Role of Electrostatic Interactions in the Design of Protein–Protein Interfaces. J. Mol. Biol. 2002, 318, 161–177. [Google Scholar] [CrossRef]
- Wang, W.; Donini, O.; Reyes, C.M.; Kollman, P.A. Biomolecular Simulations: Recent Developments in Force Fields, Simulations of Enzyme Catalysis, Protein-Ligand, Protein-Protein, and Protein-Nucleic Acid Noncovalent Interactions. Annu. Rev. Biophys. Biomol. Struct. 2001, 30, 211–243. [Google Scholar] [CrossRef]
- Durrant, J.D.; McCammon, J.A. Molecular dynamics simulations and drug discovery. BMC Biol. 2011, 9, 71. [Google Scholar] [CrossRef]
- Mobley, D.L.; Gilson, M.K. Predicting Binding Free Energies: Frontiers and Benchmarks. Annu. Rev. Biophys. 2017, 46, 531–558. [Google Scholar] [CrossRef]
- Kieslich, C.A.; Gorham, R.D.; Morikis, D. Is the rigid-body assumption reasonable?: Insights into the effects of dynamics on the electrostatic analysis of barnase–barstar. J. Non-Cryst. Solids 2011, 357, 707–716. [Google Scholar] [CrossRef]
- Hoefling, M.; Gottschalk, K.E. Barnase–Barstar: From first encounter to final complex. J. Struct. Biol. 2010, 171, 52–63. [Google Scholar] [CrossRef]
- Neumann, J.; Gottschalk, K.-E. The Effect of Different Force Applications on the Protein-Protein Complex Barnase-Barstar. Biophys. J. 2009, 97, 1687–1699. [Google Scholar] [CrossRef]
- Ishida, H.; Hayward, S. Path of Nascent Polypeptide in Exit Tunnel Revealed by Molecular Dynamics Simulation of Ribosome. Biophys. J. 2008, 95, 5962–5973. [Google Scholar] [CrossRef]
- Bui, P.T.; Hoang, T.X. Folding and escape of nascent proteins at ribosomal exit tunnel. J. Chem. Phys. 2016, 144, 095102. [Google Scholar] [CrossRef]
- Petrone, P.M.; Snow, C.D.; Lucent, D.; Pande, V.S. Side-chain recognition and gating in the ribosome exit tunnel. PNAS 2008, 105, 16549–16554. [Google Scholar] [CrossRef]
- Sattler, M.; Liang, H.; Nettesheim, D.; Meadows, R.P.; Harlan, J.E.; Eberstadt, M.; Yoon, H.S.; Shuker, S.B.; Chang, B.S.; Minn, A.J.; et al. Structure of Bcl-xL-Bak Peptide Complex: Recognition Between Regulators of Apoptosis. Science 1997, 275, 983–986. [Google Scholar] [CrossRef]
- Marimuthu, P.; Singaravelu, K. Deciphering the crucial residues involved in heterodimerization of Bak peptide and anti-apoptotic proteins for apoptosis. J. Biomol. Struct. Dyn. 2018, 36, 1637–1648. [Google Scholar] [CrossRef]
- Redler, R.L.; Shirvanyants, D.; Dagliyan, O.; Ding, F.; Kim, D.N.; Kota, P.; Proctor, E.A.; Ramachandran, S.; Tandon, A.; Dokholyan, N.V. Computational approaches to understanding protein aggregation in neurodegeneration. J. Mol. Cell Biol. 2014, 6, 104–115. [Google Scholar] [CrossRef]
- Fink, A.L. Natively unfolded proteins. Curr. Opin. Struct. Biol. 2005, 15, 35–41. [Google Scholar] [CrossRef]
- Baker, C.M.; Best, R.B. Insights into the binding of intrinsically disordered proteins from molecular dynamics simulation. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2014, 4, 182–198. [Google Scholar] [CrossRef]
- Straub, J.E.; Thirumalai, D. Principles governing oligomer formation in amyloidogenic peptides. Curr. Opin. Struct. Biol. 2010, 20, 187–195. [Google Scholar] [CrossRef]
- Sipe, J.D.; Benson, M.D.; Buxbaum, J.N.; Ikeda, S.; Merlini, G.; Saraiva, M.J.M.; Westermark, P. Amyloid fibril proteins and amyloidosis: Chemical identification and clinical classification International Society of Amyloidosis 2016 Nomenclature Guidelines. Amyloid 2016, 23, 209–213. [Google Scholar] [CrossRef]
- Lemkul, J.A.; Bevan, D.R. The Role of Molecular Simulations in the Development of Inhibitors of Amyloid β-Peptide Aggregation for the Treatment of Alzheimer’s Disease. ACS Chem. Neurosci. 2012, 3, 845–856. [Google Scholar] [CrossRef]
- Ye, W.; Wang, W.; Jiang, C.; Yu, Q.; Chen, H. Molecular dynamics simulations of amyloid fibrils: An in silico approach. Acta Biochim. Biophys. Sin. 2013, 45, 503–508. [Google Scholar] [CrossRef]
- Nasica-Labouze, J.; Nguyen, P.H.; Sterpone, F.; Berthoumieu, O.; Buchete, N.-V.; Coté, S.; De Simone, A.; Doig, A.J.; Faller, P.; Garcia, A.; et al. Amyloid β Protein and Alzheimer’s Disease: When Computer Simulations Complement Experimental Studies. Chem. Rev. 2015, 115, 3518–3563. [Google Scholar] [CrossRef]
- Coskuner-Weber, O.; Uversky, V.N. Insights into the Molecular Mechanisms of Alzheimer’s and Parkinson’s Diseases with Molecular Simulations: Understanding the Roles of Artificial and Pathological Missense Mutations in Intrinsically Disordered Proteins Related to Pathology. Int. J. Mol. Sci. 2018, 19, 336. [Google Scholar] [CrossRef]
- Bamdad, K.; Naderi-Manesh, H. Contribution of a putative salt bridge and backbone dynamics in the structural instability of human prion protein upon R208H mutation. Biochem. Biophys. Res. Commun. 2007, 364, 719–724. [Google Scholar] [CrossRef]
- Cheng, C.J.; Daggett, V. Different misfolding mechanisms converge on common conformational changes. Prion 2014, 8, 125–135. [Google Scholar] [CrossRef]
- Tao, W.; Yoon, G.; Cao, P.; Eom, K.; Park, H.S. β-sheet-like formation during the mechanical unfolding of prion protein. J. Chem. Phys. 2015, 143, 125101. [Google Scholar] [CrossRef]
- Groveman, B.R.; Kraus, A.; Raymond, L.D.; Dolan, M.A.; Anson, K.J.; Dorward, D.W.; Caughey, B. Charge Neutralization of the Central Lysine Cluster in Prion Protein (PrP) Promotes PrPSc-like Folding of Recombinant PrP Amyloids. J. Biol. Chem. 2015, 290, 1119–1128. [Google Scholar] [CrossRef]
- Xu, L.; Ma, B.; Nussinov, R.; Thompson, D. Familial Mutations May Switch Conformational Preferences in α-Synuclein Fibrils. ACS Chem. Neurosci. 2017, 8, 837–849. [Google Scholar] [CrossRef]
- Reddy, G.; Straub, J.E.; Thirumalai, D. Influence of Preformed Asp23−Lys28 Salt Bridge on the Conformational Fluctuations of Monomers and Dimers of Aβ Peptides with Implications for Rates of Fibril Formation. J. Phys. Chem. B 2009, 113, 1162–1172. [Google Scholar] [CrossRef]
- Barz, B.; Urbanc, B. Dimer Formation Enhances Structural Differences between Amyloid β-Protein (1–40) and (1–42): An Explicit-Solvent Molecular Dynamics Study. PLoS ONE 2012, 7, e34345. [Google Scholar] [CrossRef]
- žganec, M.; Kruczek, N.; Urbanc, B. Amino acid substitutions [K16A] and [K28A] distinctly affect amyloid β-protein oligomerization. J. Biol. Phys. 2016, 42, 453–476. [Google Scholar] [CrossRef]
- Huy, P.D.Q.; Vuong, Q.V.; La Penna, G.; Faller, P.; Li, M.S. Impact of Cu(II) Binding on Structures and Dynamics of Aβ42 Monomer and Dimer: Molecular Dynamics Study. ACS Chem. Neurosci. 2016, 7, 1348–1363. [Google Scholar] [CrossRef]
- Pham, D.Q.H.; Li, M.S.; La Penna, G. Copper Binding Induces Polymorphism in Amyloid-β Peptide: Results of Computational Models. J. Phys. Chem. B 2018, 122, 7243–7252. [Google Scholar] [CrossRef]
- Ono, K.; Takahashi, R.; Ikeda, T.; Yamada, M. Cross-seeding effects of amyloid β-protein and α-synuclein. J. Neurochem. 2012, 122, 883–890. [Google Scholar] [CrossRef]
- Jose, J.C.; Chatterjee, P.; Sengupta, N. Cross Dimerization of Amyloid-β and αSynuclein Proteins in Aqueous Environment: A Molecular Dynamics Simulations Study. PLoS ONE 2014, 9, e106883. [Google Scholar] [CrossRef]
- Atsmon-Raz, Y.; Miller, Y. Non-Amyloid-β Component of Human α-Synuclein Oligomers Induces Formation of New Aβ Oligomers: Insight into the Mechanisms That Link Parkinson’s and Alzheimer’s Diseases. ACS Chem. Neurosci. 2016, 7, 46–55. [Google Scholar] [CrossRef]
- Tsigelny, I.F.; Bar-On, P.; Sharikov, Y.; Crews, L.; Hashimoto, M.; Miller, M.A.; Keller, S.H.; Platoshyn, O.; Yuan, J.X.-J.; Masliah, E. Dynamics of α-synuclein aggregation and inhibition of pore-like oligomer development by β-synuclein. FEBS J. 2007, 274, 1862–1877. [Google Scholar] [CrossRef]
- Muronetz, V.I.; Melnikova, A.K.; Seferbekova, Z.N.; Barinova, K.V.; Schmalhausen, E.V. Glycation, glycolysis, and neurodegenerative diseases: Is there any connection? Biochem. Mosc. 2017, 82, 874–886. [Google Scholar] [CrossRef]
- Huang, J.; Rauscher, S.; Nawrocki, G.; Ran, T.; Feig, M.; de Groot, B.L.; Grubmüller, H.; MacKerell, A.D., Jr. CHARMM36m: An improved force field for folded and intrinsically disordered proteins. Nat. Methods 2017, 14, 71–73. [Google Scholar] [CrossRef]
- Song, D.; Luo, R.; Chen, H.-F. IDP-Specific Force Field ff14IDPSFF Improves the Conformer Sampling of Intrinsically Disordered Proteins. J. Chem. Inf. Model. 2017, 57, 1166–1178. [Google Scholar] [CrossRef]
- Robustelli, P.; Piana, S.; Shaw, D.E. Developing a molecular dynamics force field for both folded and disordered protein states. Proc. Natl. Acad. Sci. USA 2018, 115, E4758–E4766. [Google Scholar] [CrossRef]
- Johnson, L.N. The regulation of protein phosphorylation. Biochem. Soc. Trans. 2009, 37, 627–641. [Google Scholar] [CrossRef]
- Johnson, L.N.; Barford, D. The Effects of Phosphorylation on the Structure and Function of Proteins. Annu. Rev. Biophys. Biomol. Struct. 1993, 22, 199–232. [Google Scholar] [CrossRef]
- Nishi, H.; Shaytan, A.; Panchenko, A.R. Physicochemical mechanisms of protein regulation by phosphorylation. Front. Genet. 2014, 5. [Google Scholar] [CrossRef]
- Audagnotto, M.; Dal Peraro, M. Protein post-translational modifications: In silico prediction tools and molecular modeling. Comput. Struct. Biotechnol. J. 2017, 15, 307–319. [Google Scholar] [CrossRef]
- Dodson, G.G.; Lane, D.P.; Verma, C.S. Molecular simulations of protein dynamics: New windows on mechanisms in biology. EMBO Rep. 2008, 9, 144–150. [Google Scholar] [CrossRef]
- Narayanan, A.; Jacobson, M.P. Computational studies of protein regulation by post-translational phosphorylation. Curr. Opin. Struct. Biol. 2009, 19, 156–163. [Google Scholar] [CrossRef]
- Polyansky, A.A.; Zagrovic, B. Protein Electrostatic Properties Predefining the Level of Surface Hydrophobicity Change upon Phosphorylation. J. Phys. Chem. Lett. 2012, 3, 973–976. [Google Scholar] [CrossRef]
- Lee, H.J.; Srinivasan, D.; Coomber, D.; Lane, D.P.; Verma, C.S. Modulation of the p53-MDM2 Interaction by Phosphorylation of Thr18: A Computational Study. Cell Cycle 2007, 6, 2604–2611. [Google Scholar] [CrossRef] [PubMed]
- Pantano, S.; Carafoli, E. The role of phosphorylation on the structure and dynamics of phospholamban: A model from molecular simulations. Proteins Struct. Funct. Bioinform. 2007, 66, 930–940. [Google Scholar] [CrossRef] [PubMed]
- Sugita, Y.; Miyashita, N.; Yoda, T.; Ikeguchi, M.; Toyoshima, C. Structural Changes in the Cytoplasmic Domain of Phospholamban by Phosphorylation at Ser16: A Molecular Dynamics Study. Biochemistry 2006, 45, 11752–11761. [Google Scholar] [CrossRef] [PubMed]
- Homouz, D.; Joyce-Tan, K.H.; ShahirShamsir, M.; Moustafa, I.M.; Idriss, H.T. Molecular dynamics simulations suggest changes in electrostatic interactions as a potential mechanism through which serine phosphorylation inhibits DNA polymerase β activity. J. Mol. Graph. Model. 2018, 84, 236–241. [Google Scholar] [CrossRef] [PubMed]
- Meng, Y.; Roux, B. Locking the Active Conformation of c-Src Kinase through the Phosphorylation of the Activation Loop. J. Mol. Biol. 2014, 426, 423–435. [Google Scholar] [CrossRef] [PubMed]
- Espinoza-Fonseca, L.M.; Kast, D.; Thomas, D.D. Molecular Dynamics Simulations Reveal a Disorder-to-Order Transition on Phosphorylation of Smooth Muscle Myosin. Biophys. J. 2007, 93, 2083–2090. [Google Scholar] [CrossRef] [PubMed]
- Suenaga, A.; Kiyatkin, A.B.; Hatakeyama, M.; Futatsugi, N.; Okimoto, N.; Hirano, Y.; Narumi, T.; Kawai, A.; Susukita, R.; Koishi, T.; et al. Tyr-317 Phosphorylation Increases Shc Structural Rigidity and Reduces Coupling of Domain Motions Remote from the Phosphorylation Site as Revealed by Molecular Dynamics Simulations. J. Biol. Chem. 2004, 279, 4657–4662. [Google Scholar] [CrossRef] [PubMed]
- Oliveira, J.; Costa, M.; de Almeida, M.S.C.; da Cruz e Silva, O.A.B.; Henriques, A.G. Protein Phosphorylation is a Key Mechanism in Alzheimer’s Disease. J. Alzheimer’s Dis. 2017, 58, 953–978. [Google Scholar] [CrossRef] [PubMed]
- Lyons, A.J.; Gandhi, N.S.; Mancera, R.L. Molecular dynamics simulation of the phosphorylation-induced conformational changes of a tau peptide fragment. Proteins Struct. Funct. Bioinform. 2014, 82, 1907–1923. [Google Scholar] [CrossRef] [PubMed]
- Bomblies, R.; Luitz, M.P.; Zacharias, M. Molecular Dynamics Analysis of 4E-BP2 Protein Fold Stabilization Induced by Phosphorylation. J. Phys. Chem. B 2017, 121, 3387–3393. [Google Scholar] [CrossRef] [PubMed]
- Ouyang, Y.; Lane, W.S.; Moore, K.L. Tyrosylprotein sulfotransferase: Purification and molecular cloning of an enzyme that catalyzes tyrosine O-sulfation, a common posttranslational modification of eukaryotic proteins. Proc. Natl. Acad. Sci. USA 1998, 95, 2896–2901. [Google Scholar] [CrossRef] [PubMed]
- Medzihradszky, K.F.; Darula, Z.; Perlson, E.; Fainzilber, M.; Chalkley, R.J.; Ball, H.; Greenbaum, D.; Bogyo, M.; Tyson, D.R.; Bradshaw, R.A.; et al. O-Sulfonation of Serine and Threonine Mass Spectrometric Detection and Characterization of a New Posttranslational Modification in Diverse Proteins Throughout the Eukaryotes. Mol. Cell Proteom. 2004, 3, 429–440. [Google Scholar] [CrossRef]
- Semenyuk, P.I.; Muronetz, V.I.; Haertlé, T.; Izumrudov, V.A. Effect of poly(phosphate) anions on glyceraldehyde-3-phosphate dehydrogenase structure and thermal aggregation: Comparison with influence of poly(sulfoanions). Biochim. Biophys. Acta 2013, 1830, 4800–4805. [Google Scholar] [CrossRef] [PubMed]
- Huttner, W.B. Tyrosine sulfation and the secretory pathway. Annu. Rev. Physiol. 1988, 50, 363–376. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.-S.; Wang, C.-C.; Chen, B.-H.; Hou, Y.-H.; Hung, K.-S.; Mao, Y.-C. Tyrosine Sulfation as a Protein Post-Translational Modification. Molecules 2015, 20, 2138–2164. [Google Scholar] [CrossRef] [PubMed]
- Cimbro, R.; Peterson, F.C.; Liu, Q.; Guzzo, C.; Zhang, P.; Miao, H.; Van Ryk, D.; Ambroggio, X.; Hurt, D.E.; De Gioia, L.; et al. Tyrosine-sulfated V2 peptides inhibit HIV-1 infection via coreceptor mimicry. EBioMedicine 2016, 10, 45–54. [Google Scholar] [CrossRef] [PubMed]
- Rapp, C.; Klerman, H.; Levine, E.; McClendon, C.L. Hydrogen Bond Strengths in Phosphorylated and Sulfated Amino Acid Residues. PLoS ONE 2013, 8, e57804. [Google Scholar] [CrossRef]
- Rapp, C.; Snow, S.; Laufer, T.; McClendon, C.L. The role of tyrosine sulfation in the dimerization of the CXCR4:SDF-1 complex. Protein Sci. 2013, 22, 1025–1036. [Google Scholar] [CrossRef]
- Miyanabe, K.; Yamashita, T.; Abe, Y.; Akiba, H.; Takamatsu, Y.; Nakakido, M.; Hamakubo, T.; Ueda, T.; Caaveiro, J.M.M.; Tsumoto, K. Tyrosine Sulfation Restricts the Conformational Ensemble of a Flexible Peptide, Strengthening the Binding Affinity for an Antibody. Biochemistry 2018, 57, 4177–4185. [Google Scholar] [CrossRef]
- Singh, R.; Barden, A.; Mori, T.; Beilin, L. Advanced glycation end-products: A review. Diabetologia 2001, 44, 129–146. [Google Scholar] [CrossRef]
- Šebeková, K.; Somoza, V. Dietary advanced glycation endproducts (AGEs) and their health effects—PRO. Mol. Nutr. Food Res. 2007, 51, 1079–1084. [Google Scholar] [CrossRef]
- Brownlee, M. Biochemistry and molecular cell biology of diabetic complications. Nature 2001, 414, 813–820. [Google Scholar] [CrossRef]
- Li, J.; Liu, D.; Sun, L.; Lu, Y.; Zhang, Z. Advanced glycation end products and neurodegenerative diseases: Mechanisms and perspective. J. Neurol. Sci. 2012, 317, 1–5. [Google Scholar] [CrossRef]
- Muronetz, V.I.; Barinova, K.V.; Stroylova, Y.Y.; Semenyuk, P.I.; Schmalhausen, E.V. Glyceraldehyde-3-phosphate dehydrogenase: Aggregation mechanisms and impact on amyloid neurodegenerative diseases. Int. J. Biol. Macromol. 2017, 100, 55–66. [Google Scholar] [CrossRef]
- Sofronova, A.; Semenyuk, P.; Muronetz, V. The influence of β-casein glycation on its interaction with natural and synthetic polyelectrolytes. Food Hydrocoll. 2019, 89, 425–433. [Google Scholar] [CrossRef]
- Yang, Y.; Song, W. Molecular links between Alzheimer’s disease and diabetes mellitus. Neuroscience 2013, 250, 140–150. [Google Scholar] [CrossRef]
- Miranda, H.V.; El-Agnaf, O.M.A.; Outeiro, T.F. Glycation in Parkinson’s disease and Alzheimer’s disease. Mov. Disord. 2016, 31, 782–790. [Google Scholar] [CrossRef]
- Nasiri, R.; Bahrami, H.; Zahedi, M.; Moosavi-Movahedi, A.A.; Sattarahmady, N. A Theoretical Elucidation of Glucose Interaction with HSA’s Domains. J. Biomol. Struct. Dyn. 2010, 28, 211–226. [Google Scholar] [CrossRef]
- Awang, T.; Wiriyatanakorn, N.; Saparpakorn, P.; Japrung, D.; Pongprayoon, P. Understanding the effects of two bound glucose in Sudlow site I on structure and function of human serum albumin: Theoretical studies. J. Biomol. Struct. Dyn. 2017, 35, 781–790. [Google Scholar] [CrossRef]
- Pongprayoon, P.; Mori, T. The critical role of dimer formation in monosaccharides binding to human serum albumin. Phys. Chem. Chem. Phys. 2018, 20, 3249–3257. [Google Scholar] [CrossRef]
- Abidi, M.; Khan, M.S.; Ahmad, S.; Kausar, T.; Nayeem, S.M.; Islam, S.; Ali, A.; Alam, K. Moinuddin Biophysical and biochemical studies on glycoxidatively modified human low density lipoprotein. Arch. Biochem. Biophys. 2018, 645, 87–99. [Google Scholar] [CrossRef] [PubMed]
- Crabbe, M.J.C.; Cooper, L.R.; Corne, D.W. Use of essential and molecular dynamics to study γB-crystallin unfolding after non-enzymic post-translational modifications. Comput. Biol. Chem. 2003, 27, 507–510. [Google Scholar] [CrossRef]
- Saleem, A.; Azam, S.S.; Zarina, S. Docking and molecular dynamics simulation studies on glycation-induced conformational changes of human paraoxonase 1. Eur. Biophys J. 2012, 41, 241–248. [Google Scholar] [CrossRef] [PubMed]
- Gawad, A.E.-D.A. Hybrid QM/MM and classical molecular dynamics simulation of amadori product in γB-crystallin. Life Sci. J. 2013, 10, 1923–1932. [Google Scholar]
- Alizadeh-Rahrovi, J.; Shayesteh, A.; Ebrahim-Habibi, A. Structural stability of myoglobin and glycomyoglobin: A comparative molecular dynamics simulation study. J. Biol. Phys. 2015, 41, 349–366. [Google Scholar] [CrossRef] [PubMed]
- Awasthi, S.; Murugan, N.A.; Saraswathi, N.T. Advanced Glycation End Products Modulate Structure and Drug Binding Properties of Albumin. Mol. Pharm. 2015, 12, 3312–3322. [Google Scholar] [CrossRef] [PubMed]
- Yesudasan, S.; Wang, X.; Averett, R.D. Molecular dynamics simulations indicate that deoxyhemoglobin, oxyhemoglobin, carboxyhemoglobin, and glycated hemoglobin under compression and shear exhibit an anisotropic mechanical behavior. J. Biomol. Struct. Dyn. 2018, 36, 1417–1429. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, N.; Dobler, D.; Dean, M.; Thornalley, P.J. Peptide Mapping Identifies Hotspot Site of Modification in Human Serum Albumin by Methylglyoxal Involved in Ligand Binding and Esterase Activity. J. Biol. Chem. 2005, 280, 5724–5732. [Google Scholar] [CrossRef] [PubMed]
- De Rosa, M.C.; Sanna, M.T.; Messana, I.; Castagnola, M.; Galtieri, A.; Tellone, E.; Scatena, R.; Botta, B.; Botta, M.; Giardina, B. Glycated human hemoglobin (HbA1c): Functional characteristics and molecular modeling studies. Biophys. Chem. 1998, 72, 323–335. [Google Scholar] [CrossRef]
- Silva, A.M.N.; Sousa, P.R.H.; Coimbra, J.T.S.; Brás, N.F.; Vitorino, R.; Fernandes, P.A.; Ramos, M.J.; Rangel, M.; Domingues, P. The glycation site specificity of human serum transferrin is a determinant for transferrin’s functional impairment under elevated glycaemic conditions. Biochem. J. 2014, 461, 33–42. [Google Scholar] [CrossRef] [PubMed]
- Collier, T.A.; Nash, A.; Birch, H.L.; de Leeuw, N.H. Intra-molecular lysine-arginine derived advanced glycation end-product cross-linking in Type I collagen: A molecular dynamics simulation study. Biophys. Chem. 2016, 218, 42–46. [Google Scholar] [CrossRef] [PubMed]
- Johansen, M.B.; Kiemer, L.; Brunak, S. Analysis and prediction of mammalian protein glycation. Glycobiology 2006, 16, 844–853. [Google Scholar] [CrossRef] [PubMed]
- Jana, A.K.; Batkulwar, K.B.; Kulkarni, M.J.; Sengupta, N. Glycation induces conformational changes in the amyloid-β peptide and enhances its aggregation propensity: Molecular insights. Phys. Chem. Chem. Phys. 2016, 18, 31446–31458. [Google Scholar] [CrossRef] [PubMed]
- Semenyuk, P.; Barinova, K.; Muronetz, V. Glycation of α-synuclein amplifies the binding with glyceraldehyde-3-phosphate dehydrogenase. Int. J. Biol. Macromol. 2019, 127, 278–285. [Google Scholar] [CrossRef] [PubMed]
- Barford, D. The role of cysteine residues as redox-sensitive regulatory switches. Curr. Opin. Struct. Biol. 2004, 14, 679–686. [Google Scholar] [CrossRef] [PubMed]
- Jacob, C.; Battaglia, E.; Burkholz, T.; Peng, D.; Bagrel, D.; Montenarh, M. Control of Oxidative Posttranslational Cysteine Modifications: From Intricate Chemistry to Widespread Biological and Medical Applications. Chem. Res. Toxicol. 2012, 25, 588–604. [Google Scholar] [CrossRef] [PubMed]
- Chung, H.S.; Wang, S.B.; Venkatraman, V.; Murray, C.I.; Van Eyk, J.E. Cysteine Oxidative Posttranslational Modifications. Circ. Res. 2013, 112, 382–392. [Google Scholar] [CrossRef] [PubMed]
- Klomsiri, C.; Karplus, P.A.; Poole, L.B. Cysteine-Based Redox Switches in Enzymes. Antioxid. Redox Signal. 2011, 14, 1065–1077. [Google Scholar] [CrossRef] [PubMed]
- Arutyunova, E.I.; Danshina, P.V.; Domnina, L.V.; Pleten, A.P.; Muronetz, V.I. Oxidation of glyceraldehyde-3-phosphate dehydrogenase enhances its binding to nucleic acids. Biochem. Biophys. Res. Commun. 2003, 307, 547–552. [Google Scholar] [CrossRef]
- Paulech, J.; Liddy, K.A.; Engholm-Keller, K.; White, M.Y.; Cordwell, S.J. Global Analysis of Myocardial Peptides Containing Cysteines with Irreversible Sulfinic and Sulfonic Acid Post-Translational Modifications. Mol. Cell. Proteom. 2015, 14, 609–620. [Google Scholar] [CrossRef] [PubMed]
- Truong, T.H.; Ung, P.M.-U.; Palde, P.B.; Paulsen, C.E.; Schlessinger, A.; Carroll, K.S. Molecular Basis for Redox Activation of Epidermal Growth Factor Receptor Kinase. Cell Chem. Biol. 2016, 23, 837–848. [Google Scholar] [CrossRef] [PubMed]
- Petrov, D.; Daura, X.; Zagrovic, B. Effect of Oxidative Damage on the Stability and Dimerization of Superoxide Dismutase 1. Biophys. J. 2016, 110, 1499–1509. [Google Scholar] [CrossRef] [PubMed]
- Kiss, R.; Zhu, M.; Jójárt, B.; Czajlik, A.; Solti, K.; Fórizs, B.; Nagy, É.; Zsila, F.; Beke-Somfai, T.; Tóth, G. Structural features of human DJ-1 in distinct Cys106 oxidative states and their relevance to its loss of function in disease. Biochim. Et Biophys. Acta (Bba) Gen. Subj. 2017, 1861, 2619–2629. [Google Scholar] [CrossRef] [PubMed]
- Zeida, A.; Guardia, C.M.; Lichtig, P.; Perissinotti, L.L.; Defelipe, L.A.; Turjanski, A.; Radi, R.; Trujillo, M.; Estrin, D.A. Thiol redox biochemistry: Insights from computer simulations. Biophys. Rev. 2014, 6, 27–46. [Google Scholar] [CrossRef]



© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Semenyuk, P.; Muronetz, V. Protein Interaction with Charged Macromolecules: From Model Polymers to Unfolded Proteins and Post-Translational Modifications. Int. J. Mol. Sci. 2019, 20, 1252. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20051252
Semenyuk P, Muronetz V. Protein Interaction with Charged Macromolecules: From Model Polymers to Unfolded Proteins and Post-Translational Modifications. International Journal of Molecular Sciences. 2019; 20(5):1252. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20051252
Chicago/Turabian StyleSemenyuk, Pavel, and Vladimir Muronetz. 2019. "Protein Interaction with Charged Macromolecules: From Model Polymers to Unfolded Proteins and Post-Translational Modifications" International Journal of Molecular Sciences 20, no. 5: 1252. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20051252