iTRAQ-Based Quantitative Analysis of Responsive Proteins Under PEG-Induced Drought Stress in Wheat Leaves

 , ,
, ,
Abstract
:1. Introduction
2. Results
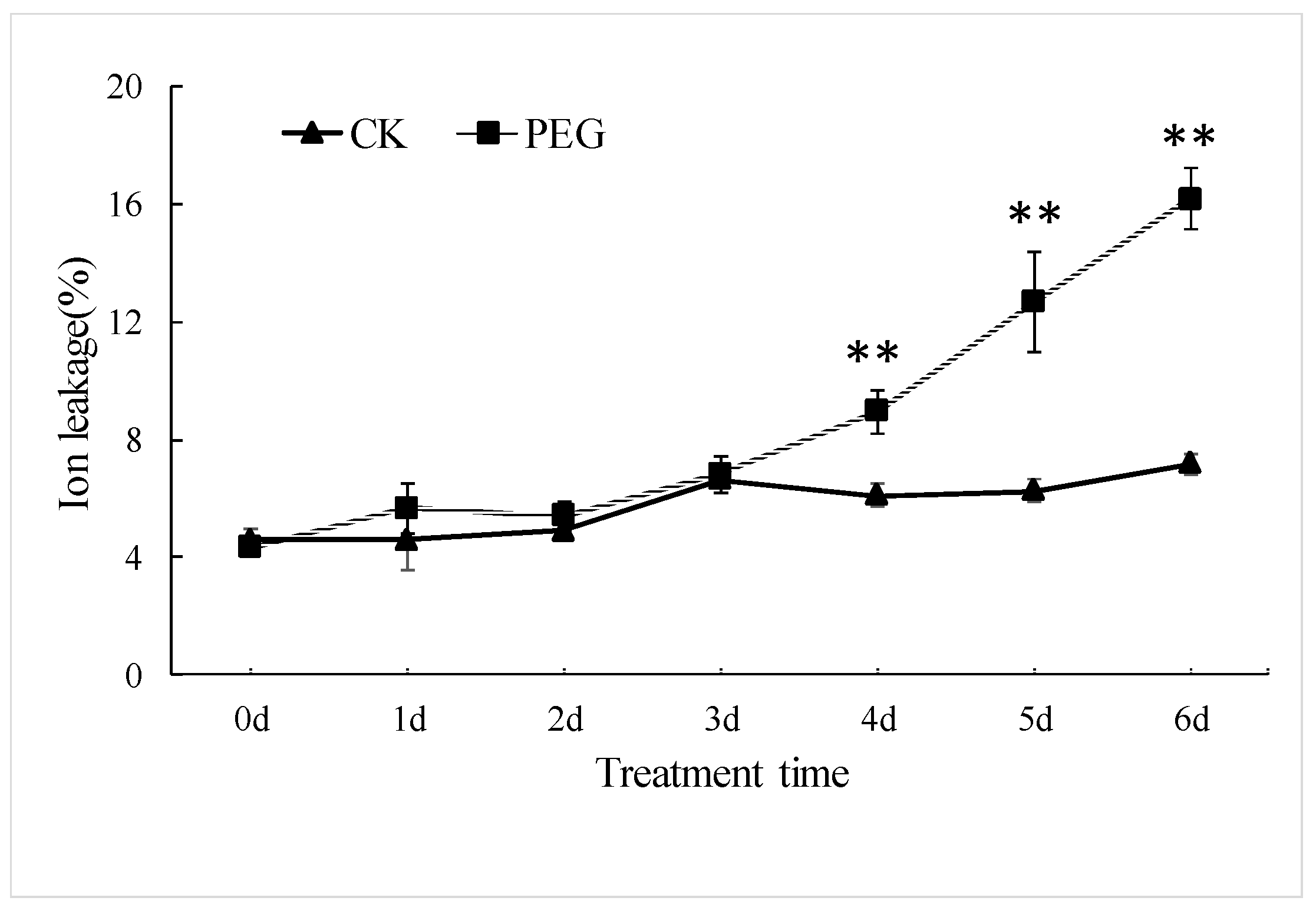
2.1. Physiological Changes in Wheat Seedlings under Drought Stress
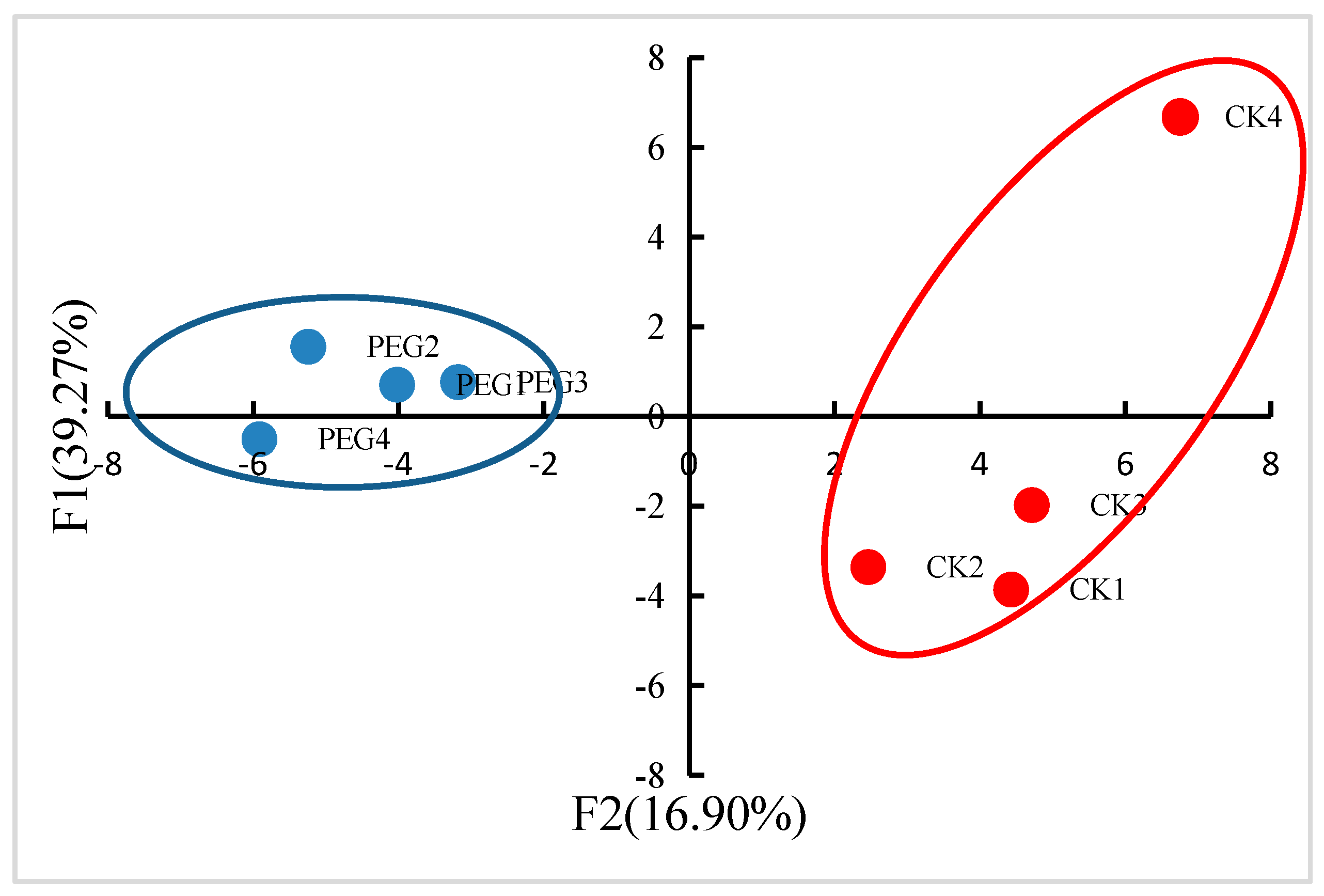
2.2. Identification of Proteins in Response to Drought Stress
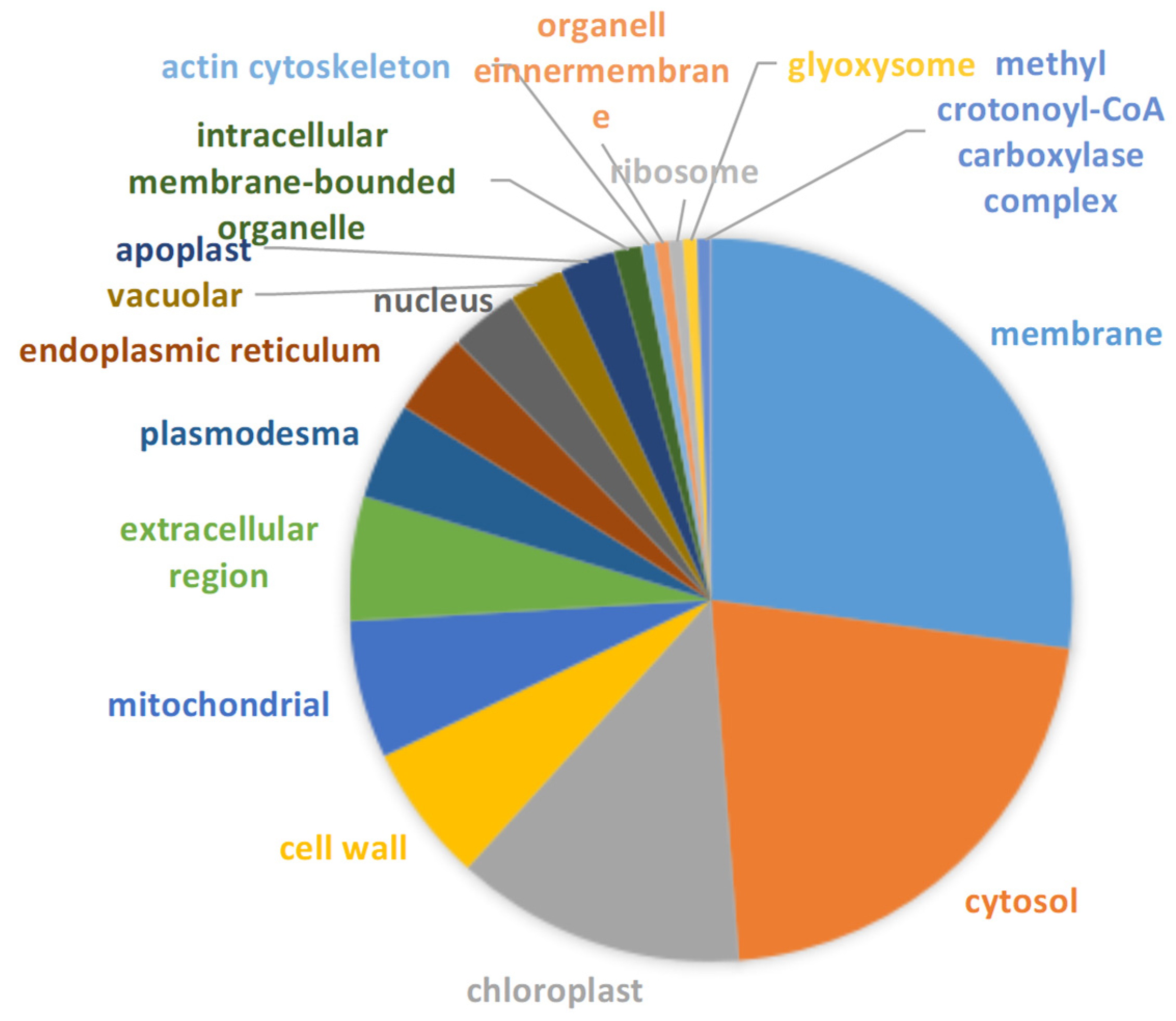
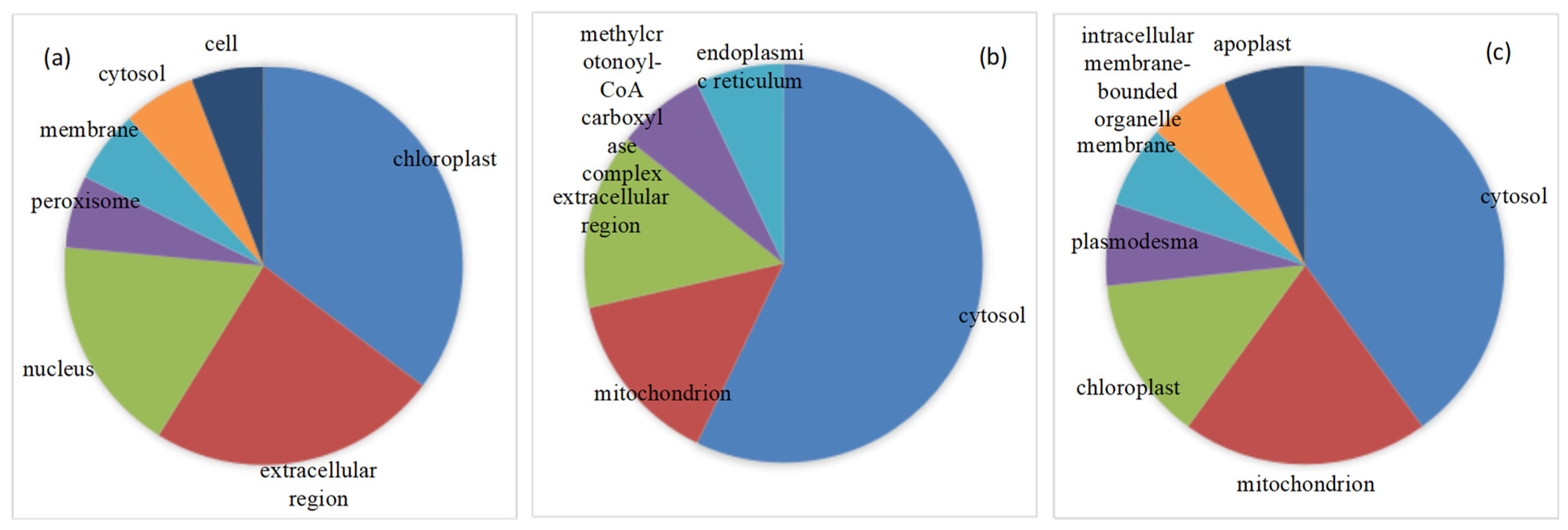
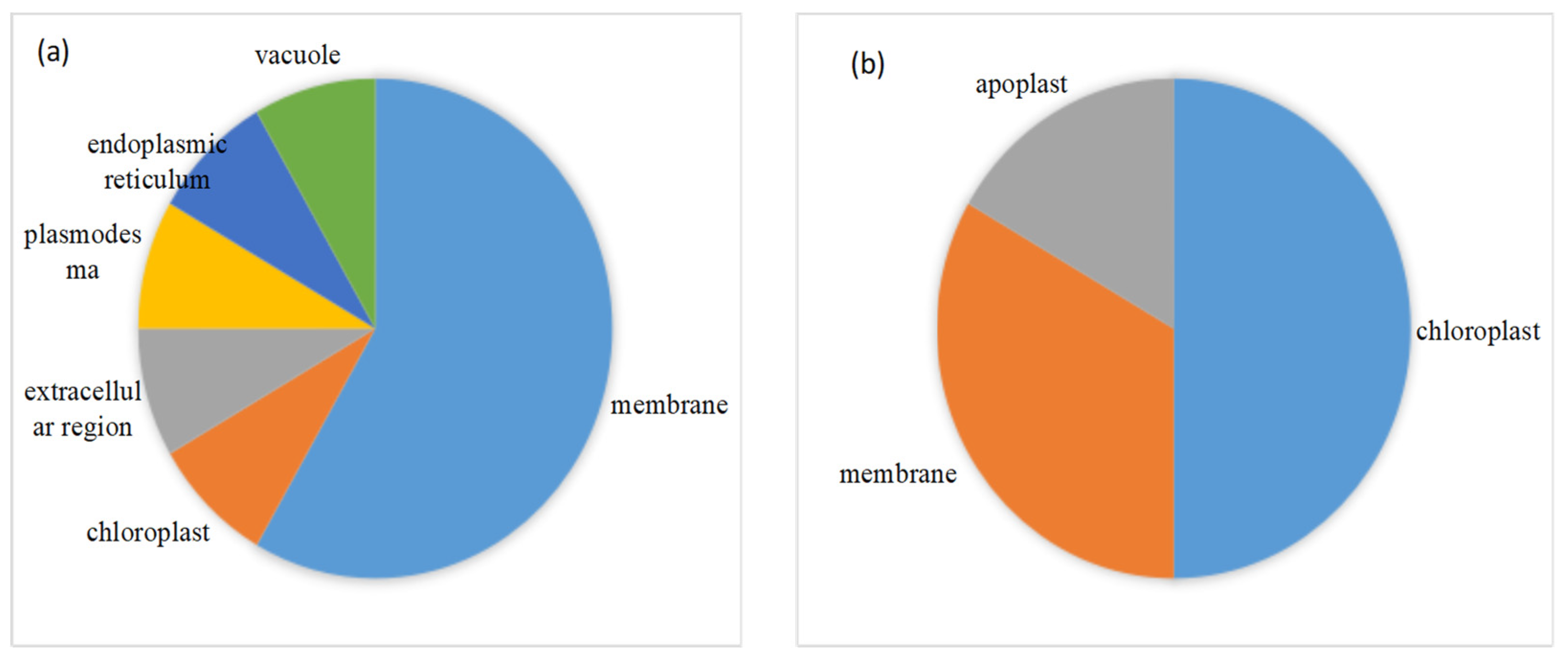
2.3. Functional Annotation, Classification and Subcellular Localization of DEPs under Drought Stress
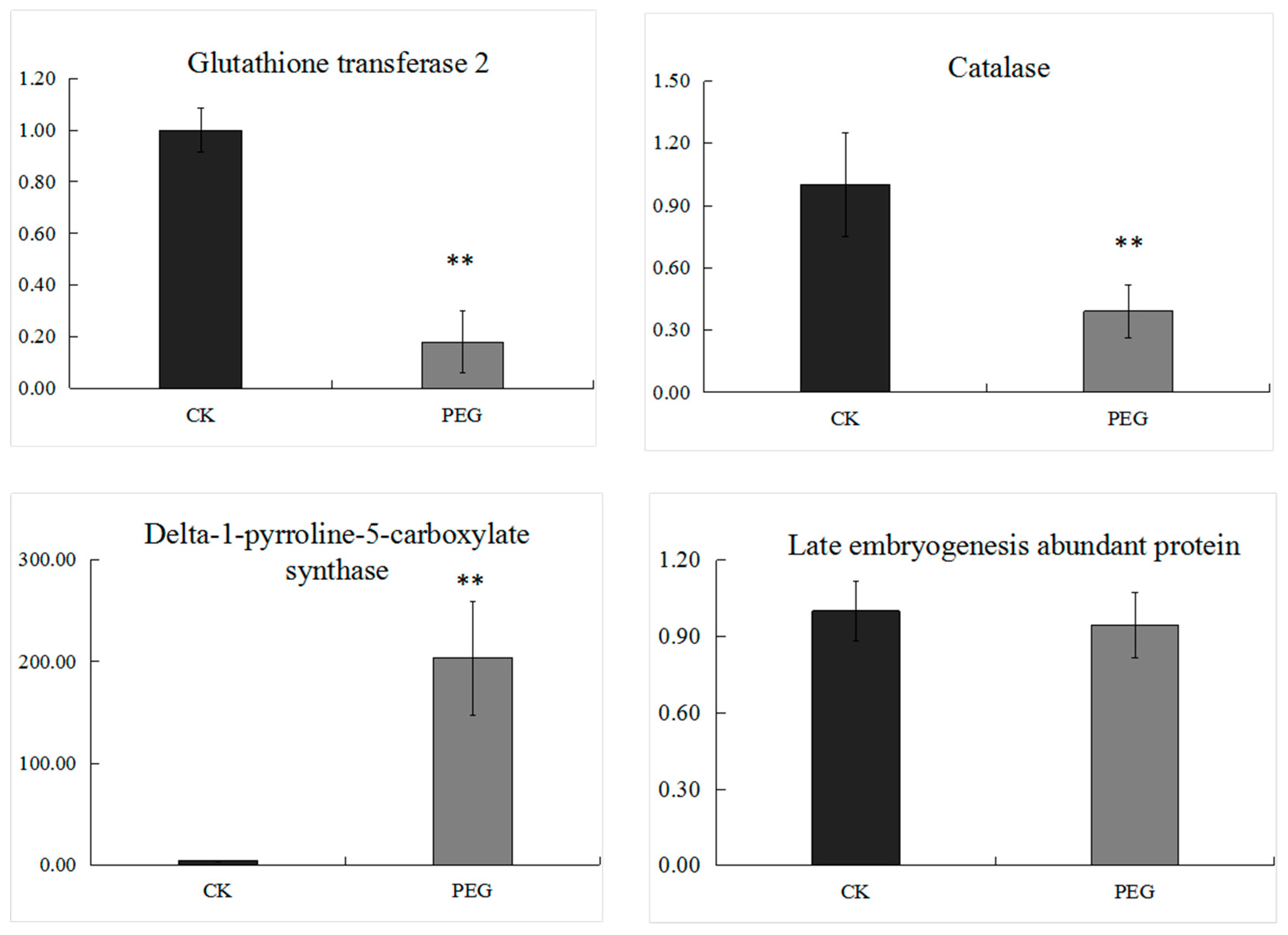
2.4. Transcriptional Expression Analysis of Genes Encoding Several Selected DEPs
3. Discussion
3.1. Carbohydrate Metabolism and Photosynthesis
3.2. Amino Acid Metabolism
3.3. Stress and Redox Regulation
3.4. Signal Transduction
3.5. Transportation
3.6. Secondary Metabolites
3.7. Other Metabolism Categories
4. Materials and Methods
4.1. Materials and Growth Conditions
4.2. Membrane Permeability Measurements
4.3. Protein Extraction
4.4. Protein Digestion and iTRAQ Labeling of Samples
4.5. High pH Reversed-Phase (HpRP) Chromatography
4.6. Mass Spectrometer Analysis
4.7. Database Searching and Protein Identification
4.8. Functional Annotation and Analysis of DEPs
4.9. qRT-PCR Analysis
4.10. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| DEP | Differentially expressed protein |
| ROS | Reactive oxygen species |
| PEG | polyethylene glycol-6000 |
| iTRAQ | Isobaric tags for relative and absolute quantitation |
| PCA | Principal component analysis |
| KEGG | KyotoEncyclopedia of Genes and Genomes |
| qRT-PCR | Quantitative real-time PCR |
References
- Raj, P.M.; Sendhil, R.; Tripathi, S.C.; Subhash, C.; Chhokar, R.S.; Sharma, R.K. Hydro-priming of seed improves the water use efficiency, grain yield and net economic return of wheat under different moisture regimes. SAARC J. Agric. 2013, 11, 149–159. [Google Scholar] [CrossRef]
- Peng, Z.; Wang, M.; Li, F.; Lv, H.; Li, C.; Xia, G. A proteomic study of the response to salinity and drought stress in an introgression strain of bread wheat. Mol. Cell Proteom. 2009, 8, 2676–2686. [Google Scholar] [CrossRef]
- Israelsson, M.; Siegel, R.S.; Young, J.; Hashimoto, M.; Iba, K.; Schroeder, J.I. Guard cell ABA and CO2 signaling network updates and Ca2+ sensor priming hypothesis. Curr. Opin. Plant Biol. 2006, 9, 654–663. [Google Scholar] [CrossRef]
- Deng, X.; Liu, Y.; Xu, X.; Liu, D.; Zhu, G.; Yan, X.; Wang, Z.; Yan, Y. Comparative proteome analysis of wheat flag leaves and developing grains under water deficit. Front. Plant Sci. 2018, 9. [Google Scholar] [CrossRef]
- Gao, H.; Wang, Y.; Xu, P.; Zhang, Z. Overexpression of a WRKY transcription factor TaWRKY2 enhances drought stress tolerance in transgenic wheat. Front. Plant Sci. 2018, 9. [Google Scholar] [CrossRef]
- Rong, W.; Qi, L.; Wang, A.; Ye, X.; Du, L.; Liang, H.; Xin, Z.; Zhang, Z. The ERF transcription factor TaERF3 promotes tolerance to salt and drought stresses in wheat. Plant Biotechnol. J. 2014, 12, 468–479. [Google Scholar] [CrossRef] [PubMed]
- Xue, G.; Way, H.M.; Richardson, T.; Drenth, J.; Joyce, P.A.; McIntyre, C.L. Overexpression of TaNAC69 leads to enhanced transcript levels of stress Up-Regulated genes and dehydration tolerance in bread wheat. Mol. Plant. 2011, 4, 697–712. [Google Scholar] [CrossRef]
- Zhang, H.; Mao, X.; Zhang, J.; Chang, X.; Jing, R. Single-nucleotide polymorphisms and association analysis of drought-resistance gene TaSnRK2.8 in common wheat. Plant Physiol. Biochem. 2013, 70, 174–181. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brenchley, R.; Spannagl, M.; Pfeifer, M.; Barker, G.L.A.; D’Amore, R.; Allen, A.M.; McKenzie, N.; Kramer, M.; Kerhornou, A.; Bolser, D.; et al. Analysis of the breadwheat genome using whole-genome shotgun sequencing. Nature 2012, 491, 705–710. [Google Scholar] [CrossRef] [PubMed]
- Appels, R.; Eversole, K.; Feuillet, C.; Keller, B.; Rogers, J.; Stein, N.; Pozniak, C.J.; Stein, N.; Choulet, F.; Distelfeld, A.; et al. Shifting the limits in wheat research and breeding using a fully annotated reference genome. Science 2018, 361, 661. [Google Scholar] [CrossRef]
- Cheng, L.; Wang, Y.; He, Q.; Li, H.; Zhang, X.; Zhang, F. Comparative proteomics illustrates the complexity of drought resistance mechanisms in two wheat (Triticum aestivum L.) cultivars under dehydration and rehydration. BMC Plant Biol. 2016, 16. [Google Scholar] [CrossRef] [PubMed]
- Ford, K.L.; Cassin, A.; Bacic, A. Quantitative proteomic analysis of wheat cultivars with differing drought stress tolerance. Front. Plant Sci. 2011, 2. [Google Scholar] [CrossRef]
- Zhang, Y.; Huang, X.; Wang, L.; Wei, L.; Wu, Z.; You, M.; Li, B. Proteomic analysis of wheat seed in response to drought stress. J. Integr. Agric. 2014, 13, 919–925. [Google Scholar] [CrossRef]
- Varshney, R.K.; Graner, A.; Sorrells, M.E. Genomics-assisted breeding for crop improvement. Trends Plant Sci. 2005, 10, 621–630. [Google Scholar] [CrossRef] [Green Version]
- Mei, X.; Zhong, X.; Vincent, V.; Liu, X. Improving water use efficiency of wheat crop varieties in the north china plain: Review and analysis. J. Integr Agr. 2013, 12, 1243–1250. [Google Scholar] [CrossRef]
- Zhang, X.Y.; Pei, D.; Hu, C.S. Conserving groundwater for irrigation in the North China Plain. Irrig. Sci. 2003, 21, 159–166. [Google Scholar] [CrossRef]
- Gygi, S.P.; Corthals, G.L.; Zhang, Y.; Rochon, Y.; Aebersold, R. Evaluation of two-dimensional gel electrophoresis-based proteome analysis technology. Proc. Natl. Acad. Sci. USA 2000, 97, 9390–9395. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Lou, H.; Guo, D.; Zhang, R.; Su, M.; Hou, Z.; Zhou, H.; Liang, R.; Xie, C.; You, M.; et al. Identifying changes in the wheat kernel proteome under heat stress using iTRAQ. CROP J. 2018, 6, 600–610. [Google Scholar] [CrossRef]
- Casado-Vela, J.; Jose Martinez-Esteso, M.; Rodriguez, E.; Borras, E.; Elortza, F.; Bru-Martinez, R. ITRAQ-based quantitative analysis of protein mixtures with large fold change and dynamic range. Proteomics 2010, 10, 343–347. [Google Scholar] [CrossRef] [PubMed]
- Pang, Q.; Chen, S.; Dai, S.; Chen, Y.; Wang, Y.; Yan, X. Comparative Proteomics of Salt Tolerance in Arabidopsis thaliana and Thellungiella halophila. J. Proteome Res. 2010, 9, 2584–2599. [Google Scholar] [CrossRef]
- Zhao, F.; Zhang, D.; Zhao, Y.; Wang, W.; Yang, H.; Tai, F.; Li, C.; Hu, X. The difference of physiological and proteomic changes in maize leaves adaptation to drought, heat, and combined both stresses. Front. Plant Sci. 2016, 7. [Google Scholar] [CrossRef]
- Zhu, M.; Simons, B.; Zhu, N.; Oppenheimer, D.G.; Chen, S. Analysis of abscisic acid responsive proteins in Brassica napus guard cells by multiplexed isobaric tagging. J. Proteom. 2010, 73, 790–805. [Google Scholar] [CrossRef]
- Koh, J.; Chen, G.; Yoo, M.; Zhu, N.; Dufresne, D.; Erickson, J.E.; Shao, H.; Chen, S. Comparative proteomic analysis of brassica napus in response to drought stress. J. Proteome Res. 2015, 14, 3068–3081. [Google Scholar] [CrossRef] [PubMed]
- Alvarez, S.; Choudhury, S.R.; Pandey, S. Comparative quantitative proteomics analysis of the ABA response of roots of Drought-Sensitive and Drought-Tolerant wheat varieties identifies proteomic signatures of drought adaptability. J. Proteome Res. 2014, 13, 1688–1701. [Google Scholar] [CrossRef]
- Xie, H.; Yang, D.; Yao, H.; Bai, G.; Zhang, Y.; Xiao, B. ITRAQ-based quantitative proteomic analysis reveals proteomic changes in leaves of cultivated tobacco (Nicotiana tabacum) in response to drought stress. Biochem. Biophys. Res. Commun. 2016, 469, 768–775. [Google Scholar] [CrossRef]
- Hossain, Z.; Komatsu, S. Contribution of proteomic studies towards understanding plant heavy metal stress response. Front. Plant Sci. 2012, 3, 310. [Google Scholar] [CrossRef]
- Coleman, H.D.; Yan, J.; Mansfield, S.D. Sucrose synthase affects carbon partitioning to increase cellulose production and altered cell wall ultrastructure. Proc. Natl. Acad. Sci. USA 2009, 106, 13118–13123. [Google Scholar] [CrossRef] [Green Version]
- Chourey, P.S.; Taliercio, E.W.; Carlson, S.J.; Ruan, Y.L. Genetic evidence that the two isozymes of sucrose synthase present in developing maize endosperm are critical, one for cell wall integrity and the other for starch biosynthesis. Mol. Gen. Genet. 1998, 259, 88–96. [Google Scholar]
- Barratt, D.H.; Derbyshire, P.; Findlay, K.; Pike, M.; Wellner, N.; Lunn, J.; Feil, R.; Simpson, C.; Maule, A.J.; Smith, A.M. Normal growth of Arabidopsis requires cytosolic invertase but not sucrose synthase. Proc. Natl. Acad. Sci. USA 2009, 106, 13124–13129. [Google Scholar] [CrossRef] [PubMed]
- Ruan, Y.L.; Jin, Y.; Huang, J. Capping invertase activity by its inhibitor: Roles and implications in sugar signaling, carbon allocation, senescence and evolution. Plant Signal. Behav. 2009, 4, 983–985. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.C.; Timasheff, S.N. The stabilization of proteins by sucrose. J. Biol. Chem. 1981, 256, 7193–7201. [Google Scholar] [PubMed]
- Andrews, T.J.; Hudson, G.S.; Mate, C.J.; von Caemmerer, S.; Evans, J.R.; Arvidsson, Y.B.C. Rubisco: The consequences of altering its expression and activation in transgenic plants. J. Exp. Bot. 1995, 46, 1293–1300. [Google Scholar] [CrossRef]
- Atkinson, N.J.; Lilley, C.J.; Urwin, P.E. Identification of genes involved in the response of arabidopsis to simultaneous biotic and abiotic stresses. Plant Physiol. 2013, 162, 2028–2041. [Google Scholar] [CrossRef] [PubMed]
- Nambara, E.; Kawaide, H.; Kamiya, Y.; Naito, S. Characterization of an Arabidopsis thaliana mutant that has a defect in ABA accumulation: ABA-dependent and ABA-independent accumulation of free amino acids during dehydration. Plant Cell Physiol. 1998, 39, 853–858. [Google Scholar] [CrossRef]
- Rebeille, F.; Jabrin, S.; Bligny, R.; Loizeau, K.; Gambonnet, B.; Van Wilder, V.; Douce, R.; Ravanel, S. Methionine catabolism in Arabidopsis cells is initiated by a gamma-cleavage process and leads to S-methylcysteine and isoleucine syntheses. Proc. Natl. Acad. Sci. USA 2006, 103, 15687–15692. [Google Scholar] [CrossRef]
- Houde, M.; Danyluk, J.; Laliberte, J.F.; Rassart, E.; Dhindsa, R.S.; Sarhan, F. Cloning, characterization, and expression of a cDNA encoding a 50-kilodalton protein specifically induced by cold acclimation in wheat. Plant Physiol. 1992, 99, 1381–1387. [Google Scholar] [CrossRef]
- Houde, M.; Diallo, A.O. Identification of genes and pathways associated with aluminum stress and tolerance using transcriptome profiling of wheat near-isogenic lines. BMC Genom. 2008, 9, 400. [Google Scholar] [CrossRef]
- Ezaki, B.; Gardner, R.C.; Ezaki, Y.; Matsumoto, H. Expression of aluminum-induced genes in transgenic arabidopsis plants can ameliorate aluminum stress and/or oxidative stress. Plant Physiol. 2000, 122, 657–665. [Google Scholar] [CrossRef]
- Ezaki, B.; Sivaguru, M.; Ezaki, Y.; Matsumoto, H.; Gardner, R.C. Acquisition of aluminum tolerance in Saccharomyces cerevisiae by expression of the BCB or NtGDI1 gene derived from plants. FEMS Microbiol. Lett. 1999, 171, 81–87. [Google Scholar] [CrossRef]
- Glijin, A.; Mita, E.; Levitchi, A.; Acciu, A.; Calmis, A.; Duca, M. Phenylalanine ammonia-lyase in normal and biotic stress conditions. Lucrari Stiintifice, Universitatea de Stiinte Agricole Si Medicina Veterinara "Ion Ionescu de la Brad" Iasi, Seria Horticultura 2011, 54, 97–102. [Google Scholar]
- Jiang, Q.; Li, X.; Niu, F.; Sun, X.; Hu, Z.; Zhang, H. ITRAQ-based quantitative proteomic analysis of wheat roots in response to salt stress. Proteomics 2017, 17, 1600265. [Google Scholar] [CrossRef] [PubMed]
- Ouellet, F.; Houde, M.; Sarhan, F. Purification, characterization and cDNA cloning of the 200 kDa protein induced by cold acclimation in wheat. Plant Cell Physiol. 1993, 34, 59–65. [Google Scholar]
- King, S.W.; Joshi, C.P.; Nguyen, H.T. DNA sequence of an ABA-responsive gene (rab 15) from water-stressed wheat roots. Plant Mol. Biol. 1992, 18, 119–121. [Google Scholar] [CrossRef]
- Labhilili, M.; Joudrier, P.; Gautier, M.O. Characterization of cDNAs encoding Triticum durum dehydrins and their expression patterns in cultivars that differ in drought tolerance. Plant Sci. 1995, 112, 219–230. [Google Scholar] [CrossRef]
- Masmoudi, K.; Brini, F.A.; Hassairi, A.; Ellouz, R. Isolation and characterization of a differentially expressed sequence tag from Triticum durum salt-stressed roots. Plant Physiol. Biochem. 2001, 39, 971–979. [Google Scholar] [CrossRef]
- Jia, X.Y.; Xu, C.Y.; Jing, R.L.; Li, R.Z.; Mao, X.G.; Wang, J.P.; Chang, X.P. Molecular cloning and characterization of wheat calreticulin (CRT) gene involved in drought-stressed responses. J. Exp. Bot. 2008, 59, 739–751. [Google Scholar] [CrossRef]
- Rahman, L.N.; Bamm, V.V.; Voyer, J.A.; Smith, G.S.; Chen, L.; Yaish, M.W.; Moffatt, B.A.; Dutcher, J.R.; Harauz, G. Zinc induces disorder-to-order transitions in free and membrane-associated Thellungiella salsuginea dehydrins TsDHN-1 and TsDHN-2: A solution CD and solid-state ATR-FTIR study. Amino Acids 2011, 40, 1485–1502. [Google Scholar] [CrossRef]
- Kovacs, D.; Kalmar, E.; Torok, Z.; Tompa, P. Chaperone activity of ERD10 and ERD14, two disordered stress-related plant proteins. Plant Physiol. 2008, 147, 381–390. [Google Scholar] [CrossRef]
- Tompa, P.; Banki, P.; Bokor, M.; Kamasa, P.; Kovacs, D.; Lasanda, G.; Tompa, K. Protein-water and protein-buffer interactions in the aqueous solution of an intrinsically unstructured plant dehydrin: NMR intensity and DSC aspects. Biophys. J. 2006, 91, 2243–2249. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Rikkerink, E.H.A.; Jones, W.T.; Uversky, V.N. Multifarious roles of intrinsic disorder in proteins illustrate its broad impact on plant biology. Plant Cell. 2013, 25, 38–55. [Google Scholar] [CrossRef]
- Boucher, V.; Buitink, J.; Lin, X.; Boudet, J.; Hoekstra, F.A.; Hundertmark, M.; Renard, D.; Leprince, O. MtPM25 is an atypical hydrophobic late embryogenesis-abundant protein that dissociates cold and desiccation-aggregated proteins. Plant Cell Environ. 2010, 33, 418–430. [Google Scholar] [CrossRef]
- Mishra, R.C.; Richa; Grover, A. Constitutive over-expression of rice ClpD1 protein enhances tolerance to salt and desiccation stresses in transgenic Arabidopsis plants. Plant Sci. 2016, 250, 69–78. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Shi, S. Physiological and Proteomic Responses of Contrasting Alfalfa (Medicago sativa L.) Varieties to PEG-Induced Osmotic Stress. Front. Plant Sci. 2018, 9. [Google Scholar] [CrossRef]
- Xu, J.; Wang, X.; Guo, W. The cytochrome P450 superfamily: Key players in plant development and defense. J. Integr. Agric. 2015, 14, 1673–1686. [Google Scholar] [CrossRef] [Green Version]
- Schweighofer, A.; Hirt, H.; Meskiene, I. Plant PP2C phosphatases: Emerging functions in stress signaling. Trends Plant Sci. 2004, 9, 236–243. [Google Scholar] [CrossRef] [PubMed]
- Pudelski, B.; Schock, A.; Hoth, S.; Radchuk, R.; Weber, H.; Hofmann, J.; Sonnewald, U.; Soll, J.; Philippar, K. The plastid outer envelope protein OEP16 affects metabolic fluxes during ABA-controlled seed development and germination. J. Exp. Bot. 2012, 63, 1919–1936. [Google Scholar] [CrossRef]
- Guo, L.; Yang, H.; Zhang, X.; Yang, S. Lipid transfer protein 3 as a target of MYB96 mediates freezing and drought stress in Arabidopsis. J. Exp. Bot. 2013, 64, 1755–1767. [Google Scholar] [CrossRef]
- Cameron, K.D.; Teece, M.A.; Smart, L.B. Increased accumulation of cuticular wax and expression of lipid transfer protein in response to periodic drying events in leaves of tree tobacco. Plant Physiol. 2006, 140, 176–183. [Google Scholar] [CrossRef]
- Pallavi, S.; Jha, A.B.; Dubey, R.S.; Pessarakli, M. Reactive oxygen species, oxidative damage, and antioxidative defense mechanism in plants under stressful conditions. J. Bot. 2012, 2012, 217037. [Google Scholar]
- Hong, Y.; Zheng, S.; Wang, X. Dual functions of phospholipase Dalpha1 in plant response to drought. Mol. Plant 2008, 1, 262–269. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| NO | 113 | 114 | 115 | 116 | 117 | 118 | 119 | 121 |
|---|---|---|---|---|---|---|---|---|
| 1 | CK1 | CK2 | PEG1 | PEG2 | POOL1 | |||
| 2 | CK3 | CK4 | PEG3 | PEG4 | POOL2 |
| Accession Number of Protein | Name of Protein | Accession Number of Related Gene | Primer Sequences (5′-3′) | Product Length (bp) |
|---|---|---|---|---|
| I7FHT3 | Glutathione transferase 2 | TraesCS1D02G190000 | F: GCCCGTGCTCATCCACAA | 220 |
| R: CAGCCCCTCCGCCTTCT | ||||
| A0A3B6NJS8 | Catalase | TraesCS6A02G041700 | F: CCCAAACTACCTGATGCTCC | 203 |
| R: TGATCCTCGTCTTCTCCCTTC | ||||
| A0A077RXE4 | Delta-1-pyrroline-5-carboxylate synthase | TraesCS3B02G395900 | F: ACCCTGAAGGCTGGAAAGATA | 176 |
| R: GCATCAGGACGAGACTCAAAA | ||||
| A0A3B5Y545 | late embryogenesis abundant protein | TraesCS1A02G364000 | F: GGACCAGACCGCCAGCAC | 261 |
| R: CCCATGCCCAGCGTGTT | ||||
| W5GYX5 | GAPDH | TraesCS6D02G196300 | F: GTTTGGCATTGTTGAGGGTT | 268 |
| R: ATCATAGGTTGCTGGCTTCG |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, Y.; Zhang, X.; Huang, G.; Feng, F.; Liu, X.; Guo, R.; Gu, F.; Zhong, X.; Mei, X. iTRAQ-Based Quantitative Analysis of Responsive Proteins Under PEG-Induced Drought Stress in Wheat Leaves. Int. J. Mol. Sci. 2019, 20, 2621. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20112621
Wang Y, Zhang X, Huang G, Feng F, Liu X, Guo R, Gu F, Zhong X, Mei X. iTRAQ-Based Quantitative Analysis of Responsive Proteins Under PEG-Induced Drought Stress in Wheat Leaves. International Journal of Molecular Sciences. 2019; 20(11):2621. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20112621
Chicago/Turabian StyleWang, Yajing, Xinying Zhang, Guirong Huang, Fu Feng, Xiaoying Liu, Rui Guo, Fengxue Gu, Xiuli Zhong, and Xurong Mei. 2019. "iTRAQ-Based Quantitative Analysis of Responsive Proteins Under PEG-Induced Drought Stress in Wheat Leaves" International Journal of Molecular Sciences 20, no. 11: 2621. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20112621