Dynamic Changes in Metabolite Accumulation and the Transcriptome during Leaf Growth and Development in Eucommia ulmoides


Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
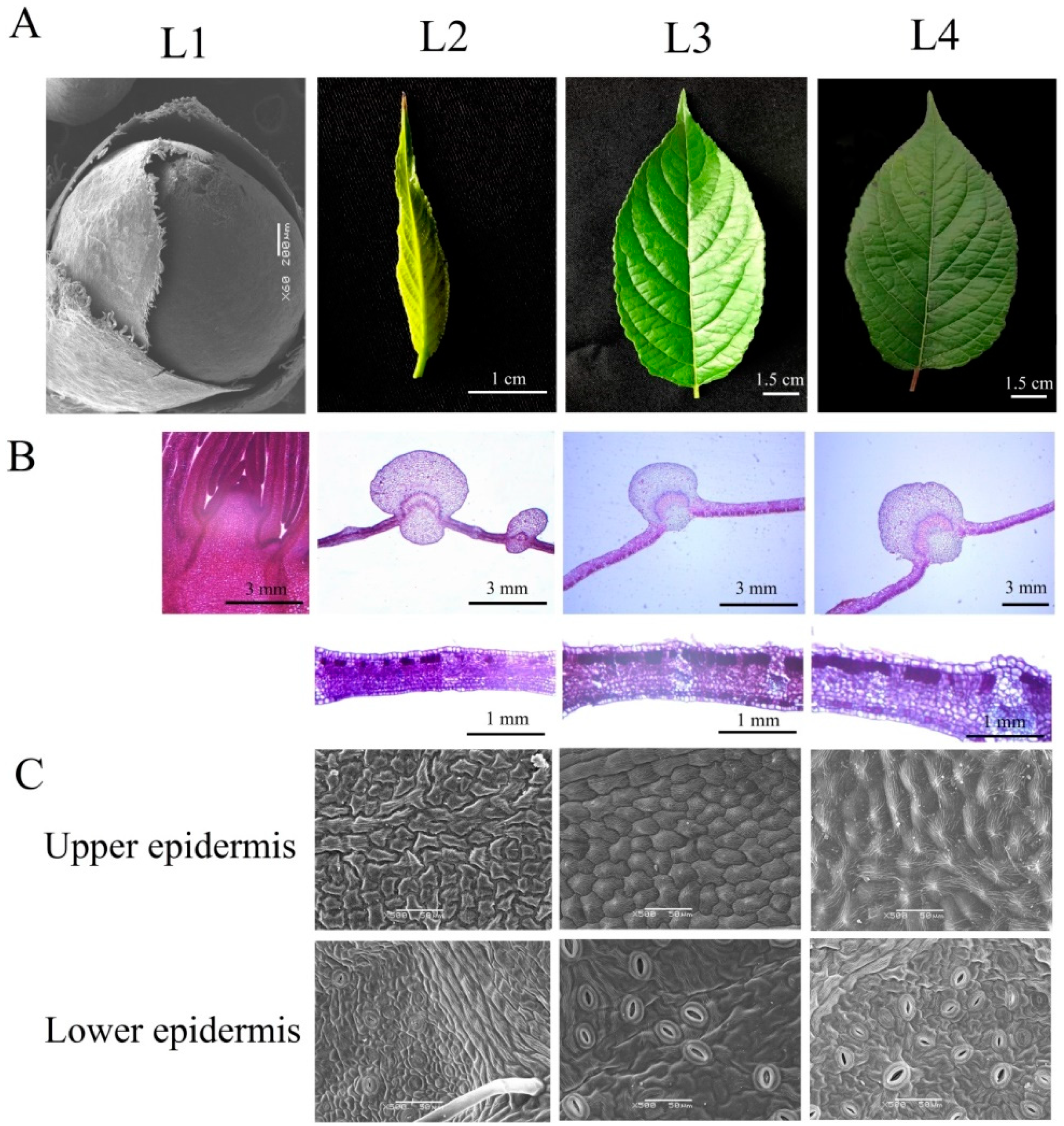
2.1. Anatomical and Physiological Analysis of E. ulmoides Leaves
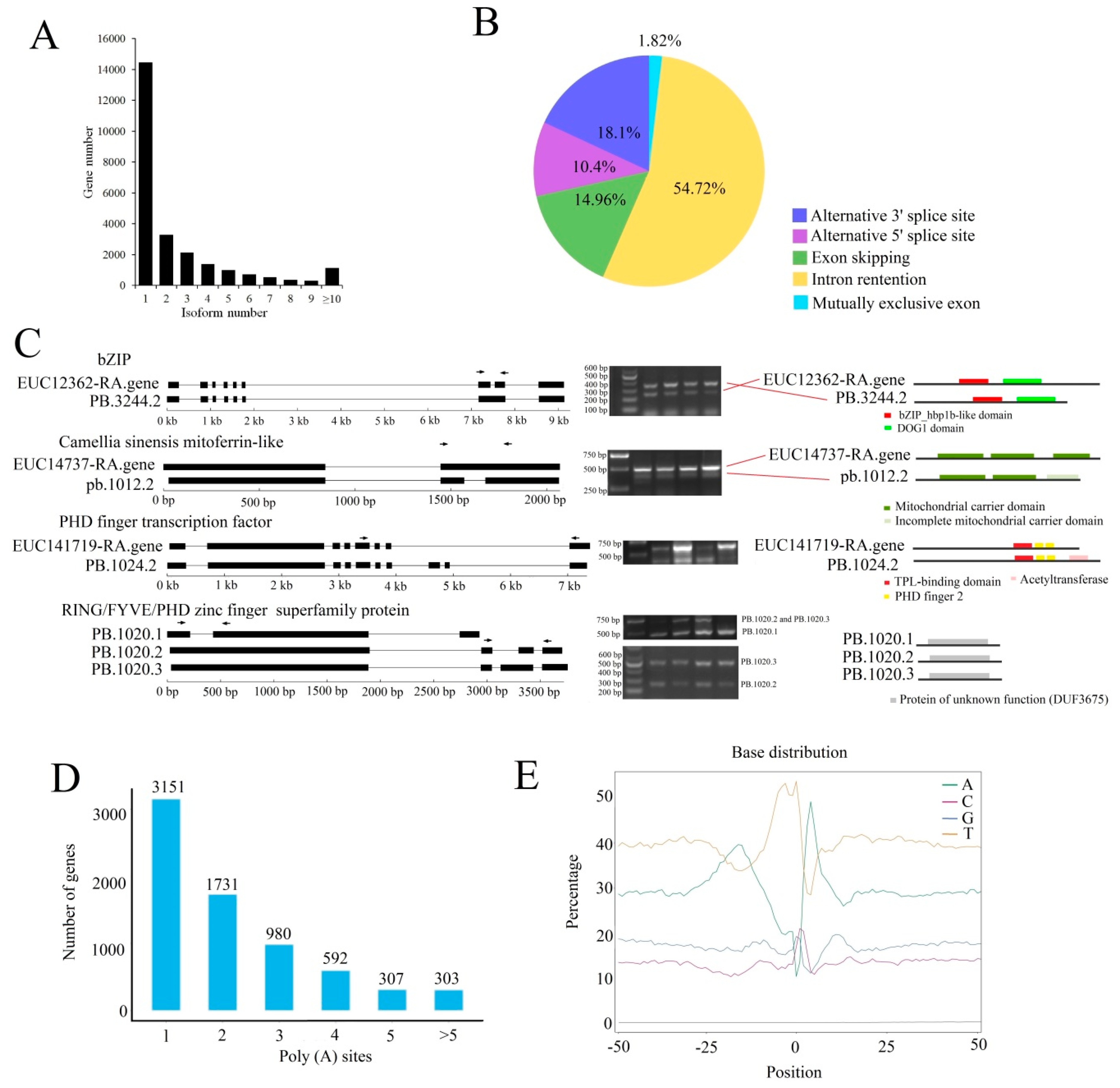
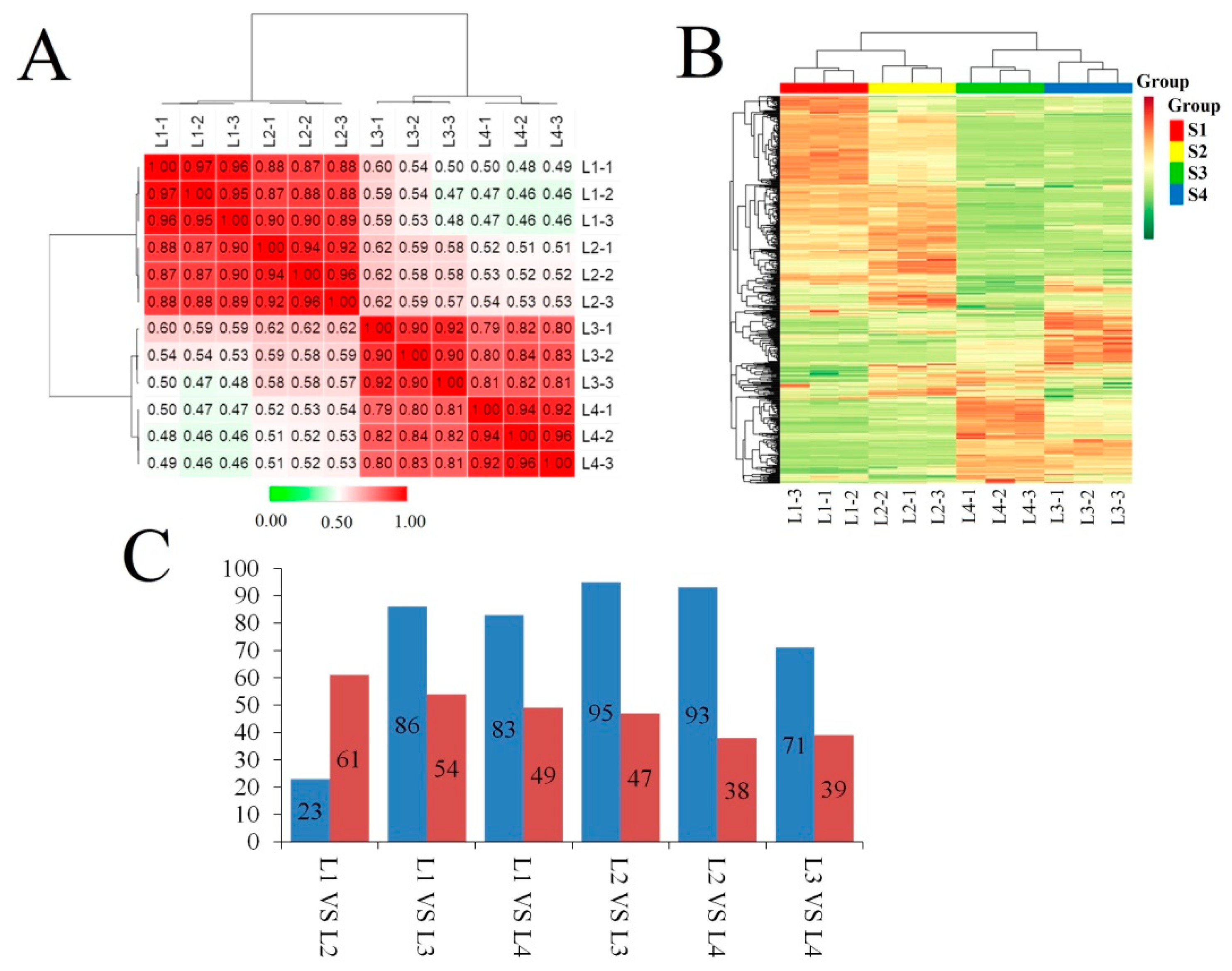
2.2. Overview of Single-Molecule Long-Read Sequencing and SGS Results
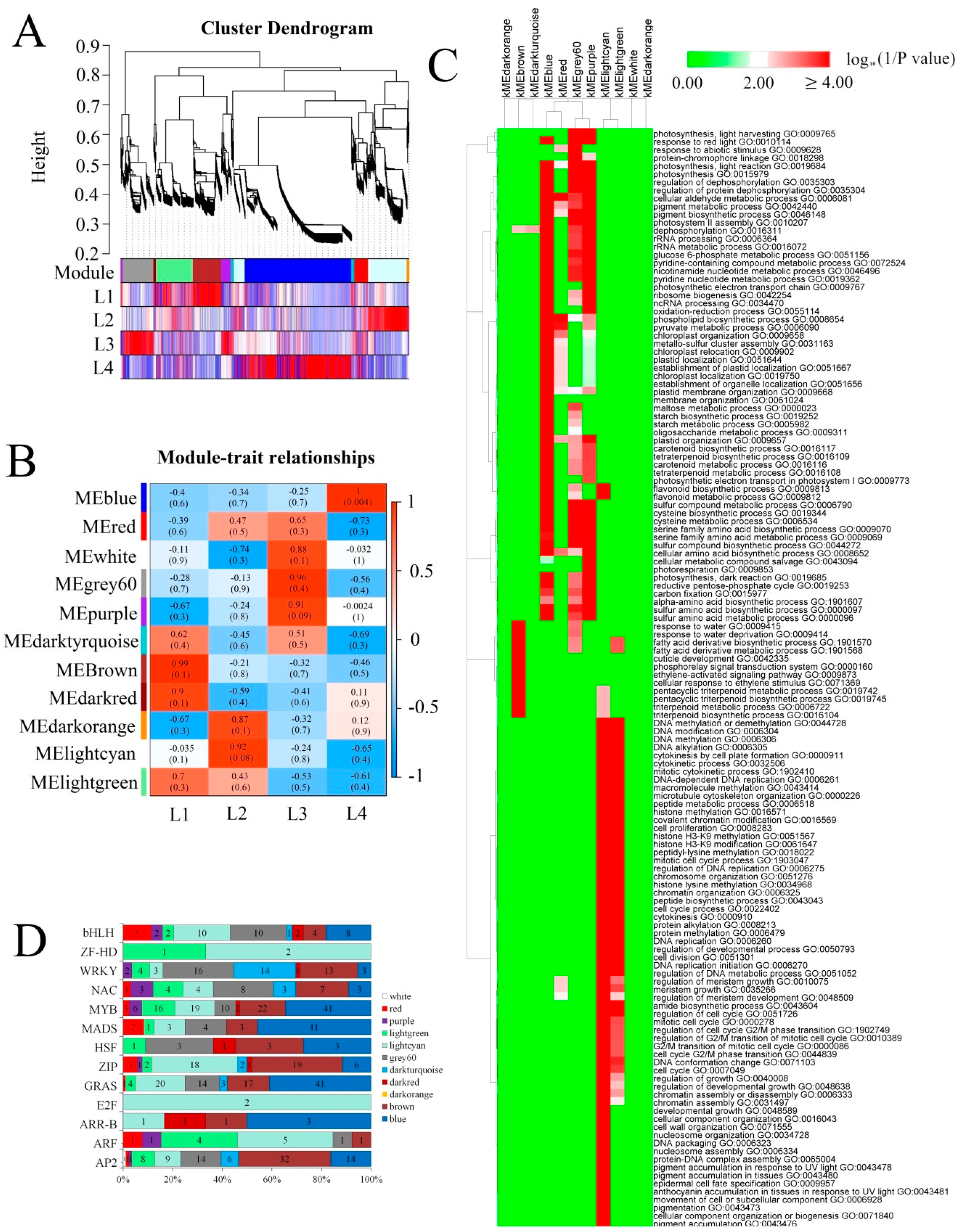
2.3. Different Regulatory Mechanisms Involved in Different E. ulmoides Growth Stages
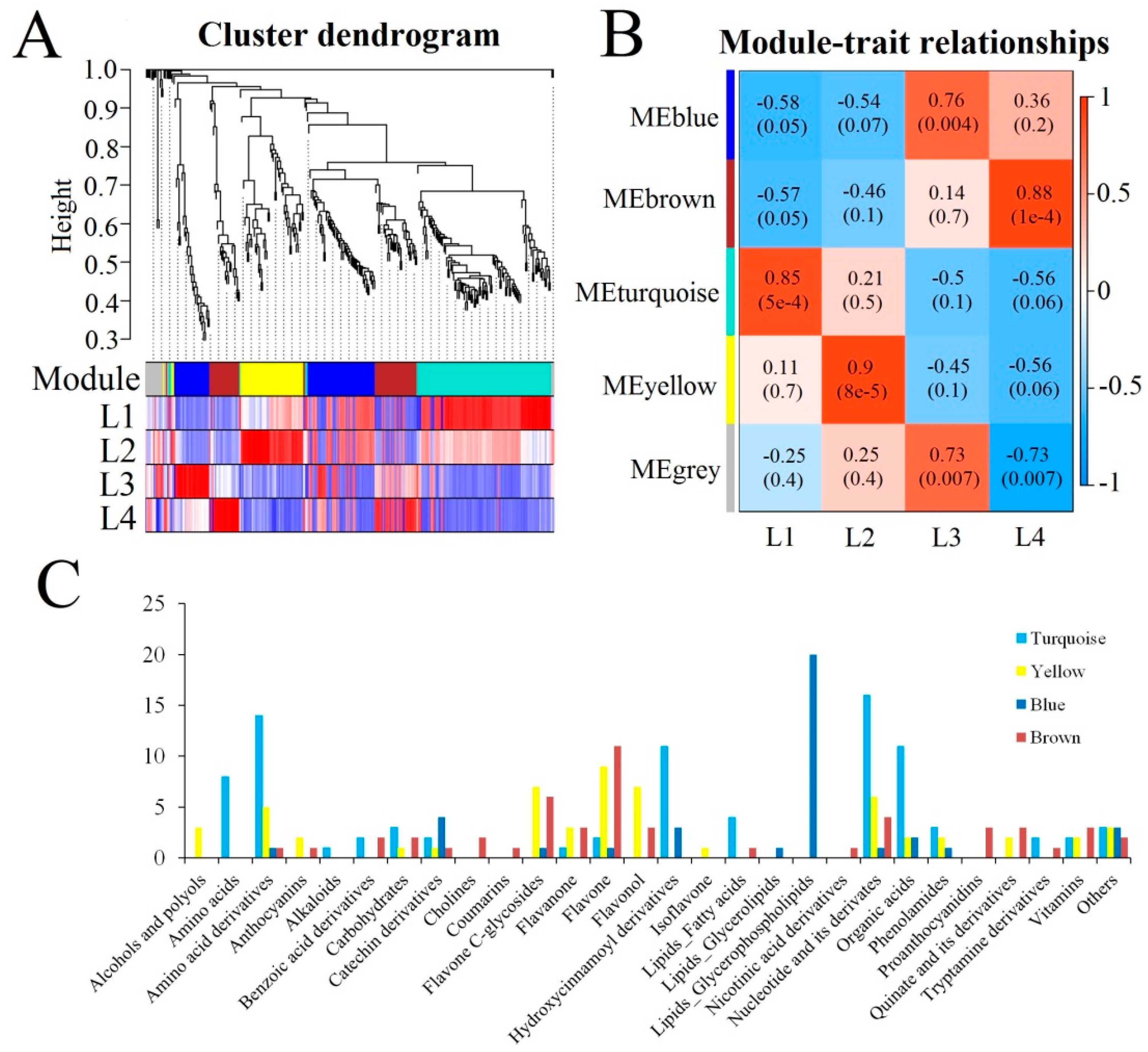
2.4. Metabolomic Analysis
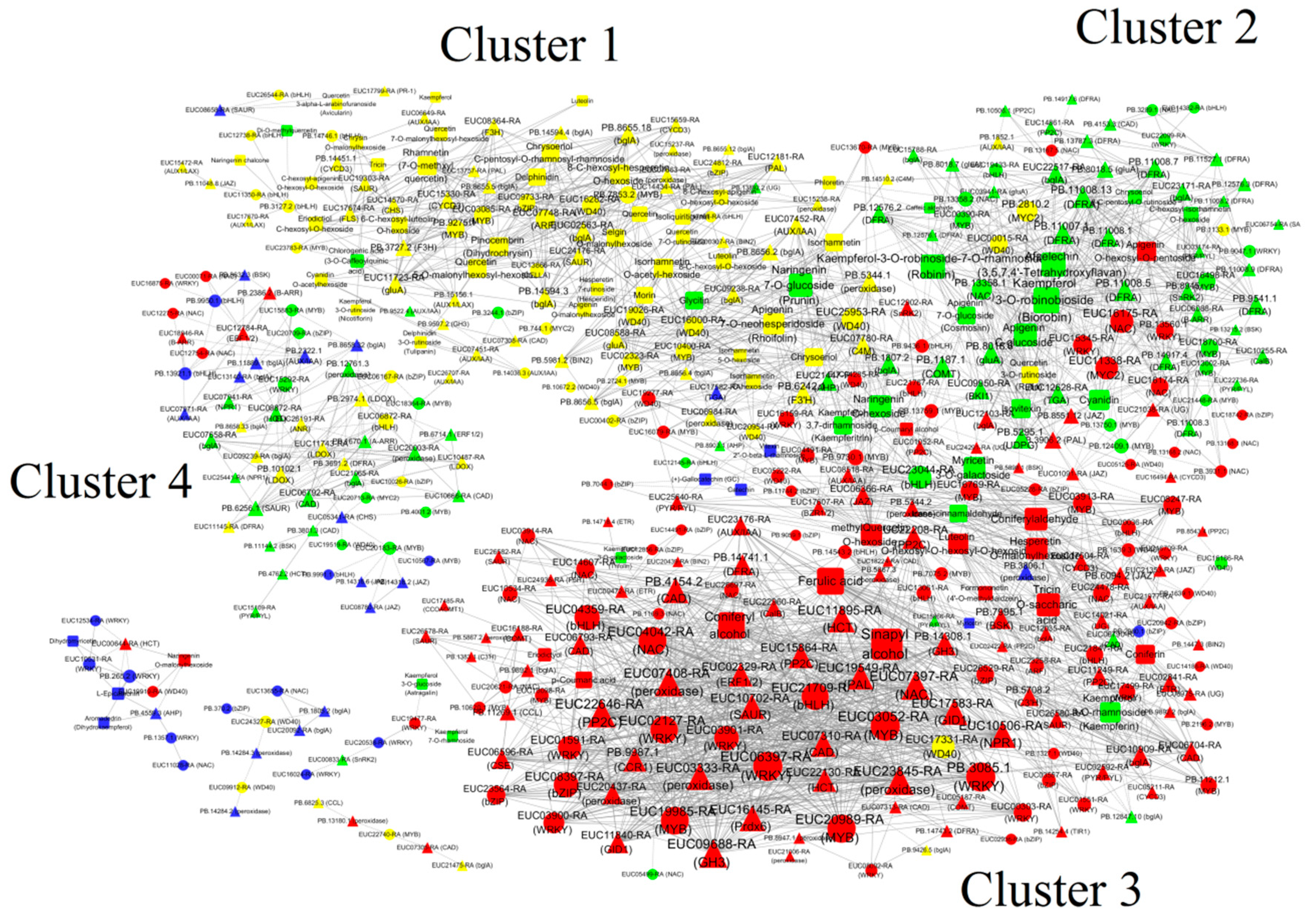
2.5. Analysis of Isoforms and Metabolites Associated with Phenylpropanoid and Flavonoid Biosynthesis
3. Discussion
3.1. The Landscape of E. ulmoides Transcript Diversity
3.2. The Molecular Mechanism Involved in Flavonoid and Phenylpropanoid Synthesis During E. ulmoides Growth and Development
3.3. The Molecular Mechanism Involved in E. ulmoides Growth and Development
4. Materials and Methods
4.1. Plant Materials
4.2. Paraffin Sectioning
4.3. SEM (Scanning Electron Microscope)
4.4. RNA Extraction and Quality Assessment
4.5. PacBio Iso-Seq Library Preparation, Sequencing and Analysis
4.6. Illumina Transcriptome Library Preparation, Sequencing and Analysis
4.7. Functional Annotation of Isoforms
4.8. Isoform Expression Analysis
4.9. Metabolite Extraction and Widely Targeted Metabolite Profiling
4.10. Determination of CGA, Rutin and Total Flavonoid Levels
4.11. Integrative Analysis of the Metabolome and Transcriptome
4.12. Semiquantitative RT-PCR Verification
4.13. Availability of Supporting Data
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Hu, S.Y. A contribution to our knowledge of Tu-chung-Eucommia ulmoides. Am. J. Chin. Med. 1979, 07, 5–37. [Google Scholar] [CrossRef]
- Wuyun, T.N.; Wang, L.; Liu, H.M.; Wang, X.W.; Zhang, L.S.; Ziang, L.; Bennetzen, J.L.; Li, T.; Yang, L.; Liu, P.; et al. The hardy rubber tree genome provides insights into the evolution of polyisoprene biosynthesis. Mol. Plant 2017, 11, 429–442. [Google Scholar] [CrossRef] [PubMed]
- Zhou, C.U.; Ai, L.Y.; Wang, M. Protection of Eucommia ulmoides polysaccharide on liver fibrosis. Chin. Tradit. Herb. Drugs 2011, 42, 324–329. [Google Scholar]
- Bamba, T.; Fukusaki, E.; Nakazawa, Y.; Kobayashi, A. In-situ chemical analyses of trans-polyisoprene by histochemical staining and fouRier transform infrared microspectroscopy in a rubber-producing plant, Eucommia ulmoides Oliver. Planta 2002, 215, 934–939. [Google Scholar] [CrossRef] [PubMed]
- Bamba, T.; Murayoshi, M.; Gyoksen, K.; Nakazawa, Y.; Okumoto, H.; Katto, H.; Fukusaki, E.; Kobayashi, A. Contribution of mevalonate and methylerythritol phosphate pathways to polyisoprenoid biosynthesis in the rubber-producing plant Eucommia ulmoides Oliver. Z. Für. Nat. C J. Biosci. 2010, 65, 363–372. [Google Scholar] [CrossRef]
- Guo, H.; Shi, F.; Li, M.J.; Liu, Q.; Yu, B. Neuroprotective effects of Eucommia ulmoides Oliv. and its bioactive constituent work via ameliorating the ubiquitin-proteasome system. BMC Complement. Altern. Med. 2015, 15, 151. [Google Scholar] [CrossRef] [PubMed]
- Zhang, R.; Pan, Y.L.; Hu, S.J.; Kong, X.H.; Juan, W.; Mei, Q.B. Effects of total lignans from Eucommia ulmoides barks prevent bone loss in vivo and in vitro. J. Ethnopharmacol. 2014, 155, 104–112. [Google Scholar] [CrossRef]
- Kwon, S.H.; Ma, S.X.; Joo, H.J.; Lee, S.Y.; Jang, C.G. Inhibitory Effects of Eucommia ulmoides Oliv. Bark on Scopolamine-Induced Learning and Memory Deficits in Mice. Biomol. Ther. 2013, 21, 462–469. [Google Scholar] [CrossRef]
- Jin, X.; Amitani, K.; Zamami, Y.; Takatori, S.; Hobara, N.; Kawamura, N.; Hirata, T.; Wada, A.; Kitamura, Y.; Kawasaki, H. Ameliorative effect of Eucommia ulmoides Oliv. leaves extract (ELE) on insulin resistance and abnormal perivascular innervation in fructose-drinking rats. J. Ethnopharmacol. 2010, 128, 672–678. [Google Scholar] [CrossRef]
- Peng, W.; Ge, S.; Li, D.; Mo, B.; Daochun, Q.; Okoshi, M. Report: Molecular basis of antibacterial activities in extracts of Eucommia ulmoides wood. Pak. J. Pharm. Sci. 2014, 27, 2133–2138. [Google Scholar]
- Fujikawa, T.; Hirata, T.; Hosoo, S.; Nakajima, K.; Wada, A. Asperuloside stimulates metabolic function in rats across several organs under high-fat diet conditions, acting like the major ingredient of Eucommia leaves with anti-obesity activity. J. Nutr. Sci. 2012, 1, e10. [Google Scholar] [CrossRef] [PubMed]
- Xie, G.P.; Jiang, N.; Wang, S.N.; Qi, R.Z.; Wang, L. Eucommia ulmoides Oliv. bark aqueous extract inhibits osteoarthritis in a rat model of osteoarthritis. J. Ethnopharmacol. 2015, 162, 148–154. [Google Scholar] [CrossRef] [PubMed]
- Ng, H.S.; Chien, T.U. Pharmacological Properties of Eucommia Ulmoides. Am. J. Chin. Med. 1957, 47, 259–300. [Google Scholar]
- Winkel-Shirley, B. Flavonoid biosynthesis. A colorful model for genetics, biochemistry, cell biology, and biotechnology. Plant Physiol. 2001, 126, 485–493. [Google Scholar] [CrossRef] [PubMed]
- Brunetti, C.; Di Ferdinando, M.; Fini, A.; Pollastr, S.; Tattini, M. Flavonoids as antioxidants and developmental regulators: relative significance in plants and humans. Int. J. Mol. Sci. 2013, 14, 3540–3555. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Y.; Qi, L.; Yang, J.; Wu, C.; Liu, Y.J.; Huang, L.Q. A Scutellaria baicalensis R2R3-MYB gene, SbMYB8, regulates flavonoid biosynthesis and improves drought stress tolerance in transgenic tobacco. Plant Cell Tiss. Org. 2015, 120, 961–972. [Google Scholar] [CrossRef]
- Ali, M.B.; McNear, D.H. Induced transcriptional profiling of phenylpropanoid pathway genes increased flavonoid and lignin content in Arabidopsisleaves in response to microbial products. BMC Plant Biol. 2014, 14, 84. [Google Scholar] [CrossRef]
- Dubos, C.; Stracke, R.; Grotewold, E.; Weisshaar, B.; Martin, C.; Lepiniec, L. MYB transcription factors in Arabidopsis. Trends Plant Sci. 2010, 15, 573–581. [Google Scholar] [CrossRef]
- Stracke, R.; Ishihara, H.; Barsch, G.H.A.; Mehrtens, F.; Niehaus, K.; Weisshaar, B. Differential regulation of closely related R2R3-MYB transcription factors controls flavonol accumulation in different parts of the Arabidopsis thaliana seedling. Plant J. 2007, 50, 660–677. [Google Scholar] [CrossRef]
- Shi, M.; Xie, D. Biosynthesis and metabolic engineeringof anthocyanins in arabidopsis. Recent Pat. Biotechnol. 2014, 8, 47–6013. [Google Scholar] [CrossRef]
- Peng, Z.H.; Han, C.Y.; Yuan, L.B.; Zhang, K.; Huang, H.M.; Ren, C.M. Brassinosteroid enhances jasmonate -induced anthocyanin accumulation in Arabidopsis seedlings. Plant Biotechnol. J. 2011, 53, 632–640. [Google Scholar] [CrossRef]
- Cho, A.S.; Jeon, S.M.; Kim, M.J.; Yeo, J.; Seo, K.I.; Choi, M.-S.; Lee, M.-K. Chlorogenic acid exhibits anti-obesity property and improves lipid metabolism in high-fat diet-induced-obese mice. Food Chem. Toxicol. 2010, 48, 937–943. [Google Scholar] [CrossRef]
- Ong, K.W.; Hsu, A.; Tan, B.K. Anti-diabetic and anti-lipidemic effects of chlorogenic acid are mediated by ampk activation. Biochem. Pharmacol. 2013, 85, 1341–1351. [Google Scholar] [CrossRef]
- Schutz, K.; Kammerer, D.; Carle, R.; Schieber, A. Identification and quantification of caffeoylquinic acids and flavonoids from artichoke (Cynara scolymus L.) heads, juice, and pomace by HPLC-DAD-ESI/MS(n). J. Agric. Food Chem. 2004, 52, 4090–4096. [Google Scholar] [CrossRef]
- Wang, M.; Simon, J.E.; Aviles, I.F.; He, K.; Zheng, Q.Y.; Tadmor, Y. Analysis of antioxidative phenolic compounds in artichoke (Cynara scolymus L.). J. Agric. Food Chem. 2003, 51, 601–608. [Google Scholar] [CrossRef]
- Payyavula, R.S.; Shakya, R.; Sengoda, V.G.; Munyaneza, J.E.; Swamy, P.; Navarre, D.A. Synthesis and regulation of chlorogenic acid in potato: Rerouting phenylpropanoid flux in HQT-silenced lines. Plant Biotechnol. J. 2015, 13, 551–564. [Google Scholar] [CrossRef]
- Chen, Z.X.; Liu, G.H.; Liu, Y.Q.; Xian, Z.Q.; Tang, N. Overexpression of the LmHQT1 gene increases chlorogenic acid production in Lonicera macranthoides Hand-Mazz. Acta Physiol. Plant 2017, 39, 27. [Google Scholar] [CrossRef]
- Chang, J.; Luo, J.; He, G. Regulation of polyphenols accumulation by combined overexpression/silencing key enzymes of phenylpropanoid pathway. Acta Biochim. Et Biophys. Sin. 2009, 41, 123–130. [Google Scholar] [CrossRef]
- Feng, Y.Z.; Wang, L.; Fu, J.M.; Wuyun, T.L.; Du, H.Y.; Tao, X. Transcriptome sequencing discovers genes related to fatty acid biosynthesis in the seeds of Eucommia Ulmoides. Genes Genom. 2016, 38, 275–283. [Google Scholar] [CrossRef]
- Feng, Y.Z.; Zhang, L.; Fu, J.M.; Li, F.D.; Wang, L.; Tan, X.; Mo, W.; Cao, H. Characterization of glycolytic pathway genes using RNA-Seq in developing kernels of Eucommia ulmoides. J. Agric. Food Chem. 2016, 64, 3712–3731. [Google Scholar] [CrossRef]
- Wang, L.; Du, H.Y.; Wuyun, T.N. Genome-wide identification of MicroRNAs and their targets in the leaves and fruits of Eucommia ulmoides using high-throughput sequencing. Front. Plant Sci. 2016, 7, 1632. [Google Scholar] [CrossRef]
- Rhoads, A.; Au, K.F. PacBio sequencing and its applications. Genom. Proteom. Bioinform. 2015, 13, 278–289. [Google Scholar] [CrossRef]
- Abdel-Ghany, S.E.; Hamilton, M.; Jacob, J.L.; Ngam, P.; Devitt, N.; Schilkey, F.; Ben-Hur, A.; Reddy, A.S.N. A survey of the sorghum transcriptome using single-molecule long reads. Nat. Commun. 2016, 7, 11706. [Google Scholar] [CrossRef] [Green Version]
- Wang, B.; Tseng, E.; Regulski, M.; Clark, T.A.; Hon, T. Unveiling the complexity of the maize transcriptome by single-molecule long-read sequencing. Nat. Commun. 2016, 7, 11708. [Google Scholar] [CrossRef] [Green Version]
- Wang, T.; Wang, H.; Cai, D.; Gao, Y.B.; Zhang, H.X.; Jiao, Y.; Lu, Z.; Olson, A.; Stein, J.C.; Ware, D. Comprehensive profiling of rhizome-associated alternative splicing and alternative polyadenylation in moso bamboo (Phyllostachys edulis). Plant J. 2017, 91, 684–699. [Google Scholar] [CrossRef]
- Wang, M.; Wang, P.; Liang, F.; Ye, Z.; Li, J.; Shen, C.; Pei, L.; Wang, F.; Hu, J.; Tu, L. A global survey of alternative splicing in allopolyploid cotton: landscape, complexity and regulation. New Phytol. 2018, 217, 163–178. [Google Scholar] [CrossRef]
- Chao, Q.; Gao, Z.F.; Zhang, D.; Zhao, B.G.; Dong, F.Q.; Fu, C.-X.; Liu, L.-J.; Wang, B.-C. The developmental dynamics of the Populus stem transcriptome. Plant Biotechnol. J. 2018, 17, 206–219. [Google Scholar] [CrossRef]
- Gordon, S.P.; Tseng, E.; Salamov, A.; Zhang, J.; Meng, X.; Zhao, Z.; Kang, D.; Underwood, J.; Grigoriev, I.V.; Figueroa, M. Widespread polycistronic transcripts in fungi revealed by single molecule mRNA sequencing. PLoS ONE 2015, 10, e0132628. [Google Scholar] [CrossRef]
- Wang, W.; Zhang, X. Identification of the sex-biased gene expression and putative sex-associated genes in Eucommia ulmoides Oliver using comparative transcriptome Analyses. Molecules 2017, 22, 2255. [Google Scholar] [CrossRef]
- Liu, H.M.; Fu, J.M.; Du, H.Y.; Hu, J.J.; Wuyun, T. De novo sequencing of Eucommia ulmoides flower bud transcriptomes for identification of genes related to floral development. Genom. Data 2016, 9, 105–110. [Google Scholar] [CrossRef]
- Shi, S.G.; Yang, M.; Zhang, M.; Wang, P.; Kang, Y.X.; Liu, J.J. Genome-wide transcriptome analysis of genes involved in flavonoid biosynthesis between red and white strains of Magnolia sprengeri pamp. BMC Genom. 2014, 15, 706. [Google Scholar] [CrossRef]
- Weitzel, C.; Petersen, M. Enzymes of phenylpropanoid metabolism in the important medicinal plant Meliss. L. Planta 2010, 232, 731–742. [Google Scholar] [CrossRef]
- Zhang, J.J.; Zhang, S.S.; Li, H.; Du, H.; Huang, H.H.; Li, Y.; Hu, Y.; Liu, H.; Liu, Y.; Yu, G. Identification of transcription factors ZmMYB111 and ZmMYB148 involved in phenylpropanoid metabolism. Front. Plant Sci. 2016, 7, 148. [Google Scholar] [CrossRef]
- Xu, W.J.; Dubos, C.; Lepiniec, L. Transcriptional control of flavonoid biosynthesis by MYB-bHLH-WDR complexes. Trends Plant Sci. 2015, 20, 3. [Google Scholar] [CrossRef]
- Xu, W.; Grain, D.; Bobet, S.; Le Gourrierec, J.; Thevenin, J.; Kelemen, Z.; Lepiniec, L.; Dubos, C. Complexity and robustness of the flavonoid transcriptional regulatory network revealed by comprehensive analyses of MYB-bHLH-WDR complexes and their targets in Arabidopsis seed. New Phytol. 2014, 202, 132–144. [Google Scholar] [CrossRef]
- Yoshida, K.; Ma, D.; Constabel, C.P. The MYB182 protein down-regulates proanthocyanidin and anthocyanin biosynthesis in Poplar by repressing both structural and regulatory flavonoid genes. Plant Physiol. 2015, 167, 693–710. [Google Scholar] [CrossRef]
- Azuma, A.; Ban, Y.; Sato, A.; Kono, A.; Shiraishi, M.; Yakushiji, H.; Kobayashi, S. MYB diplotypes at the color locus affect the ratios of tri/di-hydroxylated and methylated/non-methylated anthocyanins in grape berry skin. Tree Genet. Genom. 2015, 11, 31. [Google Scholar] [CrossRef]
- Zhang, K.; Logacheva, M.D.; Meng, Y.; Hu, J.; Wan, D.; Li, L.; Janovska, D.; Wang, Z.; Georgiev, M.I.; Yu, Z. Jasmonate-responsive MYB factors spatially repress rutin biosynthesis in Fagopyrum tataricum. J. Exp. Bot. 2018, 69, 1955–1966. [Google Scholar] [CrossRef]
- Cho, K.G.; Cho, K.S.; Sohn, H.B.; Ha, I.J.; Hong, S.Y.; Lee, H.; Kim, Y.-M.; Nam, M.H. Network analysis of the metabolome and transcriptome reveals novel regulation of potato pigmentation. J. Exp. Bot. 2016, 67, 1519–1533. [Google Scholar] [CrossRef] [Green Version]
- Qi, T.; Song, S.; Xie, D. Modified bimolecular fluorescence complementation assay to study the inhibition of transcription complex formation by JAZ proteins. Methods Mol. Biol. 2013, 1011, 187–197. [Google Scholar]
- Docimo, T.; Francese, G.; Ruggiero, A.; Batelli, G.; Palma, M.D.; Bassolino, L.; Toppino, L.; Rotino, G.L.; Mennella, G.; Tucci, M. Phenylpropanoids Accumulation in Eggplant Fruit: Characterization of Biosynthetic Genes and Regulation by a MYB Transcription Factor. Front. Plant Sci. 2016, 6, 1233. [Google Scholar] [CrossRef] [Green Version]
- Sun, T.P. The molecular mechanism and evolution of the GA-GID1-DELLA signaling module in plants. Curr. Biol. 2011, 21, R338–R345. [Google Scholar] [CrossRef]
- Zhu, X.P.; Chai, M.; Li, Y.; Sun, M.Y.; Zhang, J.Z. Global transcriptome profiling analysis of inhibitory effects of paclobutrazol on leaf growth in Lily (Lilium Longiflorum-Asiatic Hybrid). Front. Plant Sci. 2016, 7, 491. [Google Scholar] [CrossRef]
- Cao, D.; Cheng, H.; Wu, W.; Soo, H.M.; Peng, J.R. Gibberellin Mobilizes Distinct DELLA-Dependent Transcriptomes to Regulate Seed Germination and Floral Development in Arabidopsis. Plant Physiol. 2006, 142, 509–525. [Google Scholar] [CrossRef]
- Zhang, Y.Q.; Liu, Z.J.; Liu, J.P.; Lin, S.; Wang, J.F.; Lin, W.; Xu, W. GA-DELLA pathway is involved in regulation of nitrogen deficiency-induced anthocyanin accumulation. Plant Cell Rep. 2017, 36, 557–569. [Google Scholar] [CrossRef]
- Swarup, K.; Benkova, E.; Swarup, R. The auxin influx carrier LAX3 promotes lateral root emergence. Nat. Cell Biol. 2008, 10, 946–954. [Google Scholar] [CrossRef]
- Wang, Y.C.; Wang, N.; Xu, H.F.; Jiang, S.H.; Fang, H.C.; Su, M.Y.; Zhang, Z.Y.; Zhang, T.L.; Chen, X.S. Auxin regulates anthocyanin biosynthesis through the Aux/IAA–ARF signaling pathway in apple. Hortic. Res. 2018, 5, 59. [Google Scholar] [CrossRef]
- Teale, W.D.; Paponov, I.A.; Palme, K. Auxin in action: Signaling, transport and the control of plant growth and development. Nat. Rev. Mol. Cell Biol. 2006, 7, 847–859. [Google Scholar] [CrossRef]
- Foissac, S.; Sammeth, M. Astalavista: Dynamic and flexible analysis of alternative splicing events in custom gene datasets. Nucleic Acids Res. 2007, 35, W297–W299. [Google Scholar] [CrossRef]
- Peng, R.; Liu, Y.; Cai, Z.; Shen, F.; Chen, J.; Hou, R.; Zou, F. Characterization and analysis of whole transcriptome of giant panda spleens: Implying critical roles of long non-coding RNAs in immunity. Cell. Physiol. Biochem. 2018, 46, 1065–1077. [Google Scholar] [CrossRef]
- Yang, W.; Schuster, C.; Beahan, C.T.; Charoensawan, V.; Peaucelle, A.; Bacic, A.; Doblin, M.S.; Wightman, R.; Meyerowitz, E.M. Regulation of meristem morphogenesis by cell wall synthases in Arabidopsis. Curr. Biol. 2016, 26, 1404–1415. [Google Scholar] [CrossRef]
- Anders, S. Analysing RNA-Seq data with the DESeq package. J. Mol. Biol. 2010, 43, 1–17. [Google Scholar]
- Langfelder, P.; Horvath, S. WGCNA: An R package for weighted correlation network analysis. BMC Bioinform. 2008, 9, 559. [Google Scholar] [CrossRef]
- Chen, C.; Xia, R.; Xia, R.; Chen, H.C.; He, Y.H. TBtools, a Toolkit for Biologists integrating various HTS-data handling tools with a user-friendly interface. BioRxiv 2018, 289660. [Google Scholar]
- Ye, J.; Jin, C.F.; Li, N.; Liu, M.H.; Fei, Z.X.; Dong, L.-Z.; Li, L.; Li, Z.Q. Selection of suitable reference genes for qRT-PCR normalization under different experimental conditions in Eucommia ulmoides Oliv. Sci. Rep. 2018, 8, 15043. [Google Scholar] [CrossRef]
- Lisec, J.; Schauer, N.; Kopka, J.; Lothar, W.; Alisdair, R.F. Gas chromatography mass spectrometry–based metabolite profiling in plants. Nat. Protoc. 2006, 1, 387–396. [Google Scholar] [CrossRef]
- Wang, J.X.; Chen, L.; Tian, X.X.; Gao, L.J.; Niu, X.F.; Shi, M.L.; Zhang, W.W. Global metabolomic and network analysis of Escherichia coli responses to exogenous biofuels. J. Proteome Res. 2013, 12, 5302–5312. [Google Scholar] [CrossRef]
- Chen, W.; Gong, L.; Guo, Z.; Wang, W.S.; Zhang, H.Y. A novel integrated method for large-scale detection, identification, and quantification of widely targeted metabolites: Application in the study of rice metabolomics. Mol. Plant 2013, 6, 1769–1780. [Google Scholar] [CrossRef]
- Hammer, O.; Harper, D.A.T.; Ryan, P.D. PAST: Paleontological statistics software package for education and data analysis. Palaeontol. Electron. 2001, 4, 1–9. [Google Scholar]
- Wang, J.G.; Zhang, X.Q.; Shi, M.L.; Gao, L.J.; Niu, X.F.; Te, R.G.; Chen, L.; Zhang, W.W. Metabolomic analysis of the salt-saensitive mutants reveals changes in amino acid and fatty acid composition important to long-term salt stress in Synechocystis sp. PCC 6803. Funct. Integr. Genom. 2014, 14, 431–440. [Google Scholar] [CrossRef]
- Hashimoto, F.; Tanaka, M.; Maeda, H.; Shimizu, K.; Sakata, Y. Characterization of cyanic flower color of Delphinium cultivars. J. Japn. Soc. Hortic. Sci. 2000, 69, 428–434. [Google Scholar] [CrossRef]







© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, L.; Liu, M.; Shi, K.; Yu, Z.; Zhou, Y.; Fan, R.; Shi, Q. Dynamic Changes in Metabolite Accumulation and the Transcriptome during Leaf Growth and Development in Eucommia ulmoides. Int. J. Mol. Sci. 2019, 20, 4030. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20164030
Li L, Liu M, Shi K, Yu Z, Zhou Y, Fan R, Shi Q. Dynamic Changes in Metabolite Accumulation and the Transcriptome during Leaf Growth and Development in Eucommia ulmoides. International Journal of Molecular Sciences. 2019; 20(16):4030. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20164030
Chicago/Turabian StyleLi, Long, Minhao Liu, Kan Shi, Zhijing Yu, Ying Zhou, Ruishen Fan, and Qianqian Shi. 2019. "Dynamic Changes in Metabolite Accumulation and the Transcriptome during Leaf Growth and Development in Eucommia ulmoides" International Journal of Molecular Sciences 20, no. 16: 4030. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20164030