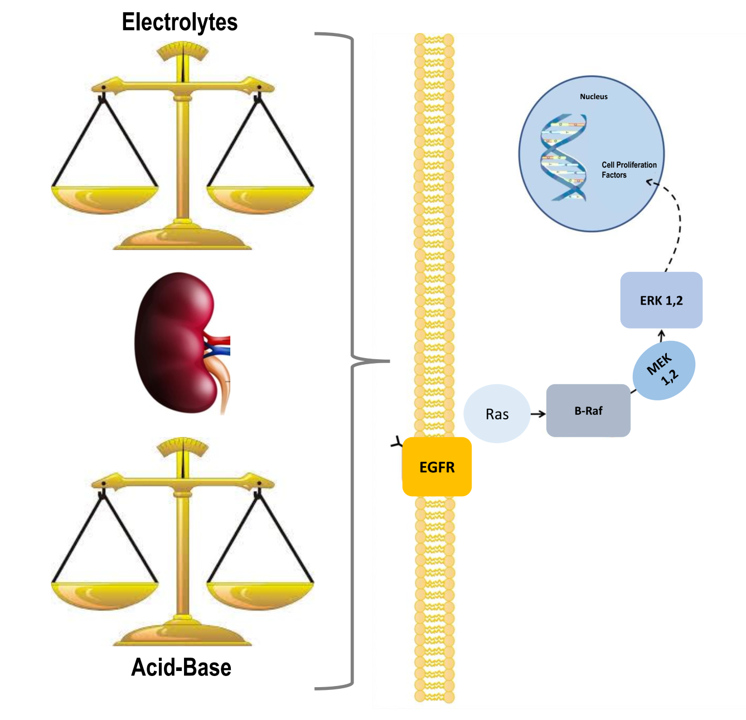
ERK1,2 Signalling Pathway along the Nephron and Its Role in Acid-base and Electrolytes Balance

Abstract
:
{kind=link}
{kind=link}
1. Introduction
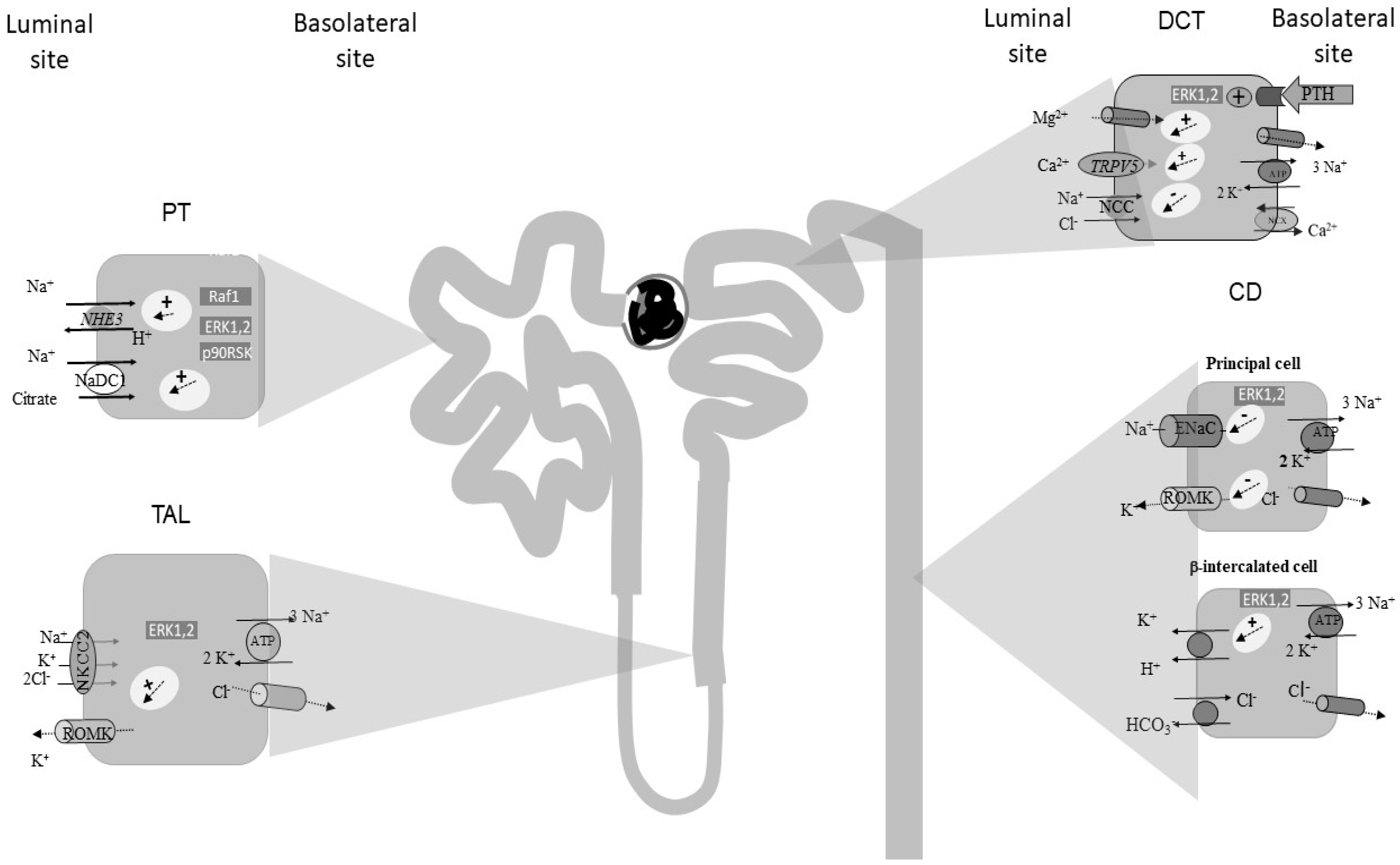
2. The Role of ERK1,2 Signalling along the Proximal Tubule (PT)
3. The Role of ERK1,2 Signalling along the Thick Ascending Limb of Henle’s Loop (TAL)
4. The Role of ERK1,2 Signalling along the Distal Convolute Tubule (DCT)
5. The Role of ERK1,2 Signalling along the Collecting Duct (CD)
5.1. ERK1,2 Signalling in Principal Cells (PCs)
5.1.1. ENaC Regulation
5.1.2. K+ Channels Regulation
5.2. ERK1,2 Signalling in Intercalated Cells (ICs)
6. Clinical Implications
6.1. ERK 1,2 and Hypomagnaesemia
6.2. ERK 1,2 Implications in Proteinuric Nephropathies and Hyperkalemia
6.3. ERk1,2 Implication in Sodium Handling and Hypertension
6.4. ERK 1,2 Implications in Cancer
6.5. ERK 1,2 Implications in Acid-Base Imbalances
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Rubinfeld, H.; Seger, R. The ERK Cascade: A Prototype of MAPK Signaling. Mol. Biotechnol. 2005, 31, 151–174. [Google Scholar] [CrossRef]
- McKay, M.M.; Morrison, D.K. Integrating signals from RTKs to ERK/MAPK. Oncogene 2007, 26, 3113–3121. [Google Scholar] [CrossRef] [Green Version]
- Kolch, W. Coordinating ERK/MAPK signalling through scaffolds and inhibitors. Nat. Rev. Mol. Cell Biol. 2005, 6, 827–837. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, A.; Burack, W.R.; Stock, J.L.; Kortum, R.; Chaika, O.V.; Afkarian, M.; Muller, W.J.; Murphy, K.M.; Morrison, D.K.; Lewis, R.E.; et al. Kinase Suppressor of Ras (KSR) Is a Scaffold Which Facilitates Mitogen-Activated Protein Kinase Activation In Vivo. Mol. Cell. Biol. 2002, 22, 3035–3045. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kurtzeborn, K.; Kwon, H.N.; Kuure, S. MAPK/ERK Signaling in Regulation of Renal Differentiation. Int. J. Mol. Sci. 2019, 20, 1779. [Google Scholar] [CrossRef]
- Hida, M.; Omori, S.; Awazu, M. ERK and p38 MAP kinase are required for rat renal development. Kidney Int. 2002, 61, 1252–1262. [Google Scholar] [CrossRef] [Green Version]
- Ihermann-Hella, A.; Hirashima, T.; Kupari, J.; Kurtzeborn, K.; Li, H.; Kwon, H.N.; Cebrian, C.; Soofi, A.; Dapkunas, A.; Miinalainen, I.; et al. Dynamic MAPK/ERK Activity Sustains Nephron Progenitors through Niche Regulation and Primes Precursors for Differentiation. Stem Cell Rep. 2018, 11, 912–928. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alderliesten, M.; De Graauw, M.; Oldenampsen, J.; Qin, Y.; Pont, C.; Van Buren, L.; Van De Water, B. Extracellular Signal-Regulated Kinase Activation during Renal Ischemia/Reperfusion Mediates Focal Adhesion Dissolution and Renal Injury. Am. J. Pathol. 2007, 171, 452–462. [Google Scholar] [CrossRef] [Green Version]
- Feliers, D.; Kasinath, B.S. Erk in kidney diseases. J. Signal. Transduct. 2011, 2011, 768512. [Google Scholar] [CrossRef] [PubMed]
- Curthoys, N.P.; Moe, O.W. Proximal tubule function and response to acidosis. Clin. J. Am. Soc. Nephrol. 2014, 9, 1627–1638. [Google Scholar] [CrossRef]
- Eid, A.; Bodin, S.; Ferrier, B.; Delage, H.; Boghossian, M.; Martin, M.; Baverel, G.; Conjard, A. Intrinsic Gluconeogenesis Is Enhanced in Renal Proximal Tubules of Zucker Diabetic Fatty Rats. J. Am. Soc. Nephrol. 2006, 17, 398–405. [Google Scholar] [CrossRef] [PubMed]
- Weiner, I.D.; Mitch, W.E.; Sands, J.M. Urea and Ammonia Metabolism and the Control of Renal Nitrogen Excretion. Clin. J. Am. Soc. Nephrol. 2015, 10, 1444–1458. [Google Scholar] [CrossRef] [PubMed]
- Hering-Smith, K.S.; Hamm, L.L. Acidosis and citrate: Provocative interactions. Ann. Transl. Med. 2018, 6, 374. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Zacchia, M.; Tian, X.; Wan, L.; Sakamoto, A.; Yanagisawa, M.; Alpern, R.J.; Preisig, P.A. Acid regulation of NaDC-1 requires a functional endothelin B receptor. Kidney Int. 2010, 78, 895–904. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsuganezawa, H.; Sato, S.; Yamaji, Y.; Preisig, P.A.; Moe, O.W.; Alpern, R.J. Role of c-SRC and ERK in acid-induced activation of NHE3. Kidney Int. 2002, 62, 41–50. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, X.C.; Hopfer, U.; Zhuo, J.L. Novel signaling mechanisms of intracellular angiotensin II-induced NHE3 expression and activation in mouse proximal tubule cells. Am. J. Physiol. Ren. Physiol. 2012, 303, F1617–F1628. [Google Scholar] [CrossRef] [Green Version]
- Zacchia, M.; Tian, X.; Zona, E.; Alpern, R.J.; Preisig, P.A. Acid Stimulation of the Citrate Transporter NaDC-1 Requires Pyk2 and ERK1/2 Signaling Pathways. J. Am. Soc. Nephrol. 2018, 29, 1720–1730. [Google Scholar] [CrossRef] [PubMed]
- Zacchia, M.; Capolongo, G.; Rinaldi, L.; Capasso, G. The importance of the thick ascending limb of Henle’s loop in renal physiology and pathophysiology. Int. J. Nephrol. Renov. Dis. 2018, 11, 81–92. [Google Scholar] [CrossRef]
- Capasso, G.; Trepiccione, F.; Zacchia, M. The Physiology of the Loop of Henle. In Critical Care Nephrology, 3rd ed.; Ronco, C., Bellomo, R., Kellum, J.A., Ricci, Z., Eds.; Elsevier: Amsterdam, The Netherlands, 2017; pp. 42–48. [Google Scholar]
- Zacchia, M.; Di Iorio, V.; Trepiccione, F.; Caterino, M.; Capasso, G. The Kidney in Bardet-Biedl Syndrome: Possible Pathogenesis of Urine Concentrating Defect. Kidney Dis. 2017, 3, 57–65. [Google Scholar] [CrossRef]
- Gamba, G.; Friedman, P.A. Thick ascending limb: The Na+/K+/2Cl− co-transporter, NKCC2, and the calcium-sensing receptor, CaSR. Pflugers Arch. 2009, 458, 61–76. [Google Scholar] [CrossRef]
- Sanada, H.; Jones, J.E.; Jose, P.A. Genetics of salt-sensitive hypertension. Curr. Hypertens. Rep. 2011, 13, 55–66. [Google Scholar] [CrossRef] [PubMed]
- Zacchia, M.; Capasso, G. The importance of uromodulin as regulator of salt reabsorption along the thick ascending limb. Nephrol. Dial. Transplant. 2015, 30, 158–160. [Google Scholar] [CrossRef] [PubMed]
- Zacchia, M.; Capolongo, G.; Rinaldi, L.; Capasso, G. Fisiopatologia dell’handling renale dell’acido urico [Renal handling of uric acid]. G. Ital. Nefrol. 2015. Available online: http://www.nephromeet.com/web/procedure/protocollo.cfm?List=WsIdEvento,WsIdRisposta,WsRelease&c1=00198&c2=4&c3=1 (accessed on 12 June 2019).
- Simeoni, M.; Damiano, S.; Capolongo, G.; Trepiccione, F.; Zacchia, M.; Fuiano, G.; Capasso, G. Rare Renal Diseases Can Be Used as Tools to Investigate Common Kidney Disorders. Kidney Dis. 2017, 3, 43–49. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Lu, M.; Balazy, M.; Hebert, S.C. Phospholipase A2 is involved in mediating the effect of extracellular Ca2+ on apical K+ channels in rat TAL. Am. J. Physiol. 1997, 273, F421–F429. [Google Scholar] [CrossRef]
- Riccardi, D.; Valenti, G. Localization and function of the renal calcium-sensing receptor. Nat. Rev. Nephrol. 2016, 12, 414–425. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tian, W.; Zhang, Z.; Cohen, D.M. MAPK signaling and the kidney. Am. J. Physiol. 2000, 279, F593–F604. [Google Scholar] [CrossRef] [Green Version]
- Terada, Y.; Tomita, K.; Homma, M.K.; Nonoguchi, H.; Yang, T.; Yamada, T.; Yuasa, Y.; Krebs, E.G.; Sasaki, S.; Marumo, F. Sequential activation of Raf-1 kinase, mitogen-activated protein (MAP) kinase kinase, MAP kinase, and S6 kinase by hyperosmolality in renal cells. J. Biol. Chem. 1994, 269, 31296–31301. [Google Scholar]
- Yoshida, T.; Sone, M.; Ogawa, T.; Nihei, H.; Ozasa, H.; Tsukada, K.; Horikawa, S. Molecular cloning of rat p38 mitogen-activated protein kinase and it’s osmotic regulation in rat kidney. Biochem. Mol. Biol. Int. 1997, 43, 63–72. [Google Scholar] [CrossRef]
- Terada, Y.; Yamada, T.; Takayama, M.; Nonoguchi, H.; Sasaki, S.; Tomita, K.; Marumo, F. Presence and regulation of Raf-1-K (Kinase), MAPK-K, MAP-K, and S6-K in rat nephron segments. J. Am. Soc. Nephrol. 1995, 6, 1565–1577. [Google Scholar]
- Kultz, D.; Burg, M.B. Intracellular signaling in response to osmotic stress. Contrib. Nephrol. 1998, 123, 94–109. [Google Scholar] [PubMed]
- Kultz, D.; Madhany, S.; Burg, M.B. Hyperosmolality causes growth arrest of murine kidney cells. Induction of GADD45 and GADD153 by osmosensing via stress-activated protein kinase. J. Biol. Chem. 1998, 273, 13645–13651. [Google Scholar] [CrossRef] [PubMed]
- Kultz, D.; Garcia-Perez, A.; Ferraris, J.D.; Burg, M.B. Distinct regulation of osmoprotective genes in yeast and mammals. Aldose reductase osmotic response element is induced independent of p38 and stress-activated protein kinase/Jun N-terminal kinase in rabbit kidney cells. J. Biol. Chem. 1997, 272, 13165–13170. [Google Scholar] [CrossRef] [PubMed]
- Gallazzini, M.; Karim, Z.; Bichara, M. Regulation of ROMK (Kir 1.1) Channel Expression in Kidney Thick Ascending Limb by Hypertonicity: Role of TonEBP and MAPK Pathways. Nephron Physiol. 2006, 104, 126–135. [Google Scholar] [CrossRef] [PubMed]
- Good, D.W. Effects of osmolality on bicarbonate absorption by medullary thick ascending limb of the rat. J. Clin. Investig. 1992, 89, 184–190. [Google Scholar] [CrossRef] [PubMed]
- Watts, B.A., III; Good, D.W. Apical membrane Na+/H+ exchange. Chem 1994, 269, 20250–20255. [Google Scholar]
- Watts, B.A., III; Di Mari, J.F.; Davis, R.J.; Good, D.W. Hypertonicity activates MAP kinases and inhibits HCO-3 absorption via distinct pathways in thick ascending limb. Am. J. Physiol. 1998, 275, F478–F486. [Google Scholar] [CrossRef] [PubMed]
- Moe, O.W. Acute regulation of proximal tubule apical membrane Na+/H+ exchange NHE-3: Role of phosphorylation, protein trafficking and regulatory factors. J. Am. Soc. Nephrol. 1999, 10, 2412–2425. [Google Scholar]
- Good, D.W.; George, T.; Watts, B.A. Aldosterone potentiates 1,25-dihydroxyvitamin D3 action in renal thick ascending limb via a nongenomic, ERK-dependent pathway. Am. J. Physiol. Cell Physiol. 2003, 285, C1122–C1130. [Google Scholar] [CrossRef]
- Bonventre, J.V.; Brezis, M.; Siegel, N.; Rosen, S.; Portilla, D.; Venkatachalam, M. Acute renal failure. I. Relative importance of proximal vs. distal tubular injury. Am. J. Physiol. Ren. Physiol. 1998, 275, F623–F632. [Google Scholar]
- Zacchia, M.; Capasso, G. Dehydration: A new modulator of klotho expression. Am. J. Physiol. Ren. Physiol. 2011, 301, F743–F744. [Google Scholar] [CrossRef] [PubMed]
- Humes, H.D.; Cieslinski, D.A.; Coimbra, T.M.; Messana, J.M.; Galvao, C. Epidermal growth factor enhances renal tubule regeneration and repair and accelerates recovery of renal function in post-ischemic renal failure. J. Clin. Investig. 1989, 84, 1757–1761. [Google Scholar] [CrossRef] [PubMed]
- Norman, J.; Tsau, Y.K.; Bacay, A.; Fine, L.G. Epidermal growth factor accelerates recovery from ischemic acute tubular necrosis: Role of EGF-R. Clin. Sci. 1990, 78, 445–449. [Google Scholar] [CrossRef] [PubMed]
- Baer, P.C.; Geiger, H. Different effects of growth factors on human renal early distal tubular cells in vitro. Kidney Blood Press. Res. 2006, 29, 225–230. [Google Scholar] [CrossRef] [PubMed]
- Stracke, S.; Ernst, F.; Jehle, D.R.; Grunewald, W.; Haller, H.; Keller, F.; Jehle, P.M. Differentiating and proliferative effects of HGF in renal proximal tubular cells are mediated via different signaling pathways. Nephrol. Dial. Transplant. 1998, 13, 1398–1405. [Google Scholar] [CrossRef] [PubMed]
- Izevbigie, E.B.; Gutkind, J.S.; Ray, P.E. Isoproterenol inhibits fibroblast growth factor-2-in-duced growth of renal epithelial cells. Pediatr. Nephrol. 2000, 14, 726–734. [Google Scholar] [CrossRef]
- Petrazzuolo, O.; Trepiccione, F.; Zacchia, M.; Capasso, G. Hypertension and renal calcium transport. J. Nephrol. 2010, 23, S112–S117. [Google Scholar]
- Zacchia, M.; Capasso, G. Parvalbumin: A key protein in early distal tubule NaCl reabsorption. Nephrol. Dial. Transplant. 2008, 23, 1109–1111. [Google Scholar] [CrossRef]
- Trepiccione, F.; Zacchia, M.; Capasso, G. The role of the kidney in salt-sensitive hypertension. Clin. Exp. Nephrol. 2012, 16, 68–72. [Google Scholar] [CrossRef]
- Ko, B.; Kamsteeg, E.J.; Cooke, L.L.; Moddes, L.N.; Deen, P.M.; Hoover, R.S. RasGRP1 stimulation enhances ubiquitination and endocytosis of the sodium-chloride cotransporter. Am. J. Physiol. Ren. Physiol. 2010, 299, F300–F309. [Google Scholar] [CrossRef] [Green Version]
- Brose, N.; Rosenmund, C. Move over protein kinase C, you’ve got company: Alternative cellular effectors of diacylglycerol and phorbol esters. J. Cell Sci. 2002, 115, 4399–4411. [Google Scholar] [CrossRef] [PubMed]
- Ko, B.; Cooke, L.L.; Hoover, R.S. Parathyroid hormone (PTH) regulates the sodium chloride cotransporter via Ras guanyl releasing protein 1 (Ras-GRP1) and extracellular signal-regulated kinase (ERK)1/2 mitogen-activated protein kinase (MAPK) pathway. Transl. Res. 2011, 158, 282–289. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Capolongo, G.; Xu, L.H.; Accardo, M.; Sanduzzi, A.; Stanziola, A.A.; Colao, A.; Agostini, C.; Zacchia, M.; Capasso, G.; Adams-Huet, B.; et al. Vitamin-D status and mineral metabolism in two ethnic populations with sarcoidosis. J. Investig. Med. 2016, 64, 1025–1034. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sneddon, W.B.; Liu, F.; Gesek, F.A.; Friedman, P.A. Obligate mitogen-activated protein kinase activation in parathyroid hormone stimulation of calcium transport but not calcium signaling. Endocrinology 2000, 141, 4185–4193. [Google Scholar] [CrossRef] [PubMed]
- Zhou, B.; Wang, D.; Feng, X.; Zhang, Y.; Wang, Y.; Zhuang, J.; Zhang, X.; Chen, G.; Delpire, E.; Gu, D.; et al. WNK4 inhibits NCC protein expression through MAPK ERK1/2 signaling pathway. Am. J. Physiol. Ren. Physiol. 2012, 302, F533–F539. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shaharabany, M.; Holtzman, E.J.; Mayan, H.; Hirschberg, K.; Seger, R.; Farfel, Z. Distinct pathways for the involvement of WNK4 in the signaling of hypertonicity and EGF. FEBS J. 2008, 275, 1631–1642. [Google Scholar] [CrossRef] [PubMed]
- Sandberg, M.B.; Maunsbach, A.B.; McDonough, A.A. Redistribution of distal tubule Na+-Cl- cotransporter (NCC) in response to a high-salt diet. Am. J. Physiol. Ren. Physiol. 2006, 291, F503–F508. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.E.; Sandberg, M.B.; Can, A.D.; Pihakaski-Maunsbach, K.; McDonough, A.A. Effects of dietary salt on renal Na+ transporter subcellular distribution, abundance, and phosphorylation status. Am. J. Physiol. Ren. Physiol. 2008, 295, F1003–F1016. [Google Scholar] [CrossRef]
- Lai, L.; Feng, X.; Liu, D.; Chen, J.; Zhang, Y.; Niu, B.; Gu, Y.; Cai, H. Dietary salt modulates the sodium chloride cotransporter expression likely through an aldosterone-mediated WNK4-ERK1/2 signaling pathway. Pflugers Arch. 2012, 463, 477–485. [Google Scholar] [CrossRef]
- Feng, X.; Zhang, Y.; Shao, N.; Wang, Y.; Zhuang, Z.; Wu, P.; Lee, M.J.; Liu, Y.; Wang, X.; Zhuang, J.; et al. Aldosterone modulates thiazide-sensitive sodium chloride cotransporter abundance via DUSP6-mediated ERK1/2 signaling pathway. Am. J. Physiol. Ren. Physiol. 2015, 308, F1119–F1127. [Google Scholar] [CrossRef] [Green Version]
- Capo-Aponte, J.E.; Wang, Z.; Bildin, V.N.; Pokorny, K.S.; Reinach, P.S. Fate of hypertonicity-stressed corneal epithelial cells depends on differential MAPK activation and p38MAPK/Na-K-2Cl cotransporter1 interaction. Exp. Eye Res. 2007, 84, 361–372. [Google Scholar] [CrossRef]
- Wallace, B.K.; Jelks, K.A.; O’Donnell, M.E. Ischemia-induced stimulation of cerebral microvascular endothelial cell Na-K-Cl cotransport involves p38 and JNK MAP kinases. Am. J. Physiol. Cell Physiol. 2012, 302, C505–C517. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.; Bildin, V.N.; Yang, H.; Capo-Aponte, J.E.; Yang, Y.; Reinach, P.S. Dependence of corneal epithelial cell proliferation on modulation of interactions between ERK1/2 and NKCC1. Cell Physiol. Biochem. 2011, 28, 703–714. [Google Scholar] [CrossRef] [PubMed]
- Grimm, P.R.; Taneja, T.K.; Liu, J.; Coleman, R.; Chen, Y.Y.; Delpire, E.; Wade, J.B.; Welling, P.A. SPAK isoforms and OSR1 regulate sodium-chloride co-transporters in a nephron-specific manner. J. Biol. Chem. 2012, 287, 37673–37690. [Google Scholar] [CrossRef] [PubMed]
- Van Abel, M.; Hoenderop, J.G.; van der Kemp, A.W.; Friedlaender, M.M.; van Leeuwen, J.P.; Bindels, R.J. Coordinated control of renal Ca(2+) transport proteins by parathyroid hormone. Kidney Int. 2005, 68, 1708–1721. [Google Scholar] [CrossRef]
- Friedman, P.A.; Gesek, F.A. Calcium transport in renal epithelial cells. Am. J. Physiol. 1993, 264, F181–F198. [Google Scholar] [CrossRef] [PubMed]
- Quamme, G.A. Renal magnesium handling: New insights in understanding old problems. Kidney Int. 1997, 52, 1180–1195. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, S.J.; Kang, H.S.; Jeong, C.W.; Park, S.Y.; Kim, I.S.; Kim, N.S.; Kim, S.Z.; Kwak, Y.G.; Kim, J.S.; Quamme, G.A. Immunosuppressants inhibit hormone-stimulated Mg2+ uptake in mouse distal convoluted tubule cells. Biochem. Biophys. Res. Commun. 2006, 341, 742–748. [Google Scholar] [CrossRef] [PubMed]
- De Baaij, J.H.; Hoenderop, J.G.; Bindels, R.J. Regulation of magnesium balance: Lessons learned from human genetic disease. Clin. Kidney J. 2012, 5, i15–i24. [Google Scholar] [CrossRef]
- Ikari, A.; Okude, C.; Sawada, H.; Yamazaki, Y.; Sugatani, J.; Miwa, M. TRPM6 expression and cell proliferation are up-regulated by phosphorylation of ERK1/2 in renal epithelial cells. Biochem. Biophys. Res. Commun. 2008, 369, 1129–1133. [Google Scholar] [CrossRef]
- Chambrey, R.; Trepiccione, F. Relative Roles of Principal and Intercalated Cells in the Regulation of Sodium Balance and Blood Pressure. Curr. Hypertens. Rep. 2015, 17, 538. [Google Scholar] [CrossRef] [PubMed]
- Trepiccione, F.; Zacchia, M.; Capasso, G. Physiopathology of Potassium Deficiency. In Seldin and Giebisch’s the Kidney, Physiology and Pathophysiology, 5th ed.; Alpern, R.J., Moe, O.W., Caplan, M., Eds.; Elsevier: San Diego, CA, USA, 2013; pp. 1713–1738. [Google Scholar]
- Zacchia, M.; Abategiovanni, M.L.; Stratigis, S.; Capasso, G. Potassium: From physiology to clinical implications. Kidney Dis. 2016, 2, 72–79. [Google Scholar] [CrossRef] [PubMed]
- Trepiccione, F.; Pisitkun, T.; Hoffert, J.D.; Poulsen, S.B.; Capasso, G.; Nielsen, S.; Knepper, M.A.; Fenton, R.A.; Christensen, B.M. Early targets of lithium in rat kidney inner medullary collecting duct include p38 and ERK1/2. Kidney Int. 2014, 86, 757–767. [Google Scholar] [CrossRef] [Green Version]
- Nielsen, J.; Hoffert, J.D.; Knepper, M.A.; Agre, P.; Nielsen, S.; Fenton, R.A. Proteomic analysis of lithium-induced nephrogenic diabetes insipidus: Mechanisms for aquaporin 2 down-regulation and cellular proliferation. Proc. Natl. Acad. Sci. USA 2008, 105, 3634–3639. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iervolino, A.; Trepiccione, F.; Petrillo, F.; Spagnuolo, M.; Scarfò, M.; Frezzetti, D.; De Vita, G.; De Felice, M.; Capasso, G. Selective dicer suppression in the kidney alters GSK3β/β-catenin pathways promoting a glomerulocystic disease. PLoS ONE 2015, 10, e0119142. [Google Scholar] [CrossRef] [PubMed]
- Vehaskari, V.M.; Hering-Smith, K.S.; Moskowitz, D.W.; Weiner, I.D.; Hamm, L.L. Effect of epidermal growth factor on sodium transport in the cortical collecting tubule. Am. J. Physiol. Ren. Physiol. 2017, 256, F803–F809. [Google Scholar] [CrossRef] [PubMed]
- Falin, R.; Veizis, I.E.; Cotton, C.U. A role for ERK1/2 in EGF- and ATP-dependent regulation of amiloride-sensitive sodium absorption. Am. J. Physiol. Cell Physiol. 2005, 288, C1003–C1011. [Google Scholar] [CrossRef] [Green Version]
- Shi, H.; Asher, C.; Chigaev, A.; Yung, Y.; Reuveny, E.; Seger, R.; Garty, H. Interactions of β and γENaC with Nedd4 can be facilitated by an ERK-mediated phosphorylation. J. Biol. Chem. 2002, 277, 13539–13547. [Google Scholar] [CrossRef]
- Rossier, B.C.; Pradervand, S.; Schild, L.; Hummler, E. Epithelial Sodium Channel and the Control of Sodium Balance: Interaction between Genetic and Environmental Factors. Annu. Rev. Physiol. 2002, 64, 877–897. [Google Scholar] [CrossRef]
- Falin, R.A.; Cotton, C.U. Acute Downregulation of ENaC by EGF Involves the PY Motif and Putative ERK Phosphorylation Site. J. Gen. Physiol. 2007, 130, 313–328. [Google Scholar] [CrossRef] [Green Version]
- Lee, I.H.; Song, S.H.; Cook, D.I.; Dinudom, A. H-ras mediates the inhibitory effect of epidermal growth factor on the epithelial Na+ channel. PLoS ONE 2015, 10, e0116938. [Google Scholar] [CrossRef] [PubMed]
- Veizis, I.E.; Cotton, C.U. Abnormal EGF-dependent regulation of sodium absorption in ARPKD collecting duct cells. Am. J. Physiol. Ren. Physiol. 2004, 288, F474–F482. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Wang, Z.; Sun, P.; Jin, Y.; Lin, D.H.; Hebert, S.C.; Giebisch, G.; Wang, W.H. Inhibition of MAPK stimulates the Ca2+-dependent big-conductance K channels in cortical collecting duct. Proc. Natl. Acad. Sci. USA 2006, 51, 19569–19574. [Google Scholar] [CrossRef] [PubMed]
- Jin, Y.; Wang, Z.; Zhang, Y.; Yang, B.; Wang, W.H. PGE 2 inhibits apical K channels in the CCD through activation of the MAPK pathway. Am. J. Physiol. Ren. Physiol. 2007, 293, F1299–F1307. [Google Scholar] [CrossRef] [PubMed]
- Droge, W. Free Radicals in the Physiological Control of Cell Function. Physiol. Rev. 2002, 82, 47–95. [Google Scholar] [CrossRef] [PubMed]
- Tang, H.; Hao, Q.; Rutherford, S.A.; Low, B.; Zhao, Z.J. Inactivation of Src family tyrosine kinases by reactive oxygen species in vivo. J. Biol. Chem. 2005, 280, 23918–23925. [Google Scholar] [CrossRef]
- Laroche-Joubert, N.; Marsy, S.; Luriau, S.; Imbert-Teboul, M.; Doucet, A. Mechanism of activation of ERK and H-K-ATPase by isoproterenol in rat cortical collecting duct. Am. J. Physiol. Ren. Physiol. 2003, 284, F948–F954. [Google Scholar] [CrossRef] [Green Version]
- Azroyan, A.; Cortez-Retamozo, V.; Bouley, R.; Liberman, R.; Ruan, Y.C.; Kiselev, E.; Jacobson, K.A.; Pittet, M.J.; Brown, D.; Breton, S. Renal intercalated cells sense and mediate inflammation via the P2Y14 receptor. PLoS ONE 2015, 10, e0121419. [Google Scholar] [CrossRef]
- Tamouza, H.; Chemouny, J.M.; Raskova Kafkova, L.; Berthelot, L.; Flamant, M.; Demion, M.; Moura, I.C. The IgA1 immune complex-mediated activation of the MAPK/ERK kinase pathway in mesangial cells is associated with glomerular damage in IgA nephropathy. Kidney Int. 2012, 82, 1284–1296. [Google Scholar] [CrossRef]
- Cianfrone, P.; Simeoni, M.; Comi, N.; Piraina, V.; Talarico, R.; Cerantonio, A.; Gentile, I.; Fabiano, F.F.; Lucisano, G.; Foti, D.; et al. How to improve duration and efficiency of the antiproteinuric response to Ramipril: RamiPROT—A prospective cohort study. J. Nephrol. 2017, 30, 95–102. [Google Scholar] [CrossRef]
- Simeoni, M.; Cianfrone, P.; Comi, N.; Gentile, I.; Fabiano, F.F.; Piraina, V.; Talarico, R.; Lucisano, G.; Rivoli, L.; Andreucci, M.; et al. Is it feasible to improve the duration and the efficiency of Ramipril anti-proteinuric response? G. Ital. Nefrol. 2015, 21, gin/32.1.9. [Google Scholar]
- Simeoni, M.; Nicotera, R.; Colao, M.; Citraro, M.L.; Pelagi, E.; Cerantonio, A.; Comi, N.; Coppolino, G.; Fuiano, G. Direct inhibition of plasmatic renin activity with aliskiren: A promising but under-investigated therapeutic option for non-diabetic glomerulonephritis. Int. Urol. Nephrol. 2016, 48, 229–237. [Google Scholar] [CrossRef]
- Simeoni, M.; Nicotera, R.; Pelagi, E.; Libri, E.; Comi, N.; Fuiano, G. Successful Use of Aliskiren in a Case of IgA-Mesangial Glomerulonephritis Unresponsive to Conventional Therapies. Rev. Recent Clin. Trials 2019, 14, 72–76. [Google Scholar] [CrossRef]
- Wei, C.; Cardarelli, M.G.; Downing, S.W.; McLaughlin, J.S. The effect of angiotensin II on mitogen-activated protein kinase in human cardiomyocytes. J. Renin Angiotensin Aldosterone Syst. 2000, 1, 379–384. [Google Scholar] [Green Version]
- Giachini, F.R.; Sullivan, J.C.; Lima, V.V.; Carneiro, F.S.; Fortes, Z.B.; Pollock, D.M.; Carvalho, M.H.; Webb, R.C.; Tostes, R.C. Extracellular signal-regulated kinase 1/2 activation, via downregulation of mitogen-activated protein kinase phosphatase 1, mediates sex differences in desoxycorticosterone acetate-salt hypertension vascular reactivity. Hypertension 2010, 55, 172–179. [Google Scholar] [CrossRef] [PubMed]
- Rossol-Haseroth, K.; Zhou, Q.; Braun, S.; Boldyreff, B.; Falkenstein, E.; Wehling, M.; Lösel, R.M. Mineralocorticoid receptor antagonists do not block rapid ERK activation by aldosterone. Biochem. Biophys. Res. Commun. 2004, 318, 281–288. [Google Scholar] [CrossRef] [PubMed]
- Lannoy, M.; Slove, S.; Louedec, L.; Choqueux, C.; Journé, C.; Michel, J.B.; Jacob, M.P. Inhibition of ERK1/2 phosphorylation: A new strategy to stimulate elastogenesis in the aorta. Hypertension 2014, 64, 423–430. [Google Scholar] [CrossRef] [PubMed]
- Morris, V.; Kopetz, S. BRAF inhibitors in clinical oncology. F1000Prime Rep. 2013, 5, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Savoia, P.; Fava, P.; Casoni, F.; Cremona, O. Targeting the ERK Signaling Pathway in Melanoma. Int. J. Mol. Sci. 2019, 20, 1483. [Google Scholar] [CrossRef] [PubMed]

© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Capolongo, G.; Suzumoto, Y.; D’Acierno, M.; Simeoni, M.; Capasso, G.; Zacchia, M. ERK1,2 Signalling Pathway along the Nephron and Its Role in Acid-base and Electrolytes Balance. Int. J. Mol. Sci. 2019, 20, 4153. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20174153
Capolongo G, Suzumoto Y, D’Acierno M, Simeoni M, Capasso G, Zacchia M. ERK1,2 Signalling Pathway along the Nephron and Its Role in Acid-base and Electrolytes Balance. International Journal of Molecular Sciences. 2019; 20(17):4153. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20174153
Chicago/Turabian StyleCapolongo, Giovanna, Yoko Suzumoto, Mariavittoria D’Acierno, Mariadelina Simeoni, Giovambattista Capasso, and Miriam Zacchia. 2019. "ERK1,2 Signalling Pathway along the Nephron and Its Role in Acid-base and Electrolytes Balance" International Journal of Molecular Sciences 20, no. 17: 4153. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20174153