Biological Rationale for Targeting MEK/ERK Pathways in Anti-Cancer Therapy and to Potentiate Tumour Responses to Radiation

{kind=link}
{kind=link}
Abstract
:1. Introduction
2. ERK1/2 Activation
3. ERK1 and ERK2: Distinct Functions or Functional Redundancy
4. ERK1/2 Substrates
5. ERK1/2 Mutants
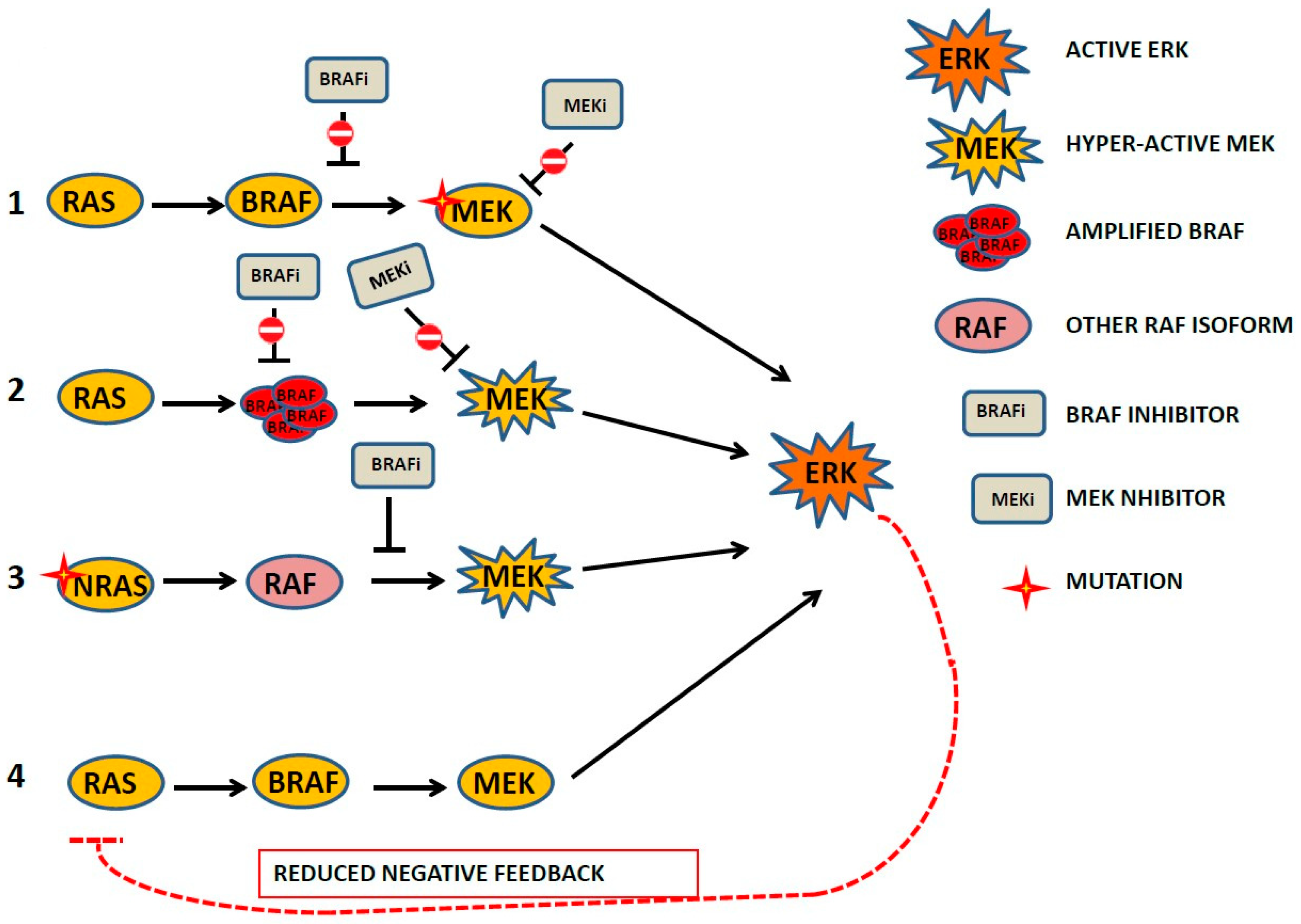
6. MEK/ERK Inhibition and ERK Reactivation
7. Impact of Mutant ERK1/2 on Anticancer Therapy Outcome
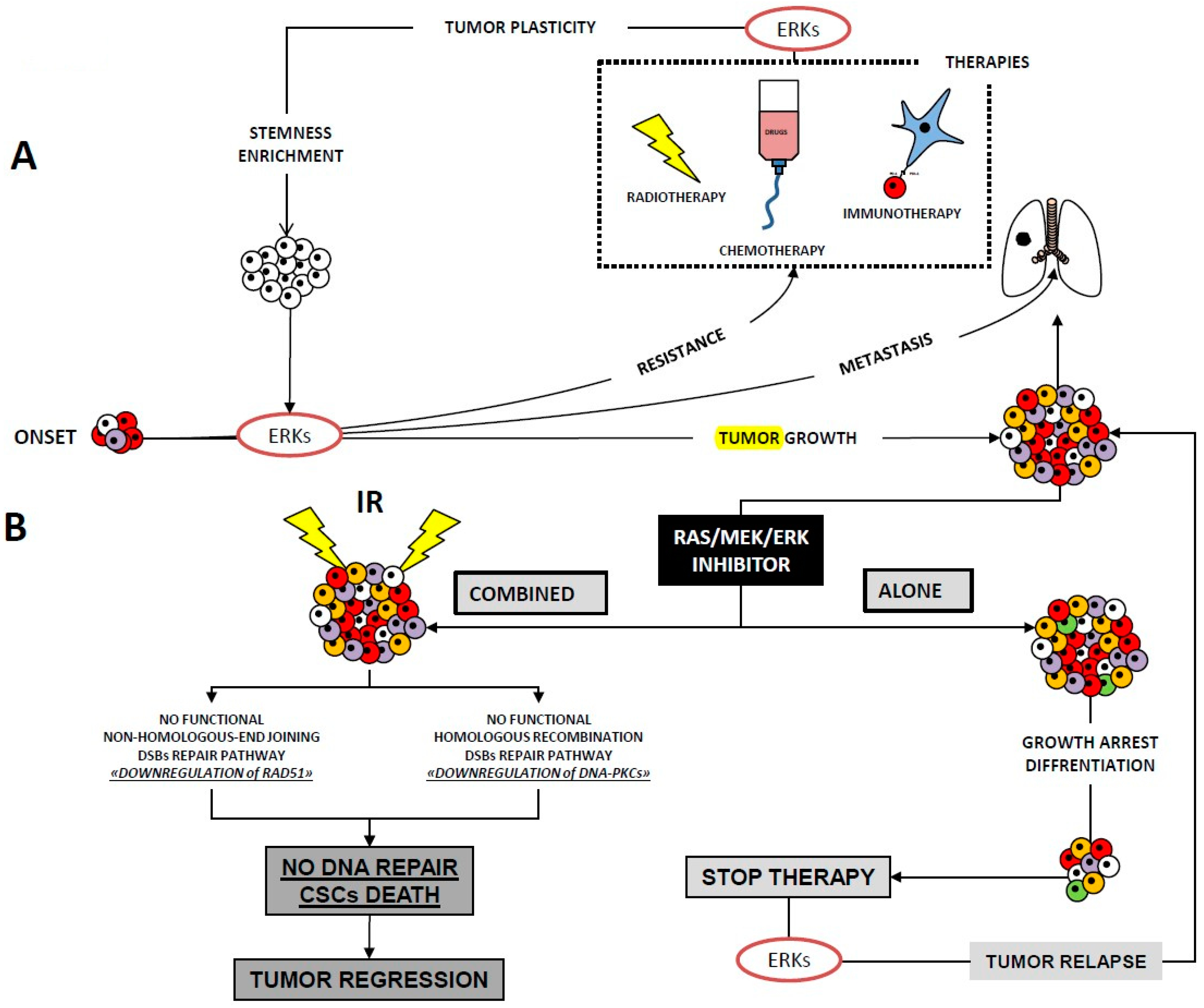
8. The Role of ERK1/2 Signalling in Cancer Cell Radioresistance
9. Concluding Remarks and Future Perspectives
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| ERK | extracellular regulated kinase |
| MAPK | mitogen-activated protein kinase |
| MEK | component of MAPK core |
| PARP | poly (ADP.ribose)polymerase |
| JNK | c.Jun N-terminal kinase |
References
- Roskoski, R., Jr. ERK1/2 MAP kinases: Structure, function, and regulation. Pharmacol. Res. 2012, 66, 105–143. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.K.; Choi, E.J. Pathological roles of MAPK signaling pathways in human diseases. Biochim. Biophys. Acta 2010, 1802, 396–405. [Google Scholar] [CrossRef] [Green Version]
- Eblen, S.T. Extracellular-Regulated Kinases: Signaling From Ras to ERK Substrates to Control Biological Outcomes. Adv. Cancer Res. 2018, 138, 99–142. [Google Scholar] [PubMed]
- Morrison, D.K.; Davis, R.J. Regulation of MAP kinase signaling modules by scaffold proteins in mammals. Annu. Rev. Cell Dev. Biol. 2003, 19, 91–118. [Google Scholar] [CrossRef] [PubMed]
- Kung, J.E.; Jura, N. Structural Basis for the Non-catalytic Functions of Protein Kinases. Structure 2016, 24, 7–24. [Google Scholar] [CrossRef] [Green Version]
- Malumbres, M.; Barbacid, M. RAS oncogenes: The first 30 years. Nat. Rev. Cancer 2003, 3, 459–465. [Google Scholar] [CrossRef]
- Fernandez-Medarde, A.; Santos, E. Ras in cancer and developmental diseases. Genes Cancer 2011, 2, 344–358. [Google Scholar] [CrossRef] [PubMed]
- Davies, H.; Bignell, G.R.; Cox, C.; Stephens, P.; Edkins, S.; Clegg, S.; Teague, J.; Woffendin, H.; Garnett, M.J.; Bottomley, W.; et al. Mutations of the BRAF gene in human cancer. Nature 2002, 417, 949–954. [Google Scholar] [CrossRef] [PubMed]
- Harding, A.; Tian, T.; Westbury, E.; Frische, E.; Hancock, J.F. Subcellular localization determines MAP kinase signal output. Curr. Biol. 2005, 15, 869–873. [Google Scholar] [CrossRef]
- Bivona, T.G.; Philips, M.R. Ras pathway signaling on endomembranes. Curr. Opin. Cell Biol. 2003, 15, 136–142. [Google Scholar] [CrossRef]
- Teis, D.; Wunderlich, W.; Huber, L.A. Localization of the MP1-MAPK scaffold complex to endosomes is mediated by p14 and required for signal transduction. Dev. Cell 2002, 3, 803–814. [Google Scholar] [CrossRef]
- Ogata, T.; Naito, D.; Nakanishi, N.; Hayashi, Y.K.; Taniguchi, T.; Miyagawa, K.; Hamaoka, T.; Maruyama, N.; Matoba, S.; Ikeda, K.; et al. MURC/Cavin-4 facilitates recruitment of ERK to caveolae and concentric cardiac hypertrophy induced by alpha1-adrenergic receptors. Proc. Natl. Acad. Sci. USA 2014, 111, 3811–3816. [Google Scholar] [CrossRef] [PubMed]
- Codenotti, S.; Faggi, F.; Ronca, R.; Chiodelli, P.; Grillo, E.; Guescini, M.; Megiorni, F.; Marampon, F.; Fanzani, A. Caveolin-1 enhances metastasis formation in a human model of embryonal rhabdomyosarcoma through Erk signaling cooperation. Cancer Lett. 2019, 449, 135–144. [Google Scholar] [CrossRef]
- Faggi, F.; Chiarelli, N.; Colombi, M.; Mitola, S.; Ronca, R.; Madaro, L.; Bouche, M.; Poliani, P.L.; Vezzoli, M.; Longhena, F.; et al. Cavin-1 and Caveolin-1 are both required to support cell proliferation, migration and anchorage-independent cell growth in rhabdomyosarcoma. Lab. Investig. 2015, 95, 585–602. [Google Scholar] [CrossRef] [Green Version]
- Ramos, J.W. The regulation of extracellular signal-regulated kinase (ERK) in mammalian cells. Int. J. Biochem. Cell Biol. 2008, 40, 2707–2719. [Google Scholar] [CrossRef] [PubMed]
- Leicht, D.T.; Balan, V.; Kaplun, A.; Singh-Gupta, V.; Kaplun, L.; Dobson, M.; Tzivion, G. Raf kinases: Function, regulation and role in human cancer. Biochim. Biophys. Acta 2007, 1773, 1196–1212. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wellbrock, C.; Karasarides, M.; Marais, R. The RAF proteins take centre stage. Nat. Rev. Mol. Cell Biol. 2004, 5, 875–885. [Google Scholar] [CrossRef]
- Frost, J.A.; Steen, H.; Shapiro, P.; Lewis, T.; Ahn, N.; Shaw, P.E.; Cobb, M.H. Cross-cascade activation of ERKs and ternary complex factors by Rho family proteins. EMBO J. 1997, 16, 6426–6438. [Google Scholar] [CrossRef] [PubMed]
- Frost, J.A.; Xu, S.; Hutchison, M.R.; Marcus, S.; Cobb, M.H. Actions of Rho family small G proteins and p21-activated protein kinases on mitogen-activated protein kinase family members. Mol. Cell Biol. 1996, 16, 3707–3713. [Google Scholar] [CrossRef] [Green Version]
- Unal, E.B.; Uhlitz, F.; Bluthgen, N. A compendium of ERK targets. FEBS Lett. 2017, 591, 2607–2615. [Google Scholar] [CrossRef] [Green Version]
- Warn-Cramer, B.J.; Cottrell, G.T.; Burt, J.M.; Lau, A.F. Regulation of connexin-43 gap junctional intercellular communication by mitogen-activated protein kinase. J. Biol. Chem. 1998, 273, 9188–9196. [Google Scholar] [CrossRef]
- Warn-Cramer, B.J.; Lampe, P.D.; Kurata, W.E.; Kanemitsu, M.Y.; Loo, L.W.; Eckhart, W.; Lau, A.F. Characterization of the mitogen-activated protein kinase phosphorylation sites on the connexin-43 gap junction protein. J. Biol. Chem. 1996, 271, 3779–3786. [Google Scholar] [CrossRef]
- Klemke, R.L.; Cai, S.; Giannini, A.L.; Gallagher, P.J.; de Lanerolle, P.; Cheresh, D.A. Regulation of cell motility by mitogen-activated protein kinase. J. Cell Biol. 1997, 137, 481–492. [Google Scholar] [CrossRef]
- Shapiro, P.S.; Whalen, A.M.; Tolwinski, N.S.; Wilsbacher, J.; Froelich-Ammon, S.J.; Garcia, M.; Osheroff, N.; Ahn, N.G. Extracellular signal-regulated kinase activates topoisomerase IIalpha through a mechanism independent of phosphorylation. Mol. Cell Biol. 1999, 19, 3551–3560. [Google Scholar] [CrossRef]
- Cohen-Armon, M.; Visochek, L.; Rozensal, D.; Kalal, A.; Geistrikh, I.; Klein, R.; Bendetz-Nezer, S.; Yao, Z.; Seger, R. DNA-independent PARP-1 activation by phosphorylated ERK2 increases Elk1 activity: A link to histone acetylation. Mol. Cell 2007, 25, 297–308. [Google Scholar] [CrossRef]
- Rodriguez, J.; Calvo, F.; Gonzalez, J.M.; Casar, B.; Andres, V.; Crespo, P. ERK1/2 MAP kinases promote cell cycle entry by rapid, kinase-independent disruption of retinoblastoma-lamin A complexes. J. Cell Biol. 2010, 191, 967–979. [Google Scholar] [CrossRef]
- Camps, M.; Nichols, A.; Gillieron, C.; Antonsson, B.; Muda, M.; Chabert, C.; Boschert, U.; Arkinstall, S. Catalytic activation of the phosphatase MKP-3 by ERK2 mitogen-activated protein kinase. Science 1998, 280, 1262–1265. [Google Scholar] [CrossRef]
- Hu, S.; Xie, Z.; Onishi, A.; Yu, X.; Jiang, L.; Lin, J.; Rho, H.S.; Woodard, C.; Wang, H.; Jeong, J.S.; et al. Profiling the human protein-DNA interactome reveals ERK2 as a transcriptional repressor of interferon signaling. Cell 2009, 139, 610–622. [Google Scholar] [CrossRef]
- Fan, L.; Yang, X.; Du, J.; Marshall, M.; Blanchard, K.; Ye, X. A novel role of p38 alpha MAPK in mitotic progression independent of its kinase activity. Cell Cycle 2005, 4, 1616–1624. [Google Scholar] [CrossRef]
- Saba-El-Leil, M.K.; Vella, F.D.; Vernay, B.; Voisin, L.; Chen, L.; Labrecque, N.; Ang, S.L.; Meloche, S. An essential function of the mitogen-activated protein kinase Erk2 in mouse trophoblast development. EMBO Rep. 2003, 4, 964–968. [Google Scholar] [CrossRef]
- Hatano, N.; Mori, Y.; Oh-hora, M.; Kosugi, A.; Fujikawa, T.; Nakai, N.; Niwa, H.; Miyazaki, J.; Hamaoka, T.; Ogata, M. Essential role for ERK2 mitogen-activated protein kinase in placental development. Genes Cells 2003, 8, 847–856. [Google Scholar] [CrossRef]
- Nekrasova, T.; Shive, C.; Gao, Y.; Kawamura, K.; Guardia, R.; Landreth, G.; Forsthuber, T.G. ERK1-deficient mice show normal T cell effector function and are highly susceptible to experimental autoimmune encephalomyelitis. J. Immunol. 2005, 175, 2374–2380. [Google Scholar] [CrossRef]
- Fischer, A.M.; Katayama, C.D.; Pages, G.; Pouyssegur, J.; Hedrick, S.M. The role of erk1 and erk2 in multiple stages of T cell development. Immunity 2005, 23, 431–443. [Google Scholar] [CrossRef]
- Mazzucchelli, C.; Vantaggiato, C.; Ciamei, A.; Fasano, S.; Pakhotin, P.; Krezel, W.; Welzl, H.; Wolfer, D.P.; Pages, G.; Valverde, O.; et al. Knockout of ERK1 MAP kinase enhances synaptic plasticity in the striatum and facilitates striatal-mediated learning and memory. Neuron 2002, 34, 807–820. [Google Scholar] [CrossRef]
- Fremin, C.; Saba-El-Leil, M.K.; Levesque, K.; Ang, S.L.; Meloche, S. Functional Redundancy of ERK1 and ERK2 MAP Kinases during Development. Cell Rep. 2015, 12, 913–921. [Google Scholar] [CrossRef] [Green Version]
- Shin, S.; Dimitri, C.A.; Yoon, S.O.; Dowdle, W.; Blenis, J. ERK2 but not ERK1 induces epithelial-to-mesenchymal transformation via DEF motif-dependent signaling events. Mol. Cell 2010, 38, 114–127. [Google Scholar] [CrossRef]
- Shin, S.; Buel, G.R.; Nagiec, M.J.; Han, M.J.; Roux, P.P.; Blenis, J.; Yoon, S.O. ERK2 regulates epithelial-to-mesenchymal plasticity through DOCK10-dependent Rac1/FoxO1 activation. Proc. Natl. Acad. Sci. USA 2019, 116, 2967–2976. [Google Scholar] [CrossRef] [Green Version]
- Mani, S.A.; Guo, W.; Liao, M.J.; Eaton, E.N.; Ayyanan, A.; Zhou, A.Y.; Brooks, M.; Reinhard, F.; Zhang, C.C.; Shipitsin, M.; et al. The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell 2008, 133, 704–715. [Google Scholar] [CrossRef]
- Goke, J.; Chan, Y.S.; Yan, J.; Vingron, M.; Ng, H.H. Genome-wide kinase-chromatin interactions reveal the regulatory network of ERK signaling in human embryonic stem cells. Mol. Cell 2013, 50, 844–855. [Google Scholar] [CrossRef]
- Busca, R.; Pouyssegur, J.; Lenormand, P. ERK1 and ERK2 Map Kinases: Specific Roles or Functional Redundancy? Front. Cell Dev. Biol. 2016, 4, 53. [Google Scholar] [CrossRef] [Green Version]
- Von Thun, A.; Birtwistle, M.; Kalna, G.; Grindlay, J.; Strachan, D.; Kolch, W.; von Kriegsheim, A.; Norman, J.C. ERK2 drives tumour cell migration in three-dimensional microenvironments by suppressing expression of Rab17 and liprin-beta2. J. Cell Sci. 2012, 125, 1465–1477. [Google Scholar] [CrossRef]
- Kosako, H.; Yamaguchi, N.; Aranami, C.; Ushiyama, M.; Kose, S.; Imamoto, N.; Taniguchi, H.; Nishida, E.; Hattori, S. Phosphoproteomics reveals new ERK MAP kinase targets and links ERK to nucleoporin-mediated nuclear transport. Nat. Struct. Mol. Biol. 2009, 16, 1026–1035. [Google Scholar] [CrossRef]
- Yoon, S.; Seger, R. The extracellular signal-regulated kinase: Multiple substrates regulate diverse cellular functions. Growth Factors 2006, 24, 21–44. [Google Scholar] [CrossRef]
- Farrell, A.S.; Sears, R.C. MYC degradation. Cold Spring Harb. Perspect. Med. 2014, 4, a014365. [Google Scholar] [CrossRef]
- Gregory, M.A.; Hann, S.R. c-Myc proteolysis by the ubiquitin-proteasome pathway: Stabilization of c-Myc in Burkitt’s lymphoma cells. Mol. Cell Biol. 2000, 20, 2423–2435. [Google Scholar] [CrossRef]
- Yohe, M.E.; Gryder, B.E.; Shern, J.F.; Song, Y.K.; Chou, H.C.; Sindiri, S.; Mendoza, A.; Patidar, R.; Zhang, X.; Guha, R.; et al. MEK inhibition induces MYOG and remodels super-enhancers in RAS-driven rhabdomyosarcoma. Sci. Transl. Med. 2018, 10, eaan4470. [Google Scholar] [CrossRef] [Green Version]
- Miner, J.H.; Wold, B.J. c-myc inhibition of MyoD and myogenin-initiated myogenic differentiation. Mol. Cell Biol. 1991, 11, 2842–2851. [Google Scholar] [CrossRef]
- La Rocca, S.A.; Crouch, D.H.; Gillespie, D.A. c-Myc inhibits myogenic differentiation and myoD expression by a mechanism which can be dissociated from cell transformation. Oncogene 1994, 9, 3499–3508. [Google Scholar]
- Falcone, G.; Tato, F.; Alema, S. Distinctive effects of the viral oncogenes myc, erb, fps, and src on the differentiation program of quail myogenic cells. Proc. Natl. Acad. Sci. USA 1985, 82, 426–430. [Google Scholar] [CrossRef]
- Ciccarelli, C.; Marampon, F.; Scoglio, A.; Mauro, A.; Giacinti, C.; De Cesaris, P.; Zani, B.M. p21WAF1 expression induced by MEK/ERK pathway activation or inhibition correlates with growth arrest, myogenic differentiation and onco-phenotype reversal in rhabdomyosarcoma cells. Mol. Cancer 2005, 4, 41. [Google Scholar] [CrossRef]
- Cheng, L.; Lopez-Beltran, A.; Massari, F.; MacLennan, G.T.; Montironi, R. Molecular testing for BRAF mutations to inform melanoma treatment decisions: A move toward precision medicine. Mod. Pathol. 2018, 31, 24–38. [Google Scholar] [CrossRef]
- Brenan, L.; Andreev, A.; Cohen, O.; Pantel, S.; Kamburov, A.; Cacchiarelli, D.; Persky, N.S.; Zhu, C.; Bagul, M.; Goetz, E.M.; et al. Phenotypic Characterization of a Comprehensive Set of MAPK1/ERK2 Missense Mutants. Cell Rep. 2016, 17, 1171–1183. [Google Scholar] [CrossRef]
- Voelkerding, K.V.; Dames, S.A.; Durtschi, J.D. Next-generation sequencing: From basic research to diagnostics. Clin. Chem. 2009, 55, 641–658. [Google Scholar] [CrossRef]
- Anderson, M.W.; Schrijver, I. Next generation DNA sequencing and the future of genomic medicine. Genes 2010, 1, 38–69. [Google Scholar] [CrossRef]
- Tucker, T.; Marra, M.; Friedman, J.M. Massively parallel sequencing: The next big thing in genetic medicine. Am. J. Hum. Genet. 2009, 85, 142–154. [Google Scholar] [CrossRef]
- Brunet, A.; Pages, G.; Pouyssegur, J. Constitutively active mutants of MAP kinase kinase (MEK1) induce growth factor-relaxation and oncogenicity when expressed in fibroblasts. Oncogene 1994, 9, 3379–3387. [Google Scholar]
- Bott, C.M.; Thorneycroft, S.G.; Marshall, C.J. The sevenmaker gain-of-function mutation in p42 MAP kinase leads to enhanced signalling and reduced sensitivity to dual specificity phosphatase action. FEBS Lett. 1994, 352, 201–205. [Google Scholar] [CrossRef] [Green Version]
- Goetz, E.M.; Ghandi, M.; Treacy, D.J.; Wagle, N.; Garraway, L.A. ERK mutations confer resistance to mitogen-activated protein kinase pathway inhibitors. Cancer Res. 2014, 74, 7079–7089. [Google Scholar] [CrossRef]
- Lawrence, M.S.; Stojanov, P.; Mermel, C.H.; Robinson, J.T.; Garraway, L.A.; Golub, T.R.; Meyerson, M.; Gabriel, S.B.; Lander, E.S.; Getz, G. Discovery and saturation analysis of cancer genes across 21 tumour types. Nature 2014, 505, 495–501. [Google Scholar] [CrossRef] [Green Version]
- Ojesina, A.I.; Lichtenstein, L.; Freeman, S.S.; Pedamallu, C.S.; Imaz-Rosshandler, I.; Pugh, T.J.; Cherniack, A.D.; Ambrogio, L.; Cibulskis, K.; Bertelsen, B.; et al. Landscape of genomic alterations in cervical carcinomas. Nature 2014, 506, 371–375. [Google Scholar] [CrossRef]
- Arvind, R.; Shimamoto, H.; Momose, F.; Amagasa, T.; Omura, K.; Tsuchida, N. A mutation in the common docking domain of ERK2 in a human cancer cell line, which was associated with its constitutive phosphorylation. Int. J. Oncol. 2005, 27, 1499–1504. [Google Scholar]
- Mahalingam, M.; Arvind, R.; Ida, H.; Murugan, A.K.; Yamaguchi, M.; Tsuchida, N. ERK2 CD domain mutation from a human cancer cell line enhanced anchorage-independent cell growth and abnormality in Drosophila. Oncol. Rep. 2008, 20, 957–962. [Google Scholar]
- Emrick, M.A.; Hoofnagle, A.N.; Miller, A.S.; Ten Eyck, L.F.; Ahn, N.G. Constitutive activation of extracellular signal-regulated kinase 2 by synergistic point mutations. J. Biol. Chem. 2001, 276, 46469–46479. [Google Scholar] [CrossRef]
- Emrick, M.A.; Lee, T.; Starkey, P.J.; Mumby, M.C.; Resing, K.A.; Ahn, N.G. The gatekeeper residue controls autoactivation of ERK2 via a pathway of intramolecular connectivity. Proc. Natl. Acad. Sci. USA 2006, 103, 18101–18106. [Google Scholar] [CrossRef] [Green Version]
- Jaiswal, B.S.; Durinck, S.; Stawiski, E.W.; Yin, J.; Wang, W.; Lin, E.; Moffat, J.; Martin, S.E.; Modrusan, Z.; Seshagiri, S. ERK Mutations and Amplification Confer Resistance to ERK-Inhibitor Therapy. Clin. Cancer Res. 2018, 24, 4044–4055. [Google Scholar] [CrossRef] [Green Version]
- Wortzel, I.; Seger, R. The ERK Cascade: Distinct Functions within Various Subcellular Organelles. Genes Cancer 2011, 2, 195–209. [Google Scholar] [CrossRef]
- Shaul, Y.D.; Seger, R. The MEK/ERK cascade: From signaling specificity to diverse functions. Biochim. Biophys. Acta 2007, 1773, 1213–1226. [Google Scholar] [CrossRef] [Green Version]
- Hong, S.K.; Wu, P.K.; Karkhanis, M.; Park, J.I. ERK1/2 can feedback-regulate cellular MEK1/2 levels. Cell Signal. 2015, 27, 1939–1948. [Google Scholar] [CrossRef] [Green Version]
- Eblen, S.T.; Slack-Davis, J.K.; Tarcsafalvi, A.; Parsons, J.T.; Weber, M.J.; Catling, A.D. Mitogen-activated protein kinase feedback phosphorylation regulates MEK1 complex formation and activation during cellular adhesion. Mol. Cell Biol. 2004, 24, 2308–2317. [Google Scholar] [CrossRef]
- Albeck, J.G.; Mills, G.B.; Brugge, J.S. Frequency-modulated pulses of ERK activity transmit quantitative proliferation signals. Mol. Cell 2013, 49, 249–261. [Google Scholar] [CrossRef]
- Shin, S.Y.; Rath, O.; Choo, S.M.; Fee, F.; McFerran, B.; Kolch, W.; Cho, K.H. Positive- and negative-feedback regulations coordinate the dynamic behavior of the Ras-Raf-MEK-ERK signal transduction pathway. J. Cell Sci. 2009, 122, 425–435. [Google Scholar] [CrossRef] [Green Version]
- Sever, R.; Brugge, J.S. Signal transduction in cancer. Cold Spring Harb. Perspect. Med. 2015, 5, a006098. [Google Scholar] [CrossRef]
- Solit, D.B.; Garraway, L.A.; Pratilas, C.A.; Sawai, A.; Getz, G.; Basso, A.; Ye, Q.; Lobo, J.M.; She, Y.; Osman, I.; et al. BRAF mutation predicts sensitivity to MEK inhibition. Nature 2006, 439, 358–362. [Google Scholar] [CrossRef]
- Bollag, G.; Hirth, P.; Tsai, J.; Zhang, J.; Ibrahim, P.N.; Cho, H.; Spevak, W.; Zhang, C.; Zhang, Y.; Habets, G.; et al. Clinical efficacy of a RAF inhibitor needs broad target blockade in BRAF-mutant melanoma. Nature 2010, 467, 596–599. [Google Scholar] [CrossRef]
- Van Allen, E.M.; Wagle, N.; Sucker, A.; Treacy, D.J.; Johannessen, C.M.; Goetz, E.M.; Place, C.S.; Taylor-Weiner, A.; Whittaker, S.; Kryukov, G.V.; et al. The genetic landscape of clinical resistance to RAF inhibition in metastatic melanoma. Cancer Discov. 2014, 4, 94–109. [Google Scholar] [CrossRef]
- Emery, C.M.; Vijayendran, K.G.; Zipser, M.C.; Sawyer, A.M.; Niu, L.; Kim, J.J.; Hatton, C.; Chopra, R.; Oberholzer, P.A.; Karpova, M.B.; et al. MEK1 mutations confer resistance to MEK and B-RAF inhibition. Proc. Natl. Acad. Sci. USA 2009, 106, 20411–20416. [Google Scholar] [CrossRef] [Green Version]
- Corcoran, R.B.; Dias-Santagata, D.; Bergethon, K.; Iafrate, A.J.; Settleman, J.; Engelman, J.A. BRAF gene amplification can promote acquired resistance to MEK inhibitors in cancer cells harboring the BRAF V600E mutation. Sci. Signal. 2010, 3, ra84. [Google Scholar] [CrossRef]
- Wang, J.; Yao, Z.; Jonsson, P.; Allen, A.N.; Qin, A.C.R.; Uddin, S.; Dunkel, I.J.; Petriccione, M.; Manova, K.; Haque, S.; et al. A Secondary Mutation in BRAF Confers Resistance to RAF Inhibition in a BRAF(V600E)-Mutant Brain Tumor. Cancer Discov. 2018, 8, 1130–1141. [Google Scholar] [CrossRef]
- Hoogstraat, M.; Gadellaa-van Hooijdonk, C.G.; Ubink, I.; Besselink, N.J.; Pieterse, M.; Veldhuis, W.; van Stralen, M.; Meijer, E.F.; Willems, S.M.; Hadders, M.A.; et al. Detailed imaging and genetic analysis reveal a secondary BRAF(L505H) resistance mutation and extensive intrapatient heterogeneity in metastatic BRAF mutant melanoma patients treated with vemurafenib. Pigment. Cell Melanoma Res. 2015, 28, 318–323. [Google Scholar] [CrossRef]
- Little, A.S.; Balmanno, K.; Sale, M.J.; Newman, S.; Dry, J.R.; Hampson, M.; Edwards, P.A.; Smith, P.D.; Cook, S.J. Amplification of the driving oncogene, KRAS or BRAF, underpins acquired resistance to MEK1/2 inhibitors in colorectal cancer cells. Sci. Signal. 2011, 4, ra17. [Google Scholar] [CrossRef]
- Hatzivassiliou, G.; Haling, J.R.; Chen, H.; Song, K.; Price, S.; Heald, R.; Hewitt, J.F.; Zak, M.; Peck, A.; Orr, C.; et al. Mechanism of MEK inhibition determines efficacy in mutant KRAS- versus BRAF-driven cancers. Nature 2013, 501, 232–236. [Google Scholar] [CrossRef]
- Manchado, E.; Weissmueller, S.; Morris, J.P.; Chen, C.C.; Wullenkord, R.; Lujambio, A.; de Stanchina, E.; Poirier, J.T.; Gainor, J.F.; Corcoran, R.B.; et al. A combinatorial strategy for treating KRAS-mutant lung cancer. Nature 2016, 534, 647–651. [Google Scholar] [CrossRef] [Green Version]
- Merchant, M.; Moffat, J.; Schaefer, G.; Chan, J.; Wang, X.; Orr, C.; Cheng, J.; Hunsaker, T.; Shao, L.; Wang, S.J.; et al. Correction: Combined MEK and ERK inhibition overcomes therapy-mediated pathway reactivation in RAS mutant tumors. PLoS ONE 2018, 13, e0192059. [Google Scholar] [CrossRef]
- Villanueva, J.; Vultur, A.; Lee, J.T.; Somasundaram, R.; Fukunaga-Kalabis, M.; Cipolla, A.K.; Wubbenhorst, B.; Xu, X.; Gimotty, P.A.; Kee, D.; et al. Acquired resistance to BRAF inhibitors mediated by a RAF kinase switch in melanoma can be overcome by cotargeting MEK and IGF-1R/PI3K. Cancer Cell 2010, 18, 683–695. [Google Scholar] [CrossRef]
- Long, G.V.; Fung, C.; Menzies, A.M.; Pupo, G.M.; Carlino, M.S.; Hyman, J.; Shahheydari, H.; Tembe, V.; Thompson, J.F.; Saw, R.P.; et al. Increased MAPK reactivation in early resistance to dabrafenib/trametinib combination therapy of BRAF-mutant metastatic melanoma. Nat. Commun. 2014, 5, 5694. [Google Scholar] [CrossRef] [Green Version]
- Qin, J.; Xin, H.; Nickoloff, B.J. Specifically targeting ERK1 or ERK2 kills melanoma cells. J. Transl. Med. 2012, 10, 15. [Google Scholar] [CrossRef]
- Yadav, V.; Zhang, X.; Liu, J.; Estrem, S.; Li, S.; Gong, X.Q.; Buchanan, S.; Henry, J.R.; Starling, J.J.; Peng, S.B. Reactivation of mitogen-activated protein kinase (MAPK) pathway by FGF receptor 3 (FGFR3)/Ras mediates resistance to vemurafenib in human B-RAF V600E mutant melanoma. J. Biol. Chem. 2012, 287, 28087–28098. [Google Scholar] [CrossRef]
- Jha, S.; Morris, E.J.; Hruza, A.; Mansueto, M.S.; Schroeder, G.K.; Arbanas, J.; McMasters, D.; Restaino, C.R.; Dayananth, P.; Black, S.; et al. Dissecting Therapeutic Resistance to ERK Inhibition. Mol. Cancer Ther. 2016, 15, 548–559. [Google Scholar] [CrossRef]
- Delaney, G.; Jacob, S.; Featherstone, C.; Barton, M. The role of radiotherapy in cancer treatment: Estimating optimal utilization from a review of evidence-based clinical guidelines. Cancer 2005, 104, 1129–1137. [Google Scholar] [CrossRef]
- Begg, A.C.; Stewart, F.A.; Vens, C. Strategies to improve radiotherapy with targeted drugs. Nat. Rev. Cancer 2011, 11, 239–253. [Google Scholar] [CrossRef]
- Barnett, G.C.; West, C.M.; Dunning, A.M.; Elliott, R.M.; Coles, C.E.; Pharoah, P.D.; Burnet, N.G. Normal tissue reactions to radiotherapy: Towards tailoring treatment dose by genotype. Nat. Rev. Cancer 2009, 9, 134–142. [Google Scholar] [CrossRef] [PubMed]
- Barker, H.E.; Paget, J.T.; Khan, A.A.; Harrington, K.J. The tumour microenvironment after radiotherapy: Mechanisms of resistance and recurrence. Nat. Rev. Cancer 2015, 15, 409–425. [Google Scholar] [CrossRef] [PubMed]
- Makena, M.R.; Ranjan, A.; Thirumala, V.; Reddy, A.P. Cancer stem cells: Road to therapeutic resistance and strategies to overcome resistance. Biochim. Biophys. Acta Mol. Basis Dis. 2018. [Google Scholar] [CrossRef] [PubMed]
- Chang, L.; Graham, P.; Hao, J.; Ni, J.; Deng, J.; Bucci, J.; Malouf, D.; Gillatt, D.; Li, Y. Cancer stem cells and signaling pathways in radioresistance. Oncotarget 2016, 7, 11002–11017. [Google Scholar] [CrossRef] [PubMed]
- Santivasi, W.L.; Xia, F. Ionizing radiation-induced DNA damage, response, and repair. Antioxid. Redox. Signal. 2014, 21, 251–259. [Google Scholar] [CrossRef] [PubMed]
- Baskar, R.; Lee, K.A.; Yeo, R.; Yeoh, K.W. Cancer and radiation therapy: Current advances and future directions. Int. J. Med. Sci. 2012, 9, 193–199. [Google Scholar] [CrossRef]
- Walle, T.; Martinez Monge, R.; Cerwenka, A.; Ajona, D.; Melero, I.; Lecanda, F. Radiation effects on antitumor immune responses: Current perspectives and challenges. Ther. Adv. Med. Oncol. 2018, 10, 1758834017742575. [Google Scholar] [CrossRef]
- Pajonk, F.; Vlashi, E.; McBride, W.H. Radiation resistance of cancer stem cells: The 4 R’s of radiobiology revisited. Stem Cells 2010, 28, 639–648. [Google Scholar] [CrossRef]
- West, C.M.; Davidson, S.E.; Elyan, S.A.; Swindell, R.; Roberts, S.A.; Orton, C.J.; Coyle, C.A.; Valentine, H.; Wilks, D.P.; Hunter, R.D.; et al. The intrinsic radiosensitivity of normal and tumour cells. Int. J. Radiat. Biol. 1998, 73, 409–413. [Google Scholar]
- Balmukhanov, S.B.; Yefimov, M.L.; Kleinbock, T.S. Acquired radioresistance of tumour cells. Nature 1967, 216, 709–711. [Google Scholar] [CrossRef]
- Steel, G.G.; McMillan, T.J.; Peacock, J.H. The 5Rs of radiobiology. Int. J. Radiat. Biol. 1989, 56, 1045–1048. [Google Scholar] [CrossRef]
- Dent, P.; Yacoub, A.; Contessa, J.; Caron, R.; Amorino, G.; Valerie, K.; Hagan, M.P.; Grant, S.; Schmidt-Ullrich, R. Stress and radiation-induced activation of multiple intracellular signaling pathways. Radiat. Res. 2003, 159, 283–300. [Google Scholar] [CrossRef]
- Schmidt-Ullrich, R.K.; Dent, P.; Grant, S.; Mikkelsen, R.B.; Valerie, K. Signal transduction and cellular radiation responses. Radiat. Res. 2000, 153, 245–257. [Google Scholar] [CrossRef]
- Dent, P.; Yacoub, A.; Fisher, P.B.; Hagan, M.P.; Grant, S. MAPK pathways in radiation responses. Oncogene 2003, 22, 5885–5896. [Google Scholar] [CrossRef] [Green Version]
- Sklar, M.D. The ras oncogenes increase the intrinsic resistance of NIH 3T3 cells to ionizing radiation. Science 1988, 239, 645–647. [Google Scholar] [CrossRef]
- Bernhard, E.J.; Stanbridge, E.J.; Gupta, S.; Gupta, A.K.; Soto, D.; Bakanauskas, V.J.; Cerniglia, G.J.; Muschel, R.J.; McKenna, W.G. Direct evidence for the contribution of activated N-ras and K-ras oncogenes to increased intrinsic radiation resistance in human tumor cell lines. Cancer Res. 2000, 60, 6597–6600. [Google Scholar]
- Ciccarelli, C.; Di Rocco, A.; Gravina, G.L.; Mauro, A.; Festuccia, C.; Del Fattore, A.; Berardinelli, P.; De Felice, F.; Musio, D.; Bouche, M.; et al. Disruption of MEK/ERK/c-Myc signaling radiosensitizes prostate cancer cells in vitro and in vivo. J. Cancer Res. Clin. Oncol. 2018, 144, 1685–1699. [Google Scholar] [CrossRef]
- Ciccarelli, C.; Vulcano, F.; Milazzo, L.; Gravina, G.L.; Marampon, F.; Macioce, G.; Giampaolo, A.; Tombolini, V.; Di Paolo, V.; Hassan, H.J.; et al. Key role of MEK/ERK pathway in sustaining tumorigenicity and in vitro radioresistance of embryonal rhabdomyosarcoma stem-like cell population. Mol. Cancer 2016, 15, 16. [Google Scholar] [CrossRef]
- Marampon, F.; Gravina, G.L.; Festuccia, C.; Popov, V.M.; Colapietro, E.A.; Sanita, P.; Musio, D.; De Felice, F.; Lenzi, A.; Jannini, E.A.; et al. Vitamin D protects endothelial cells from irradiation-induced senescence and apoptosis by modulating MAPK/SirT1 axis. J. Endocrinol. Investig. 2016, 39, 411–422. [Google Scholar] [CrossRef]
- Marampon, F.; Gravina, G.L.; Zani, B.M.; Popov, V.M.; Fratticci, A.; Cerasani, M.; Di Genova, D.; Mancini, M.; Ciccarelli, C.; Ficorella, C.; et al. Hypoxia sustains glioblastoma radioresistance through ERKs/DNA-PKcs/HIF-1alpha functional interplay. Int. J. Oncol. 2014, 44, 2121–2131. [Google Scholar] [CrossRef]
- Marampon, F.; Gravina, G.L.; Popov, V.M.; Scarsella, L.; Festuccia, C.; La Verghetta, M.E.; Parente, S.; Cerasani, M.; Bruera, G.; Ficorella, C.; et al. Close correlation between MEK/ERK and Aurora-B signaling pathways in sustaining tumorigenic potential and radioresistance of gynecological cancer cell lines. Int. J. Oncol. 2014, 44, 285–294. [Google Scholar] [CrossRef]
- Marampon, F.; Gravina, G.L.; Di Rocco, A.; Bonfili, P.; Di Staso, M.; Fardella, C.; Polidoro, L.; Ciccarelli, C.; Festuccia, C.; Popov, V.M.; et al. MEK/ERK inhibitor U0126 increases the radiosensitivity of rhabdomyosarcoma cells in vitro and in vivo by downregulating growth and DNA repair signals. Mol. Cancer Ther. 2011, 10, 159–168. [Google Scholar] [CrossRef]
- Munshi, A.; Ramesh, R. Mitogen-activated protein kinases and their role in radiation response. Genes Cancer 2013, 4, 401–408. [Google Scholar] [CrossRef]
- Lee, H.C.; An, S.; Lee, H.; Woo, S.H.; Jin, H.O.; Seo, S.K.; Choe, T.B.; Yoo, D.H.; Lee, S.J.; Hong, Y.J.; et al. Activation of epidermal growth factor receptor and its downstream signaling pathway by nitric oxide in response to ionizing radiation. Mol. Cancer Res. 2008, 6, 996–1002. [Google Scholar] [CrossRef]
- Yan, Y.; Black, C.P.; Cowan, K.H. Irradiation-induced G2/M checkpoint response requires ERK1/2 activation. Oncogene 2007, 26, 4689–4698. [Google Scholar] [CrossRef] [Green Version]
- Wei, F.; Yan, J.; Tang, D. Extracellular signal-regulated kinases modulate DNA damage response—A contributing factor to using MEK inhibitors in cancer therapy. Curr. Med. Chem. 2011, 18, 5476–5482. [Google Scholar] [CrossRef]
- Haber, J.E. Partners and pathwaysrepairing a double-strand break. Trends Genet. 2000, 16, 259–264. [Google Scholar] [CrossRef]
- Lieber, M.R. The mechanism of human nonhomologous DNA end joining. J. Biol. Chem. 2008, 283, 1–5. [Google Scholar] [CrossRef]
- Thompson, L.H.; Schild, D. Homologous recombinational repair of DNA ensures mammalian chromosome stability. Mutat. Res. 2001, 477, 131–153. [Google Scholar] [CrossRef]
- Shibata, A.; Conrad, S.; Birraux, J.; Geuting, V.; Barton, O.; Ismail, A.; Kakarougkas, A.; Meek, K.; Taucher-Scholz, G.; Lobrich, M.; et al. Factors determining DNA double-strand break repair pathway choice in G2 phase. EMBO J. 2011, 30, 1079–1092. [Google Scholar] [CrossRef]
- Zhou, B.B.; Elledge, S.J. The DNA damage response: Putting checkpoints in perspective. Nature 2000, 408, 433–439. [Google Scholar] [CrossRef] [PubMed]
- Golding, S.E.; Rosenberg, E.; Neill, S.; Dent, P.; Povirk, L.F.; Valerie, K. Extracellular signal-related kinase positively regulates ataxia telangiectasia mutated, homologous recombination repair, and the DNA damage response. Cancer Res. 2007, 67, 1046–1053. [Google Scholar] [CrossRef] [PubMed]
- Wei, F.; Xie, Y.; Tao, L.; Tang, D. Both ERK1 and ERK2 kinases promote G2/M arrest in etoposide-treated MCF7 cells by facilitating ATM activation. Cell Signal. 2010, 22, 1783–1789. [Google Scholar] [CrossRef]
- Toulany, M.; Iida, M.; Keinath, S.; Iyi, F.F.; Mueck, K.; Fehrenbacher, B.; Mansour, W.Y.; Schaller, M.; Wheeler, D.L.; Rodemann, H.P. Dual targeting of PI3K and MEK enhances the radiation response of K-RAS mutated non-small cell lung cancer. Oncotarget 2016, 7, 43746–43761. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gravina, G.L.; Festuccia, C.; Popov, V.M.; Di Rocco, A.; Colapietro, A.; Sanita, P.; Monache, S.D.; Musio, D.; De Felice, F.; Di Cesare, E.; et al. c-Myc Sustains Transformed Phenotype and Promotes Radioresistance of Embryonal Rhabdomyosarcoma Cell Lines. Radiat. Res. 2016, 185, 411–422. [Google Scholar] [CrossRef] [PubMed]
- Schick, U.; Kyula, J.; Barker, H.; Patel, R.; Zaidi, S.; Gregory, C.; Hafsi, H.; Roulstone, V.; Deutsch, E.; McLaughlin, M.; et al. Trametinib radiosensitises RAS- and BRAF-mutated melanoma by perturbing cell cycle and inducing senescence. Radiother Oncol. 2015, 117, 364–375. [Google Scholar] [CrossRef]
- Yu, W.; Gu, K.; Yu, Z.; Yuan, D.; He, M.; Ma, N.; Lai, S.; Zhao, J.; Ren, Z.; Zhang, X.; et al. Sorafenib potentiates irradiation effect in hepatocellular carcinoma in vitro and in vivo. Cancer Lett. 2013, 329, 109–117. [Google Scholar] [CrossRef]
- Tomiyama, A.; Tachibana, K.; Suzuki, K.; Seino, S.; Sunayama, J.; Matsuda, K.I.; Sato, A.; Matsumoto, Y.; Nomiya, T.; Nemoto, K.; et al. MEK-ERK-dependent multiple caspase activation by mitochondrial proapoptotic Bcl-2 family proteins is essential for heavy ion irradiation-induced glioma cell death. Cell Death Dis. 2010, 1, e60. [Google Scholar] [CrossRef]
- Golding, S.E.; Morgan, R.N.; Adams, B.R.; Hawkins, A.J.; Povirk, L.F.; Valerie, K. Pro-survival AKT and ERK signaling from EGFR and mutant EGFRvIII enhances DNA double-strand break repair in human glioma cells. Cancer Biol. Ther. 2009, 8, 730–738. [Google Scholar] [CrossRef] [Green Version]
- Wei, F.; Yan, J.; Tang, D.; Lin, X.; He, L.; Xie, Y.; Tao, L.; Wang, S. Inhibition of ERK activation enhances the repair of double-stranded breaks via non-homologous end joining by increasing DNA-PKcs activation. Biochim. Biophys. Acta 2013, 1833, 90–100. [Google Scholar] [CrossRef] [Green Version]
- Yacoub, A.; McKinstry, R.; Hinman, D.; Chung, T.; Dent, P.; Hagan, M.P. Epidermal growth factor and ionizing radiation up-regulate the DNA repair genes XRCC1 and ERCC1 in DU145 and LNCaP prostate carcinoma through MAPK signaling. Radiat. Res. 2003, 159, 439–452. [Google Scholar] [CrossRef]
- Chambard, J.C.; Lefloch, R.; Pouyssegur, J.; Lenormand, P. ERK implication in cell cycle regulation. Biochim. Biophys. Acta 2007, 1773, 1299–1310. [Google Scholar] [CrossRef] [PubMed]
- Strasser-Wozak, E.M.; Hartmann, B.L.; Geley, S.; Sgonc, R.; Bock, G.; Santos, A.J.; Hattmannstorfer, R.; Wolf, H.; Pavelka, M.; Kofler, R. Irradiation induces G2/M cell cycle arrest and apoptosis in p53-deficient lymphoblastic leukemia cells without affecting Bcl-2 and Bax expression. Cell Death Differ. 1998, 5, 687–693. [Google Scholar] [CrossRef] [PubMed]
- Pawlik, T.M.; Keyomarsi, K. Role of cell cycle in mediating sensitivity to radiotherapy. Int. J. Radiat. Oncol. Biol. Phys. 2004, 59, 928–942. [Google Scholar] [CrossRef]
- Sinclair, W.K. Cyclic x-ray responses in mammalian cells in vitro. Radiat. Res. 1968, 33, 620–643. [Google Scholar] [CrossRef]
- Bernhard, E.J.; Maity, A.; Muschel, R.J.; McKenna, W.G. Effects of ionizing radiation on cell cycle progression. A review. Radiat. Environ. Biophys. 1995, 34, 79–83. [Google Scholar] [CrossRef]
- Abbott, D.W.; Holt, J.T. Mitogen-activated protein kinase kinase 2 activation is essential for progression through the G2/M checkpoint arrest in cells exposed to ionizing radiation. J. Biol. Chem. 1999, 274, 2732–2742. [Google Scholar] [CrossRef]
- Baskar, R.; Dai, J.; Wenlong, N.; Yeo, R.; Yeoh, K.W. Biological response of cancer cells to radiation treatment. Front. Mol. Biosci. 2014, 1, 24. [Google Scholar] [CrossRef]
- Estrada-Bernal, A.; Chatterjee, M.; Haque, S.J.; Yang, L.; Morgan, M.A.; Kotian, S.; Morrell, D.; Chakravarti, A.; Williams, T.M. MEK inhibitor GSK1120212-mediated radiosensitization of pancreatic cancer cells involves inhibition of DNA double-strand break repair pathways. Cell Cycle 2015, 14, 3713–3724. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ng, W.L.; Huang, Q.; Liu, X.; Zimmerman, M.; Li, F.; Li, C.Y. Molecular mechanisms involved in tumor repopulation after radiotherapy. Transl. Cancer Res. 2013, 2, 442–448. [Google Scholar]
- Huang, Q.; Li, F.; Liu, X.; Li, W.; Shi, W.; Liu, F.F.; O’Sullivan, B.; He, Z.; Peng, Y.; Tan, A.C.; et al. Caspase 3-mediated stimulation of tumor cell repopulation during cancer radiotherapy. Nat. Med. 2011, 17, 860–866. [Google Scholar] [CrossRef]
- Rahmanian, N.; Hosseinimehr, S.J.; Khalaj, A. The paradox role of caspase cascade in ionizing radiation therapy. J. Biomed. Sci. 2016, 23, 88. [Google Scholar] [CrossRef]
- Liu, J.; Mao, W.; Ding, B.; Liang, C.S. ERKs/p53 signal transduction pathway is involved in doxorubicin-induced apoptosis in H9c2 cells and cardiomyocytes. Am. J. Physiol. Heart Circ. Physiol. 2008, 295, H1956–H1965. [Google Scholar] [CrossRef] [Green Version]
- Cheng, J.; Tian, L.; Ma, J.; Gong, Y.; Zhang, Z.; Chen, Z.; Xu, B.; Xiong, H.; Li, C.; Huang, Q. Dying tumor cells stimulate proliferation of living tumor cells via caspase-dependent protein kinase Cdelta activation in pancreatic ductal adenocarcinoma. Mol. Oncol. 2015, 9, 105–114. [Google Scholar] [CrossRef]
- Seta, K.A.; Spicer, Z.; Yuan, Y.; Lu, G.; Millhorn, D.E. Responding to hypoxia: Lessons from a model cell line. Sci. STKE 2002, 2002, re11. [Google Scholar] [CrossRef]
- Minet, E.; Arnould, T.; Michel, G.; Roland, I.; Mottet, D.; Raes, M.; Remacle, J.; Michiels, C. ERK activation upon hypoxia: Involvement in HIF-1 activation. FEBS Lett 2000, 468, 53–58. [Google Scholar] [CrossRef]
- Vaupel, P.; Kelleher, D.K.; Hockel, M. Oxygen status of malignant tumors: Pathogenesis of hypoxia and significance for tumor therapy. Semin. Oncol. 2001, 28, 29–35. [Google Scholar] [CrossRef]
- Cross, T.G.; Scheel-Toellner, D.; Henriquez, N.V.; Deacon, E.; Salmon, M.; Lord, J.M. Serine/threonine protein kinases and apoptosis. Exp. Cell Res. 2000, 256, 34–41. [Google Scholar] [CrossRef] [PubMed]
- Xia, Z.; Dickens, M.; Raingeaud, J.; Davis, R.J.; Greenberg, M.E. Opposing effects of ERK and JNK-p38 MAP kinases on apoptosis. Science 1995, 270, 1326–1331. [Google Scholar] [CrossRef] [PubMed]
- Krause, M.; Dubrovska, A.; Linge, A.; Baumann, M. Cancer stem cells: Radioresistance, prediction of radiotherapy outcome and specific targets for combined treatments. Adv. Drug Deliv. Rev. 2017, 109, 63–73. [Google Scholar] [CrossRef]
- Vlashi, E.; Pajonk, F. Cancer stem cells, cancer cell plasticity and radiation therapy. Semin. Cancer Biol. 2015, 31, 28–35. [Google Scholar] [CrossRef]
- Raghav, K.P.; Gonzalez-Angulo, A.M.; Blumenschein, G.R., Jr. Role of HGF/MET axis in resistance of lung cancer to contemporary management. Transl. Lung Cancer Res. 2012, 1, 179–193. [Google Scholar]
- Oskarsson, T.; Batlle, E.; Massague, J. Metastatic stem cells: Sources, niches, and vital pathways. Cell Stem Cell 2014, 14, 306–321. [Google Scholar] [CrossRef]
- Kyjacova, L.; Hubackova, S.; Krejcikova, K.; Strauss, R.; Hanzlikova, H.; Dzijak, R.; Imrichova, T.; Simova, J.; Reinis, M.; Bartek, J.; et al. Radiotherapy-induced plasticity of prostate cancer mobilizes stem-like non-adherent, Erk signaling-dependent cells. Cell Death Differ. 2015, 22, 898–911. [Google Scholar] [CrossRef]
- Megiorni, F.; Gravina, G.L.; Camero, S.; Ceccarelli, S.; Del Fattore, A.; Desiderio, V.; Papaccio, F.; McDowell, H.P.; Shukla, R.; Pizzuti, A.; et al. Pharmacological targeting of the ephrin receptor kinase signalling by GLPG1790 in vitro and in vivo reverts oncophenotype, induces myogenic differentiation and radiosensitizes embryonal rhabdomyosarcoma cells. J. Hematol. Oncol. 2017, 10, 161. [Google Scholar] [CrossRef] [PubMed]
- Zheng, H.C. The molecular mechanisms of chemoresistance in cancers. Oncotarget 2017, 8, 59950–59964. [Google Scholar] [CrossRef]
- Holohan, C.; Van Schaeybroeck, S.; Longley, D.B.; Johnston, P.G. Cancer drug resistance: An evolving paradigm. Nat. Rev. Cancer 2013, 13, 714–726. [Google Scholar] [CrossRef]


© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Marampon, F.; Ciccarelli, C.; Zani, B.M. Biological Rationale for Targeting MEK/ERK Pathways in Anti-Cancer Therapy and to Potentiate Tumour Responses to Radiation. Int. J. Mol. Sci. 2019, 20, 2530. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20102530
Marampon F, Ciccarelli C, Zani BM. Biological Rationale for Targeting MEK/ERK Pathways in Anti-Cancer Therapy and to Potentiate Tumour Responses to Radiation. International Journal of Molecular Sciences. 2019; 20(10):2530. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20102530
Chicago/Turabian StyleMarampon, Francesco, Carmela Ciccarelli, and Bianca Maria Zani. 2019. "Biological Rationale for Targeting MEK/ERK Pathways in Anti-Cancer Therapy and to Potentiate Tumour Responses to Radiation" International Journal of Molecular Sciences 20, no. 10: 2530. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20102530