EGF Receptor Stalls upon Activation as Evidenced by Complementary Fluorescence Correlation Spectroscopy and Fluorescence Recovery after Photobleaching Measurements

 ,
,
Abstract
:1. Introduction
2. Results
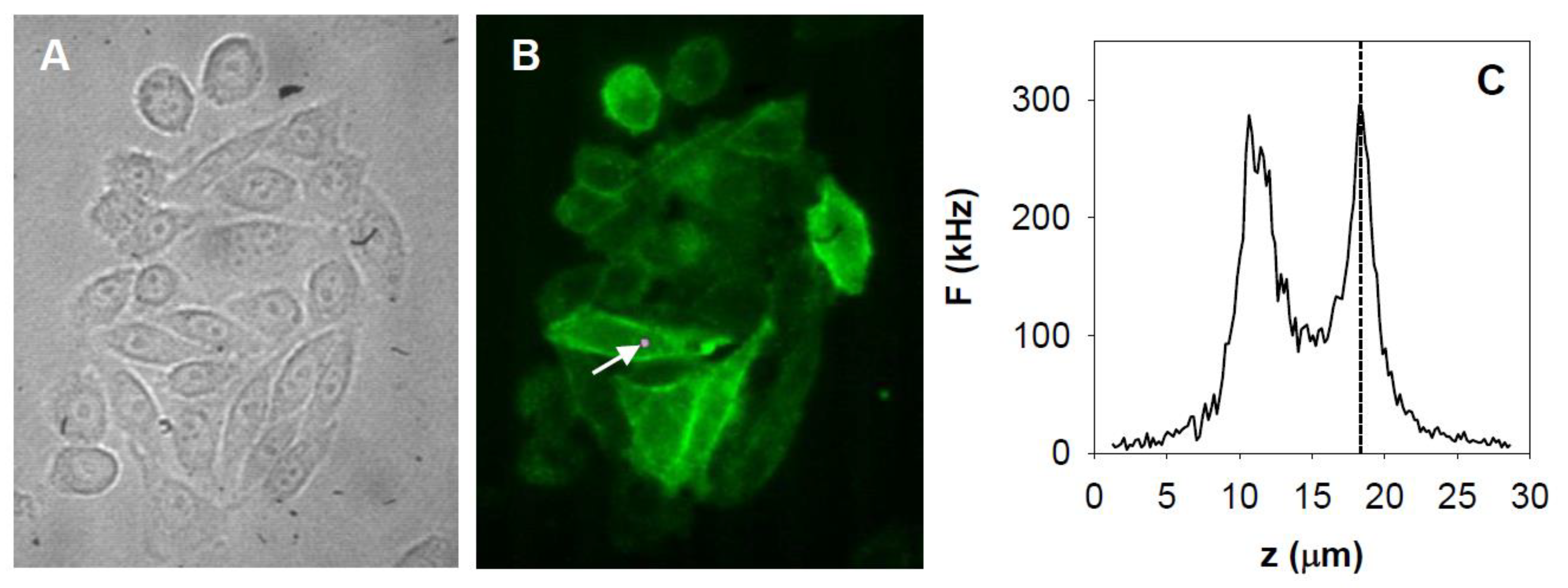
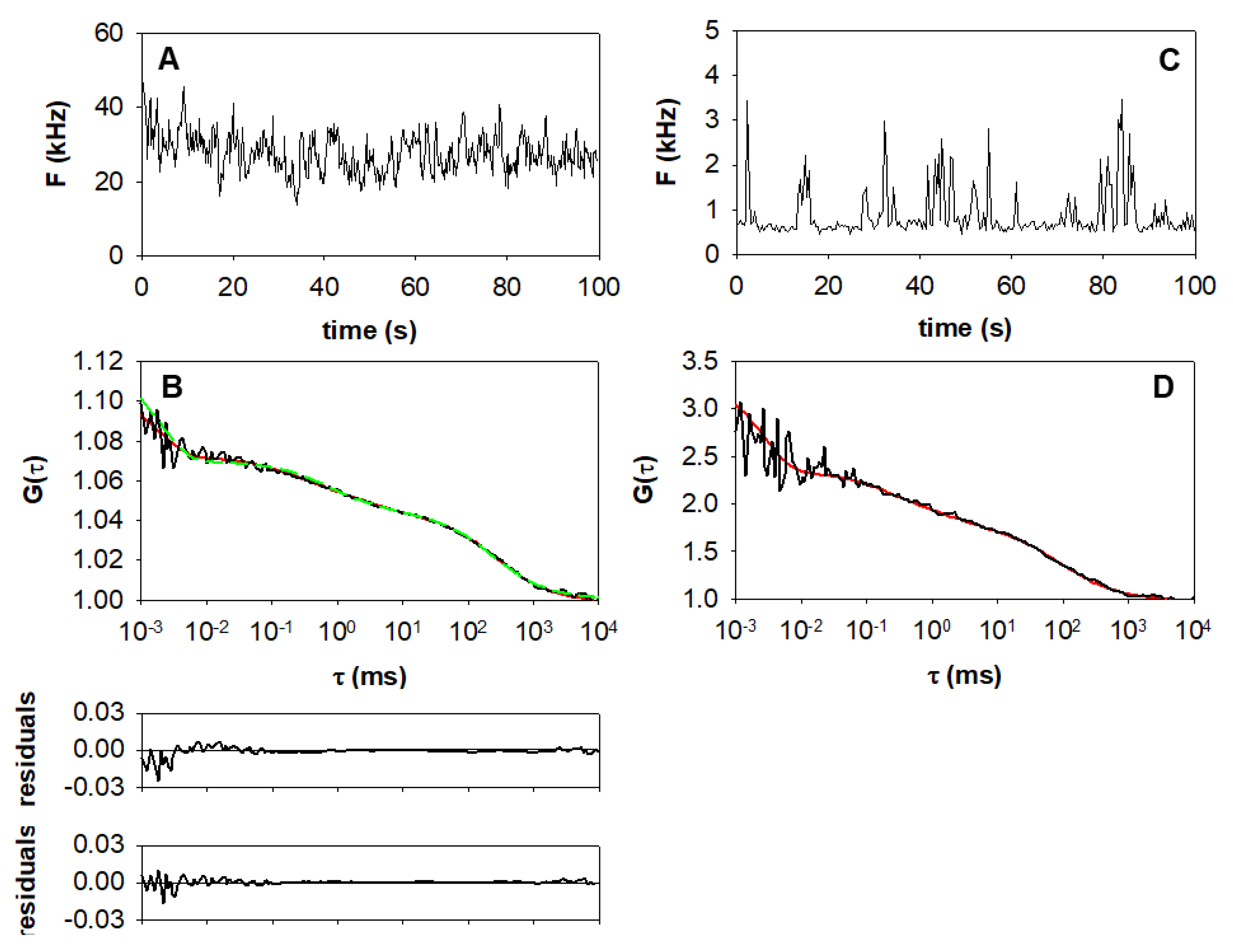
2.1. Measurement and Analysis of Autocorrelation Functions
2.2. FCS Measurements on EGF-Stimulated Cells
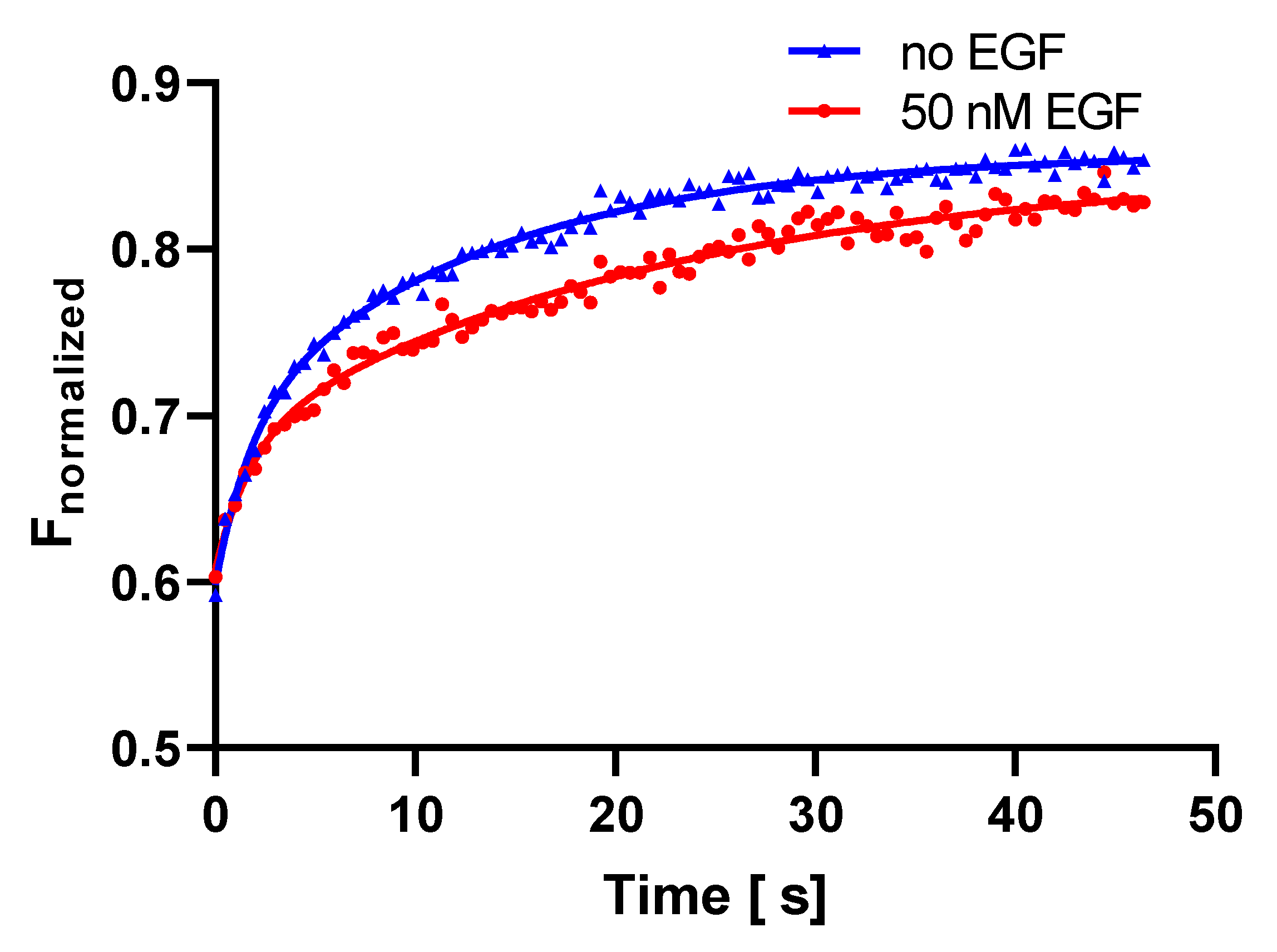
2.3. FRAP Measurements
2.4. Signaling Competence of EGFR–eGFP
3. Discussion
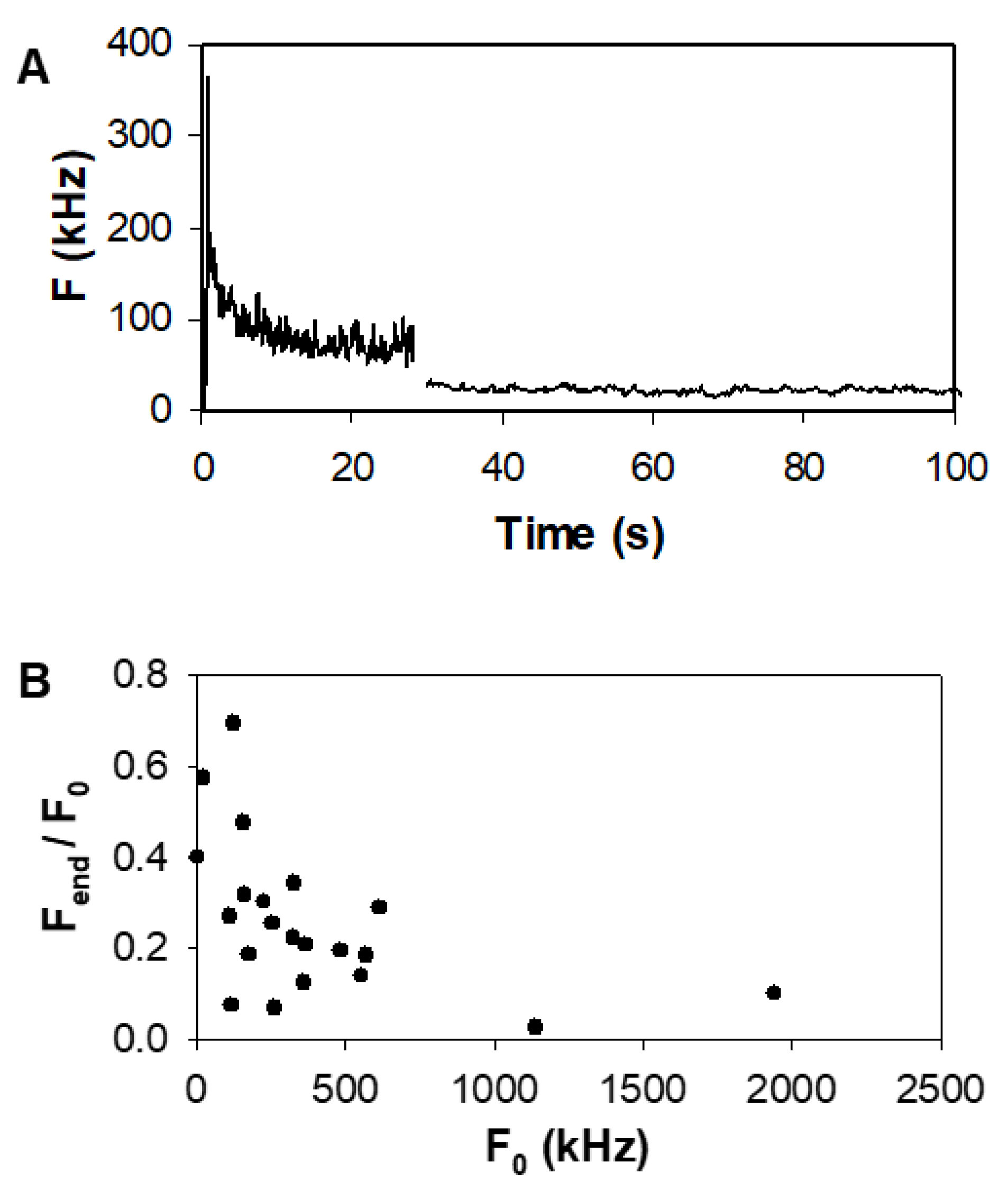
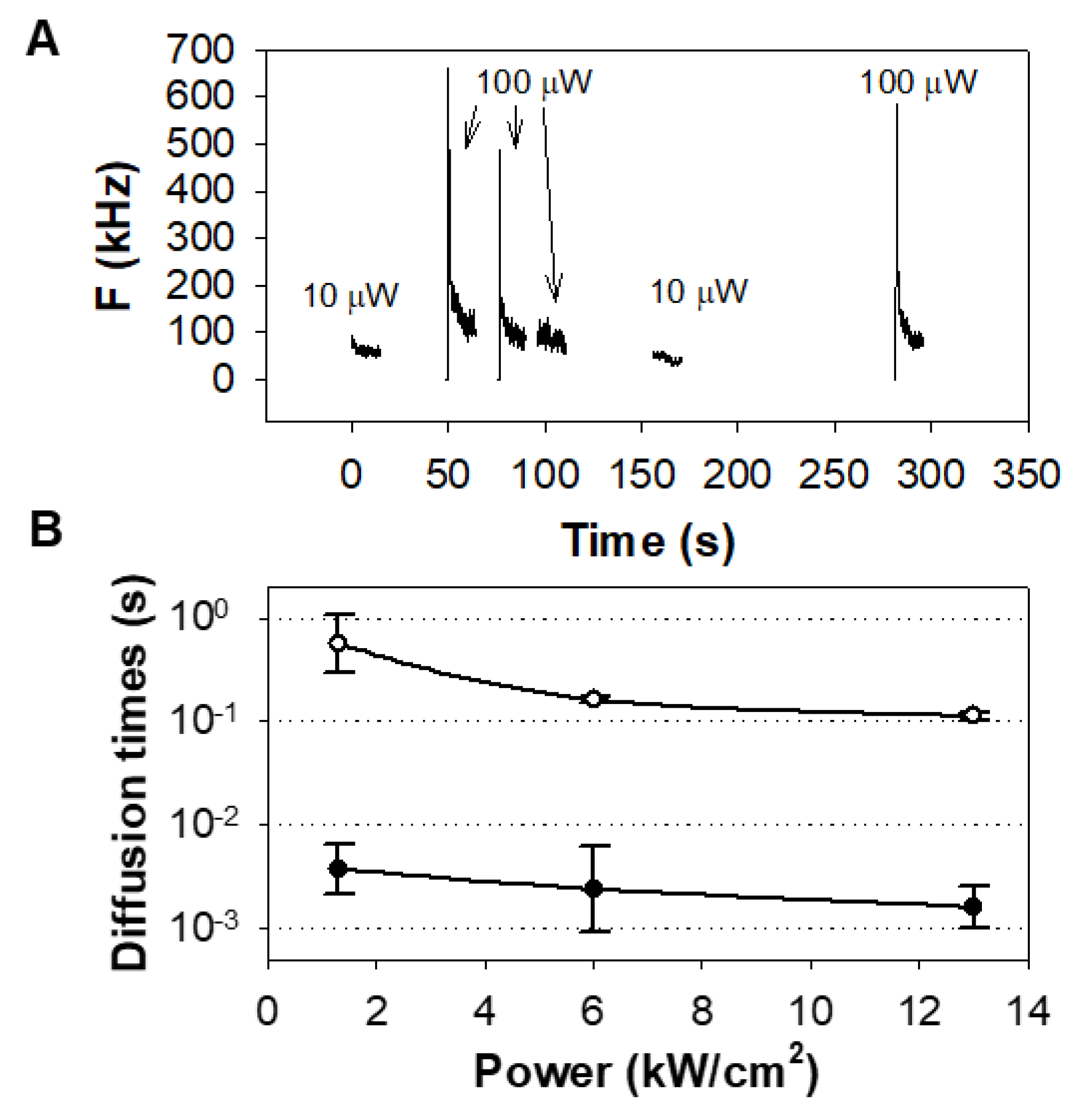
3.1. Effect of Bleaching/Light-Induced Blinking on Autocorrelation Measurements
3.2. Effects of Receptor Stimulation on Autocorrelation Measurements
3.3. FRAP Analysis Indicates Slowing Down of Long-Range Diffusion
4. Materials and Methods
4.1. Cell Culture
4.2. Fluorescence Correlation Spectroscopy: Measurement of Fluorescence Autocorrelations
4.3. Evaluation of Autocorrelation Functions
4.4. Background Correction
4.5. Experimental Determination of the Dimensions of the Ellipsoid of the Confocal Detection Volume, and Calculation of the Diffusion Coefficient of EGFR–eGFP
4.6. Statistical Analysis of FCS Data
4.7. Fluorescence Recovery after Photobleaching (FRAP)
4.8. Western Blot Analysis of Tyrosine Phosphorylation after Receptor Stimulation with EGF
4.9. Measurement of Ca2+ Responses upon EGF Stimulation
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| EGF | Epidermal growth factor |
| EGFR | Epidermal growth factor receptor |
| FCS | Fluorescence correlation spectroscopy |
| FRAP | Fluorescence recovery after photobleaching |
Appendix A
Appendix A.1. Characterization of the Bleachable Fraction of EGFR Molecules

Appendix A.2. Effect of EGF Stimulation on F1-10 Cells—Individual Data

Appendix A.3. Fluorescence Recovery after Photobleaching Curves Measured with F1-10 Cells

References
- Wee, P.; Wang, Z. Epidermal Growth Factor Receptor Cell Proliferation Signaling Pathways. Cancers 2017, 9, 52. [Google Scholar] [Green Version]
- Bublil, E.M.; Yarden, Y. The EGF receptor family: Spearheading a merger of signaling and therapeutics. Curr. Opin. Cell Biol. 2007, 19, 124–134. [Google Scholar] [CrossRef]
- Marmor, M.D.; Skaria, K.B.; Yarden, Y. Signal transduction and oncogenesis by ErbB/HER receptors. Int. J. Radiat. Oncol. Biol. Phys. 2004, 58, 903–913. [Google Scholar] [CrossRef] [PubMed]
- Yarden, Y.; Sliwkowski, M.X. Untangling the ErbB signalling network. Nat. Rev. Mol. Cell Biol. 2001, 2, 127–137. [Google Scholar] [CrossRef]
- Ogiso, H.; Ishitani, R.; Nureki, O.; Fukai, S.; Yamanaka, M.; Kim, J.H.; Saito, K.; Sakamoto, A.; Inoue, M.; Shirouzu, M.; et al. Crystal structure of the complex of human epidermal growth factor and receptor extracellular domains. Cell 2002, 110, 775–787. [Google Scholar] [CrossRef]
- Jovin, T.M. Pinning down the EGF receptor. Biophys. J. 2014, 107, 2486–2488. [Google Scholar] [CrossRef] [PubMed]
- Kovacs, E.; Zorn, J.A.; Huang, Y.; Barros, T.; Kuriyan, J. A structural perspective on the regulation of the epidermal growth factor receptor. Annu. Rev. Biochem. 2015, 84, 739–764. [Google Scholar] [CrossRef] [PubMed]
- Tynan, C.J.; Lo Schiavo, V.; Zanetti-Domingues, L.; Needham, S.R.; Roberts, S.K.; Hirsch, M.; Rolfe, D.J.; Korovesis, D.; Clarke, D.T.; Martin-Fernandez, M.L. A tale of the epidermal growth factor receptor: The quest for structural resolution on cells. Methods 2016, 95, 86–93. [Google Scholar] [CrossRef]
- Ward, C.W.; Lawrence, M.C.; Streltsov, V.A.; Adams, T.E.; McKern, N.M. The insulin and EGF receptor structures: New insights into ligand-induced receptor activation. Trends Biochem. Sci. 2007, 32, 129–137. [Google Scholar] [CrossRef]
- Yarden, Y.; Schlessinger, J. Epidermal growth factor induces rapid, reversible aggregation of the purified epidermal growth factor receptor. Biochemistry 1987, 26, 1443–1451. [Google Scholar] [CrossRef]
- Yarden, Y.; Schlessinger, J. Self-phosphorylation of epidermal growth factor receptor: Evidence for a model of intermolecular allosteric activation. Biochemistry 1987, 26, 1434–1442. [Google Scholar] [CrossRef] [PubMed]
- Van Belzen, N.; Rijken, P.J.; Hage, W.J.; de Laat, S.W.; Verkleij, A.J.; Boonstra, J. Direct visualization and quantitative analysis of epidermal growth factor-induced receptor clustering. J. Cell. Physiol. 1988, 134, 413–420. [Google Scholar] [CrossRef] [PubMed]
- Lemmon, M.A.; Bu, Z.; Ladbury, J.E.; Zhou, M.; Pinchasi, D.; Lax, I.; Engelman, D.M.; Schlessinger, J. Two EGF molecules contribute additively to stabilization of the EGFR dimer. EMBO J. 1997, 16, 281–294. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gadella, T.W., Jr.; Jovin, T.M. Oligomerization of epidermal growth factor receptors on A431 cells studied by time-resolved fluorescence imaging microscopy. A stereochemical model for tyrosine kinase receptor activation. J. Cell Biol. 1995, 129, 1543–1558. [Google Scholar] [CrossRef] [PubMed]
- Stern, D.F.; Kamps, M.P.; Cao, H. Oncogenic activation of p185neu stimulates tyrosine phosphorylation in vivo. Mol. Cell. Biol. 1988, 8, 3969–3973. [Google Scholar] [CrossRef] [PubMed]
- Moriki, T.; Maruyama, H.; Maruyama, I.N. Activation of preformed EGF receptor dimers by ligand-induced rotation of the transmembrane domain. J. Mol. Biol. 2001, 311, 1011–1026. [Google Scholar] [CrossRef] [PubMed]
- Saffarian, S.; Li, Y.; Elson, E.L.; Pike, L.J. Oligomerization of the EGF receptor investigated by live cell fluorescence intensity distribution analysis. Biophys. J. 2007, 93, 1021–1031. [Google Scholar] [CrossRef] [PubMed]
- Bocharov, E.V.; Bragin, P.E.; Pavlov, K.V.; Bocharova, O.V.; Mineev, K.S.; Polyansky, A.A.; Volynsky, P.E.; Efremov, R.G.; Arseniev, A.S. The Conformation of the Epidermal Growth Factor Receptor Transmembrane Domain Dimer Dynamically Adapts to the Local Membrane Environment. Biochemistry 2017, 56, 1697–1705. [Google Scholar] [CrossRef] [PubMed]
- Kaplan, M.; Narasimhan, S.; de Heus, C.; Mance, D.; van Doorn, S.; Houben, K.; Popov-Celeketic, D.; Damman, R.; Katrukha, E.A.; Jain, P.; et al. EGFR Dynamics Change during Activation in Native Membranes as Revealed by NMR. Cell 2016, 167, 1241–1251. [Google Scholar] [CrossRef]
- Nagy, P.; Claus, J.; Jovin, T.M.; Arndt-Jovin, D.J. Distribution of resting and ligand-bound ErbB1 and ErbB2 receptor tyrosine kinases in living cells using number and brightness analysis. Proc. Natl. Acad. Sci. USA 2010, 107, 16524–16529. [Google Scholar] [CrossRef] [Green Version]
- Day, C.A.; Kraft, L.J.; Kang, M.; Kenworthy, A.K. Analysis of protein and lipid dynamics using confocal fluorescence recovery after photobleaching (FRAP). Curr. Protoc. Cytom. 2012, 62, 2–19. [Google Scholar] [CrossRef] [PubMed]
- Kusumi, A.; Tsunoyama, T.A.; Hirosawa, K.M.; Kasai, R.S.; Fujiwara, T.K. Tracking single molecules at work in living cells. Nat. Chem. Biol. 2014, 10, 524–532. [Google Scholar] [CrossRef] [PubMed]
- Valley, C.C.; Arndt-Jovin, D.J.; Karedla, N.; Steinkamp, M.P.; Chizhik, A.I.; Hlavacek, W.S.; Wilson, B.S.; Lidke, K.A.; Lidke, D.S. Enhanced dimerization drives ligand-independent activity of mutant epidermal growth factor receptor in lung cancer. Mol. Biol. Cell 2015, 26, 4087–4099. [Google Scholar] [CrossRef] [PubMed]
- Low-Nam, S.T.; Lidke, K.A.; Cutler, P.J.; Roovers, R.C.; van Bergen en Henegouwen, P.M.; Wilson, B.S.; Lidke, D.S. ErbB1 dimerization is promoted by domain co-confinement and stabilized by ligand binding. Nat. Struct. Mol. Biol. 2011, 18, 1244–1249. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brock, R.; Vamosi, G.; Vereb, G.; Jovin, T.M. Rapid characterization of green fluorescent protein fusion proteins on the molecular and cellular level by fluorescence correlation microscopy. Proc. Natl. Acad. Sci. USA 1999, 96, 10123–10128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haupts, U.; Maiti, S.; Schwille, P.; Webb, W.W. Dynamics of fluorescence fluctuations in green fluorescent protein observed by fluorescence correlation spectroscopy. Proc. Natl. Acad. Sci. USA 1998, 95, 13573–13578. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saxton, M.J. Anomalous diffusion due to obstacles: A Monte Carlo study. Biophys. J. 1994, 66 Pt 1, 394–401. [Google Scholar] [CrossRef]
- Wachsmuth, M.; Waldeck, W.; Langowski, J. Anomalous diffusion of fluorescent probes inside living cell nuclei investigated by spatially-resolved fluorescence correlation spectroscopy. J. Mol. Biol. 2000, 298, 677–689. [Google Scholar] [CrossRef] [Green Version]
- Nagle, J.F. Long tail kinetics in biophysics? Biophys. J. 1992, 63, 366–370. [Google Scholar] [CrossRef] [Green Version]
- Brock, R.; Jovin, T.M. Heterogeneity of signal transduction at the subcellular level: Microsphere-based focal EGF receptor activation and stimulation of Shc translocation. J. Cell Sci. 2001, 114 Pt 13, 2437–2447. [Google Scholar]
- Sunada, H.; Peacock, J.; Mendelsohn, J. Ligand induced internalization of epidermal growth factor receptors by A431 cells decreases at high cell densities in culture. Growth Factors 1991, 5, 45–55. [Google Scholar] [CrossRef] [PubMed]
- Margolis, B.; Rhee, S.G.; Felder, S.; Mervic, M.; Lyall, R.; Levitzki, A.; Ullrich, A.; Zilberstein, A.; Schlessinger, J. EGF induces tyrosine phosphorylation of phospholipase C-II: A potential mechanism for EGF receptor signaling. Cell 1989, 57, 1101–1107. [Google Scholar] [CrossRef]
- Garcia-Parajo, M.F.; Segers-Nolten, G.M.; Veerman, J.A.; Greve, J.; van Hulst, N.F. Real-time light-driven dynamics of the fluorescence emission in single green fluorescent protein molecules. Proc. Natl. Acad. Sci. USA 2000, 97, 7237–7242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peterman, E.J.; Brasselet, S.; Moerner, W.E. The Fluorescence Dynamics of Single Molecules of Green Fluorescent Protein. J. Phys. Chem. A 1999, 103, 10553–10560. [Google Scholar] [CrossRef]
- Widengren, J.; Terry, B.; Rigler, R. Protonation kinetics of GFP and FITC investigated by FCS-aspects of the use of fluorescent indicators for measuring pH. Chem. Phys. 1999, 249, 259–271. [Google Scholar] [CrossRef]
- Wachsmuth, M.; Weidemann, T.; Muller, G.; Hoffmann-Rohrer, U.W.; Knoch, T.A.; Waldeck, W.; Langowski, J. Analyzing intracellular binding and diffusion with continuous fluorescence photobleaching. Biophys. J. 2003, 84, 3353–3363. [Google Scholar] [CrossRef]
- Van Bergen en Henegouwen, P.M.; Defize, L.H.; de Kroon, J.; van Damme, H.; Verkleij, A.J.; Boonstra, J. Ligand-induced association of epidermal growth factor receptor to the cytoskeleton of A431 cells. J. Cell. Biochem. 1989, 39, 455–465. [Google Scholar] [CrossRef]
- Chung, Y.D.; Sinzinger, M.D.; Bovee-Geurts, P.; Krause, M.; Dinkla, S.; Joosten, I.; Koopman, W.J.; Adjobo-Hermans, M.J.; Brock, R. Analyzing the homeostasis of signaling proteins by a combination of Western blot and fluorescence correlation spectroscopy. Biophys. J. 2011, 101, 2807–2815. [Google Scholar] [CrossRef]
- Huang, Y.; Bharill, S.; Karandur, D.; Peterson, S.M.; Marita, M.; Shi, X.; Kaliszewski, M.J.; Smith, A.W.; Isacoff, E.Y.; Kuriyan, J. Molecular basis for multimerization in the activation of the epidermal growth factor receptor. Elife 2016, 5, e14107. [Google Scholar] [CrossRef]
- Clayton, A.H.; Orchard, S.G.; Nice, E.C.; Posner, R.G.; Burgess, A.W. Predominance of activated EGFR higher-order oligomers on the cell surface. Growth Factors 2008, 26, 316–324. [Google Scholar] [CrossRef]
- Kozer, N.; Barua, D.; Henderson, C.; Nice, E.C.; Burgess, A.W.; Hlavacek, W.S.; Clayton, A.H. Recruitment of the adaptor protein Grb2 to EGFR tetramers. Biochemistry 2014, 53, 2594–2604. [Google Scholar] [CrossRef] [PubMed]
- Liu, P.; Sudhaharan, T.; Koh, R.M.; Hwang, L.C.; Ahmed, S.; Maruyama, I.N.; Wohland, T. Investigation of the dimerization of proteins from the epidermal growth factor receptor family by single wavelength fluorescence cross-correlation spectroscopy. Biophys. J. 2007, 93, 684–698. [Google Scholar] [CrossRef] [PubMed]
- Ma, X.; Ahmed, S.; Wohland, T. EGFR activation monitored by SW-FCCS in live cells. Front. Biosci. 2011, 3, 22–32. [Google Scholar]
- Bag, N.; Huang, S.; Wohland, T. Plasma Membrane Organization of Epidermal Growth Factor Receptor in Resting and Ligand-Bound States. Biophys. J. 2015, 109, 1925–1936. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rijken, P.J.; Hage, W.J.; van Bergen en Henegouwen, P.M.; Verkleij, A.J.; Boonstra, J. Epidermal growth factor induces rapid reorganization of the actin microfilament system in human A431 cells. J. Cell Sci. 1991, 100 Pt 3, 491–499. [Google Scholar] [PubMed]
- Rijken, P.J.; Post, S.M.; Hage, W.J.; van Bergen en Henegouwen, P.M.; Verkleij, A.J.; Boonstra, J. Actin polymerization localizes to the activated epidermal growth factor receptor in the plasma membrane, independent of the cytosolic free calcium transient. Exp. Cell Res. 1995, 218, 223–232. [Google Scholar] [CrossRef] [PubMed]
- Ibach, J.; Radon, Y.; Gelleri, M.; Sonntag, M.H.; Brunsveld, L.; Bastiaens, P.I.; Verveer, P.J. Single Particle Tracking Reveals that EGFR Signaling Activity Is Amplified in Clathrin-Coated Pits. PLoS ONE 2015, 10, e0143162. [Google Scholar] [CrossRef] [PubMed]
- Brock, R.; Jovin, T.M. Fluorescence correlation microscopy (FCM)-fluorescence correlation spectroscopy (FCS) taken into the cell. Cell. Mol. Biol. 1998, 44, 847–856. [Google Scholar] [PubMed]
- Widengren, J.; Mets, Ü. Triplet state monitoring by fluorescence correlation spectroscopy. J. Fluoresc. 1994, 4, 255–258. [Google Scholar] [CrossRef]
- Widengren, J.; Mets, Ü.; Rigler, R. Fluorescence correlation spectroscopy of triplet state in solution: A theoretical and experimental study. J. Phys. Chem. 1995, 99, 13368–13379. [Google Scholar] [CrossRef]
- Koppel, D.E.; Axelrod, D.; Schlessinger, J.; Elson, E.; Webb, W.W. Dynamics of fluorescence marker concentrations as a probe of mobility. Biophys. J. 1976, 16, 1315–1329. [Google Scholar] [CrossRef]
- Majewski, V. Fluoreszenz-Korrelations-Spektroskopie-FRET und Kreuz-Korrelationsanalyse an Biologischen Makromolekülen. Master’s Thesis, Universität Hannover, Hannover, Germany, 1998. [Google Scholar]
- Grynkiewicz, G.; Poenie, M.; Tsien, R.Y. A new generation of Ca2+ indicators with greatly improved fluorescence properties. J. Biol. Chem. 1985, 260, 3440–3450. [Google Scholar] [PubMed]
- Yavas, S.; Machan, R.; Wohland, T. The Epidermal Growth Factor Receptor Forms Location-Dependent Complexes in Resting Cells. Biophys. J. 2016, 111, 2241–2254. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Widengren, J.; Rigler, R. Mechanisms of photobleaching investigated by fluorescence correlation spectroscopy. Bioimaging 1996, 4, 149–157. [Google Scholar] [CrossRef]
- Vámosi, G.; Mücke, N.; Müller, G.; Krieger, J.W.; Curth, U.; Langowski, J.; Tóth, K. EGFP oligomers as natural fluorescence and hydrodynamic standards. Sci. Rep. 2016, 6, 33022. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| FCS | Model 1 2-Component Free | Model 2 Blinking + 2-Comp. Anomalous | ||
|---|---|---|---|---|
| Before EGF | After EGF | Before EGF | After EGF | |
| D1 (μm2/s) | 24.9 ± 8.7 * | 18.8 ± 9.1 * | 19.7 ± 8.3 | 16.3 ± 9.4 |
| D2 (μm2/s) | 0.107 ± 0.037 * | 0.057 ± 0.023 * | 0.117 ± 0.051 * | 0.066 ± 0.03 * |
| w2 | 0.77 ± 0.05 | 0.63 ± 0.12 | 0.70 ± 0.06 | 0.63 ± 0.12 |
| α1 | n.a. | n.a. | 0.94 ± 0.17 | 0.82 ± 0.12 ** |
| α2 | n.a. | n.a. | 0.99 ± 0.07 | 1.02 ± 0.08 |
| Θc | n.a. | n.a. | 0.09 ± 0.01 | 0.09 ± 0.01 |
| τc (μs) | n.a. | n.a. | 181 ± 21 | 187 ± 22 |
| T | 0.56 ± 0.11 | 0.58 ± 0.12 | 0.47 ± 0.08 | 0.51 ± 0.14 |
| τtr (μs) | 1.7 ± 0.4 | 2.1 ± 0.8 | 2.3 ± 0.5 | 2.5 ± 0.9 |
| mse | 3.9 | 6.5 | 3.5 | 3.5 |
| Fluorescence recovery after photobleaching (FRAP) | Before EGF | After EGF | ||
| D2 (μm2/s) from τ2 | 0.067 | 0.040 | ||
| w2 | 0.64 | 0.69 | ||
| Immobile fraction | 0.14 | 0.15 | ||
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vámosi, G.; Friedländer-Brock, E.; Ibrahim, S.M.; Brock, R.; Szöllősi, J.; Vereb, G. EGF Receptor Stalls upon Activation as Evidenced by Complementary Fluorescence Correlation Spectroscopy and Fluorescence Recovery after Photobleaching Measurements. Int. J. Mol. Sci. 2019, 20, 3370. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20133370
Vámosi G, Friedländer-Brock E, Ibrahim SM, Brock R, Szöllősi J, Vereb G. EGF Receptor Stalls upon Activation as Evidenced by Complementary Fluorescence Correlation Spectroscopy and Fluorescence Recovery after Photobleaching Measurements. International Journal of Molecular Sciences. 2019; 20(13):3370. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20133370
Chicago/Turabian StyleVámosi, György, Elza Friedländer-Brock, Shehu M. Ibrahim, Roland Brock, János Szöllősi, and György Vereb. 2019. "EGF Receptor Stalls upon Activation as Evidenced by Complementary Fluorescence Correlation Spectroscopy and Fluorescence Recovery after Photobleaching Measurements" International Journal of Molecular Sciences 20, no. 13: 3370. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20133370