The Role of Extracellular Matrix Expression, ERK1/2 Signaling and Cell Cohesiveness for Cartilage Yield from iPSCs

 ,
,
Abstract
:1. Introduction
2. Results
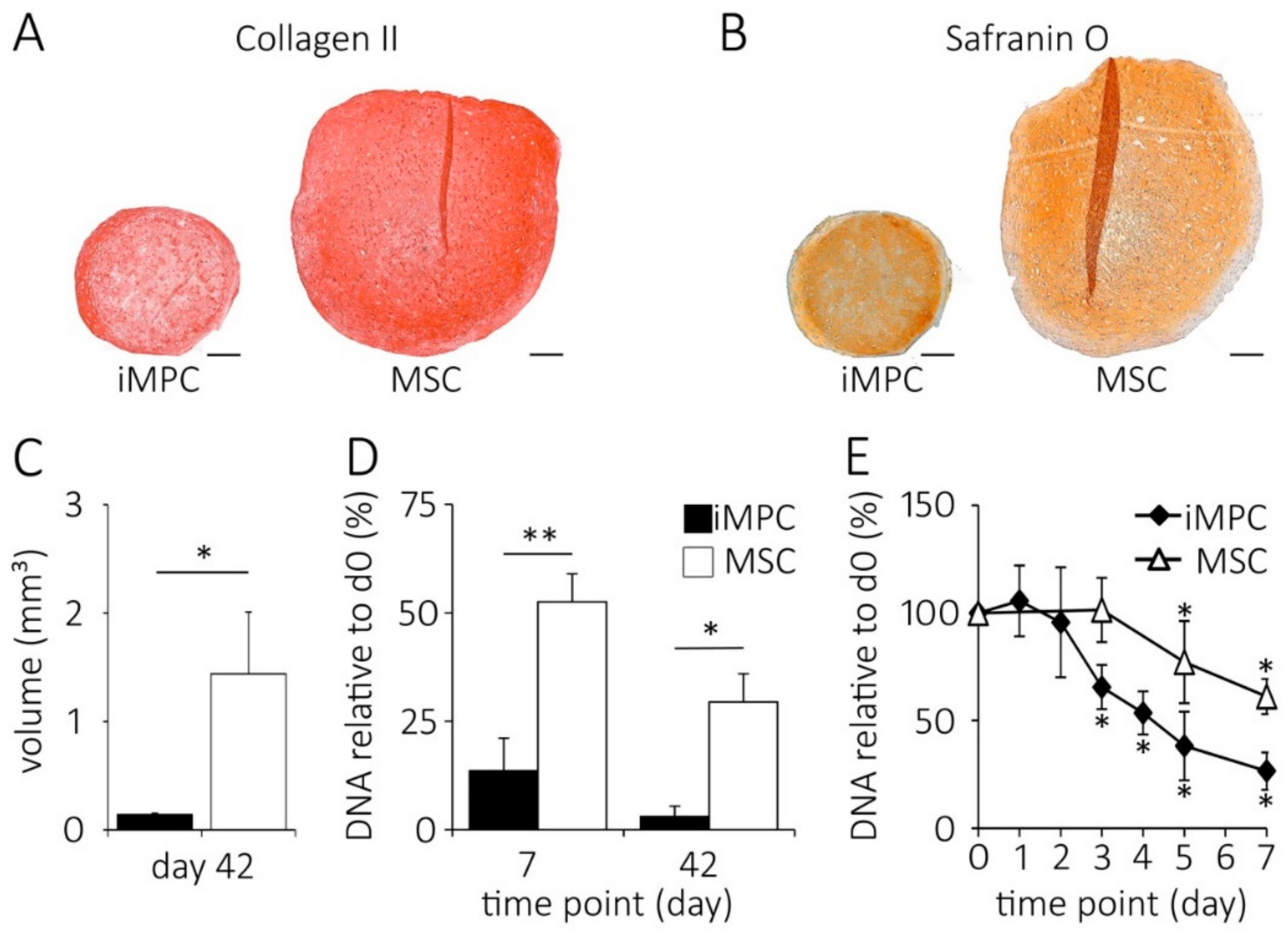
2.1. High Cell Loss Occurs at the Onset of In Vitro Chondrogenesis of iMPCs
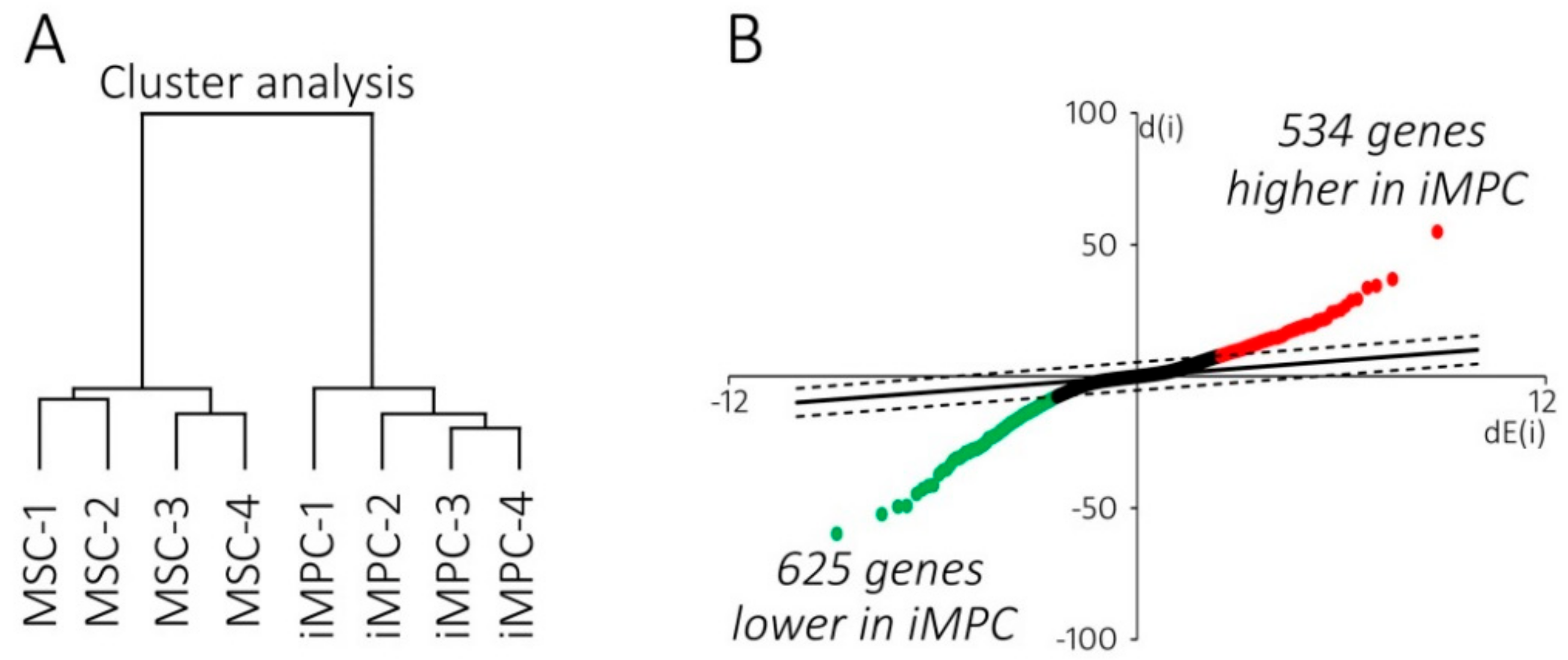
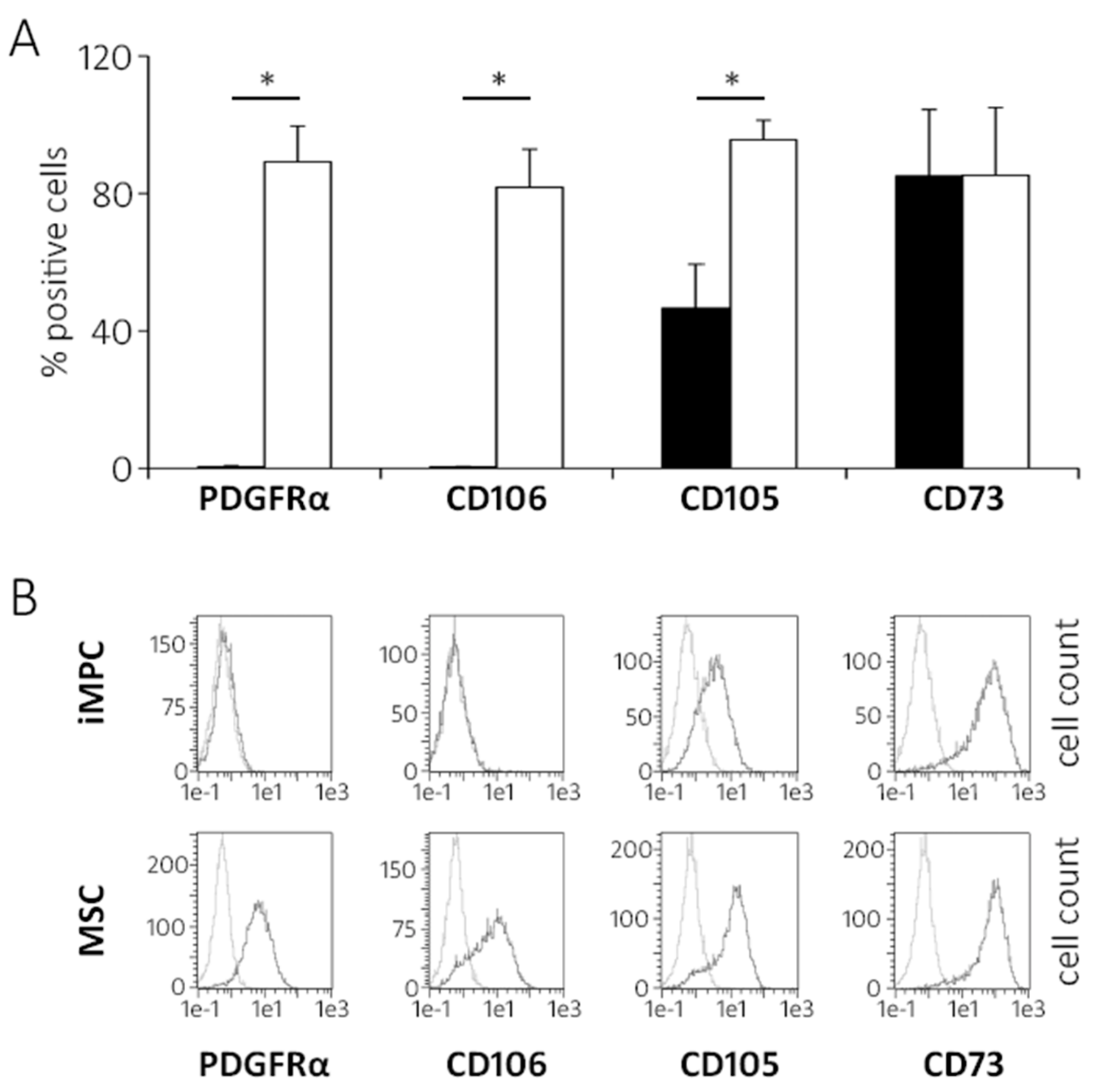
2.2. IMPCs Are More Juvenile Mesenchymal Progenitors than MSCs
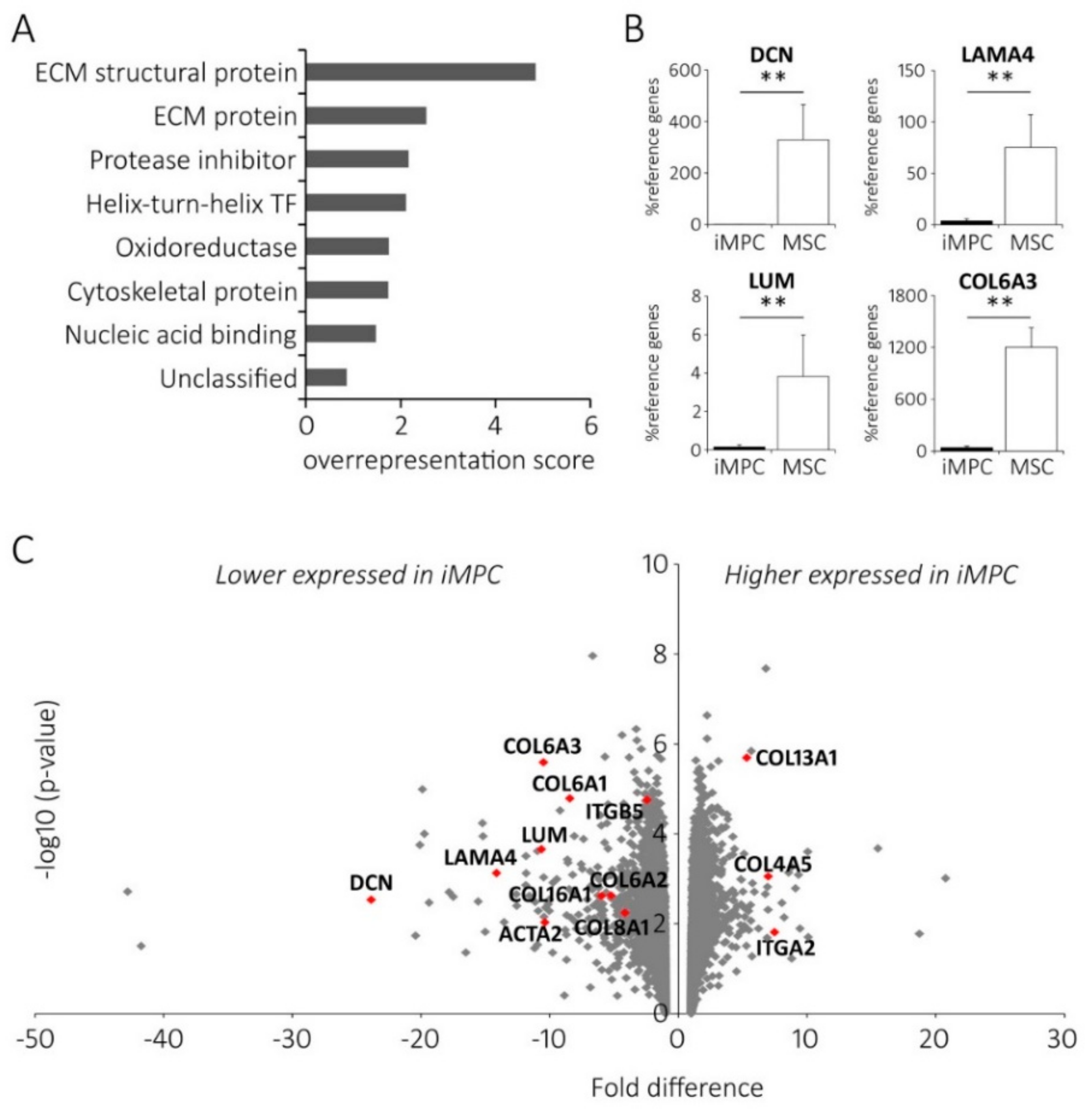
2.3. Distinct ECM Expression Profiles of iMPCs and MSCs
2.4. Differential Expression of Integrin Signaling Network Members
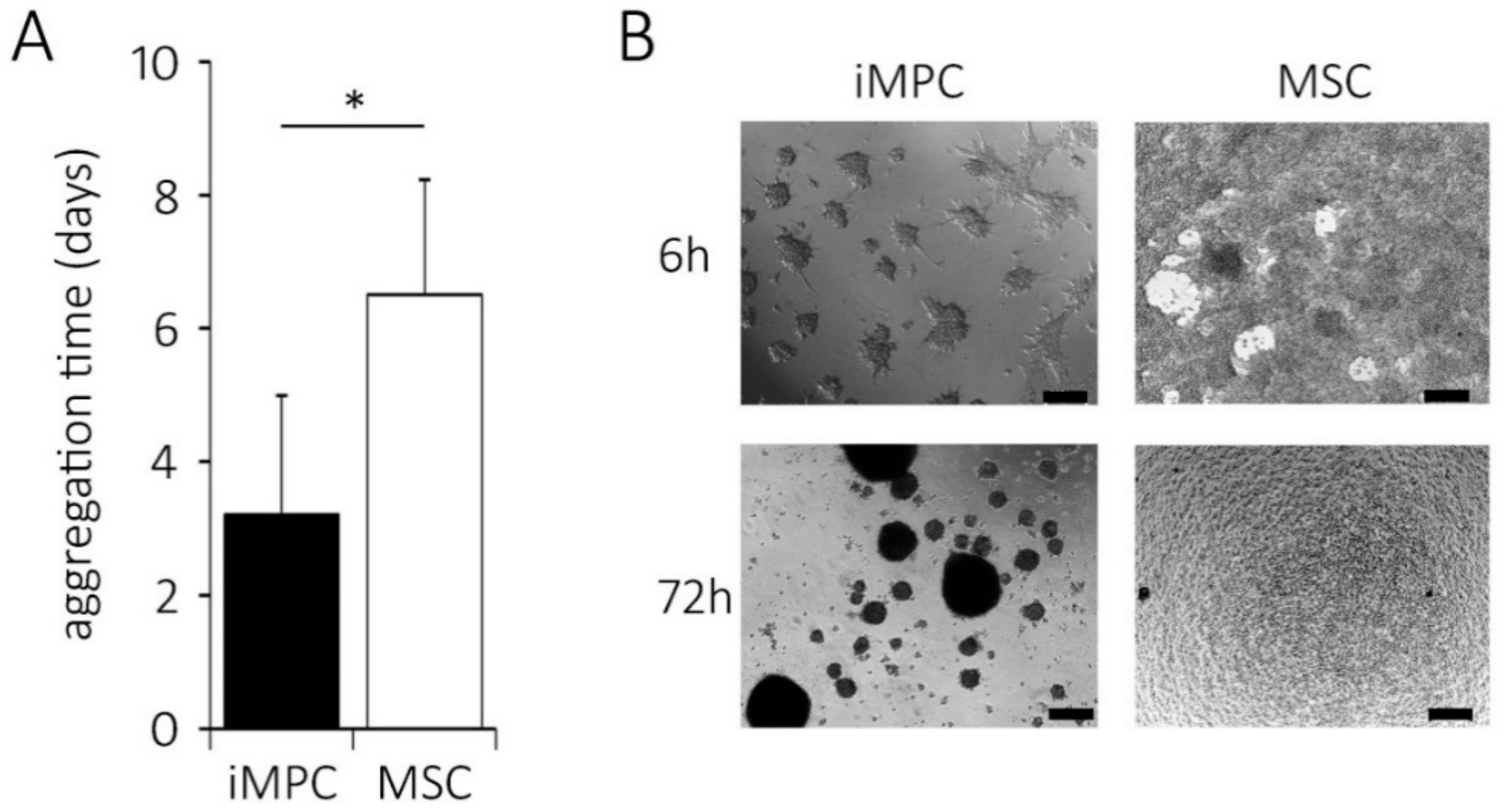
2.5. Distinct Aggregation Behavior of iMPCs versus MSCs
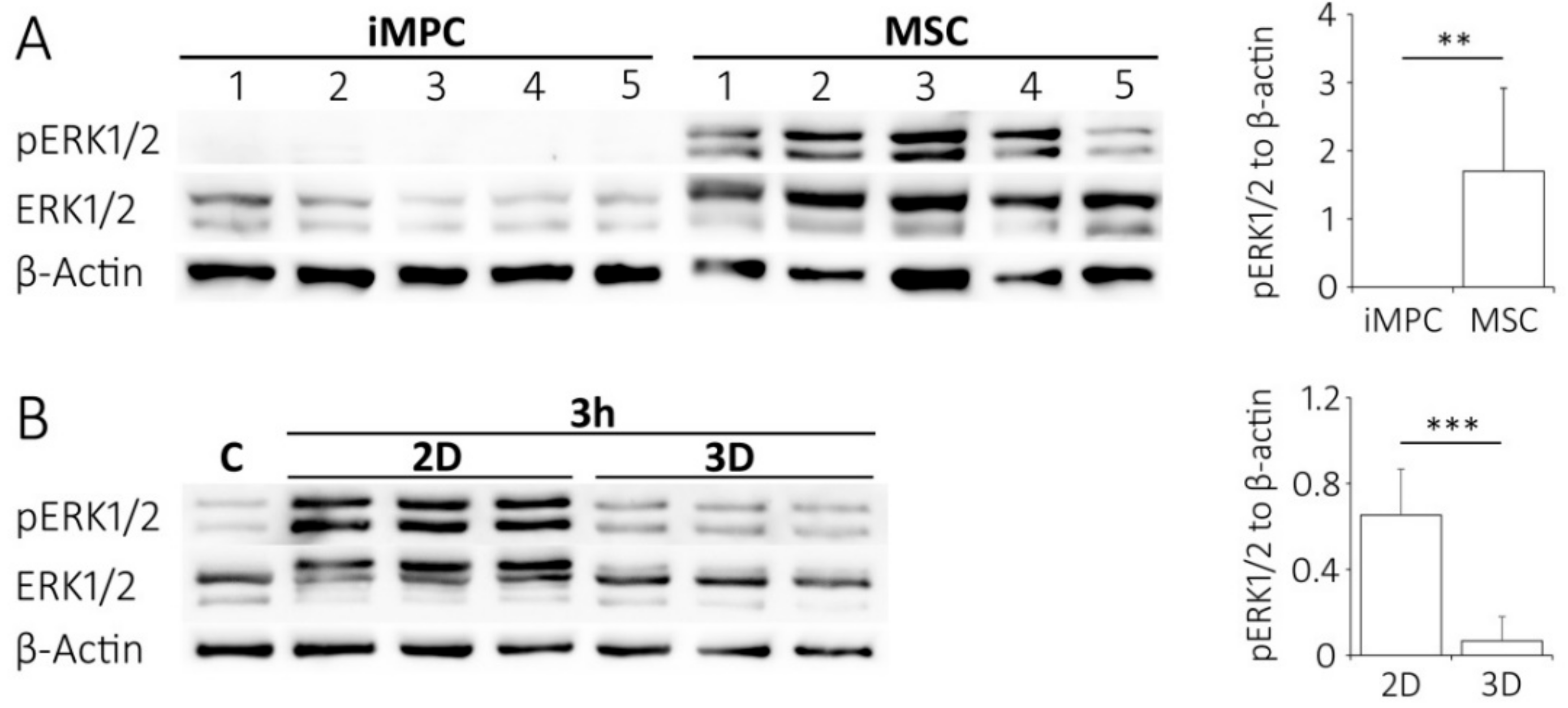
2.6. Lower ERK1/2 Signaling in iMPCs versus MSCs
3. Discussion
4. Materials and Methods
4.1. MSC Isolation and Expansion
4.2. IPSC Culture
4.3. Transcriptome Analysis
4.4. Bioinformatic Analysis
4.5. Chondrogenic Induction
4.6. Aggregation Assay
4.7. 3D agarose Culture
4.8. Quantitative Gene Expression Analysis
4.9. Quantification of DNA Content
4.10. Flow Cytometry
4.11. Histology
4.12. Histomorphometry
4.13. Western Blotting
4.14. Statistical Analyses
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| DEGs | differentially expressed genes |
| ECM | extracellular matrix |
| ERK | extracellular signal-regulated kinase |
| iMPCs | iPSC-derived mesenchymal progenitor cells |
| iPSCs | induced pluripotent stem cells |
| MSCs | mesenchymal stromal cells |
| SAM | significance analysis of microarrays |
References
- Yoo, J.U.; Barthel, T.S.; Nishimura, K.; Solchaga, L.; Caplan, A.I.; Goldberg, V.M.; Johnstone, B. The chondrogenic potential of human bone-marrow-derived mesenchymal progenitor cells. J. Bone Joint Surg. Am. 1998, 80, 1745–1757. [Google Scholar] [CrossRef] [PubMed]
- Winter, A.; Breit, S.; Parsch, D.; Benz, K.; Steck, E.; Hauner, H.; Weber, R.M.; Ewerbeck, V.; Richter, W. Cartilage-like gene expression in differentiated human stem cell spheroids: A comparison of bone marrow-derived and adipose tissue-derived stromal cells. Arthr. Rheum. 2003, 48, 418–429. [Google Scholar] [CrossRef] [PubMed]
- Pelttari, K.; Steck, E.; Richter, W. The use of mesenchymal stem cells for chondrogenesis. Injury 2008, 39 (Suppl. S1), S58–S65. [Google Scholar] [CrossRef] [PubMed]
- Jiang, T.; Xu, G.; Wang, Q.; Yang, L.; Zheng, L.; Zhao, J.; Zhang, X. In vitro expansion impaired the stemness of early passage mesenchymal stem cells for treatment of cartilage defects. Cell Death Dis. 2017, 8, e2851. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.K.; Ogando, C.R.; Wang See, C.; Chang, T.Y.; Barabino, G.A. Changes in phenotype and differentiation potential of human mesenchymal stem cells aging in vitro. Stem Cell Res. Ther. 2018, 9, 131. [Google Scholar] [CrossRef]
- Pelttari, K.; Winter, A.; Steck, E.; Goetzke, K.; Hennig, T.; Ochs, B.G.; Aigner, T.; Richter, W. Premature induction of hypertrophy during in vitro chondrogenesis of human mesenchymal stem cells correlates with calcification and vascular invasion after ectopic transplantation in SCID mice. Arthr. Rheum. 2006, 54, 3254–3266. [Google Scholar] [CrossRef] [PubMed]
- Goldberg, A.; Mitchell, K.; Soans, J.; Kim, L.; Zaidi, R. The use of mesenchymal stem cells for cartilage repair and regeneration: A systematic review. J. Orthop. Surg. Res. 2017, 12, 39. [Google Scholar] [CrossRef]
- Galipeau, J.; Sensebe, L. Mesenchymal stromal cells: Clinical challenges and therapeutic opportunities. Cell Stem Cell 2018, 22, 824–833. [Google Scholar] [CrossRef]
- Yamashita, A.; Morioka, M.; Yahara, Y.; Okada, M.; Kobayashi, T.; Kuriyama, S.; Matsuda, S.; Tsumaki, N. Generation of scaffoldless hyaline cartilaginous tissue from human iPSCs. Stem Cell Rep. 2015, 4, 404–418. [Google Scholar] [CrossRef]
- Diederichs, S.; Gabler, J.; Autenrieth, J.; Kynast, K.L.; Merle, C.; Walles, H.; Utikal, J.; Richter, W. Differential regulation of SOX9 protein during chondrogenesis of induced pluripotent stem cells versus mesenchymal stromal cells: A shortcoming for cartilage formation. Stem Cells Dev. 2016, 25, 598–609. [Google Scholar] [CrossRef]
- Adkar, S.S.; Wu, C.L.; Willard, V.P.; Dicks, A.; Ettyreddy, A.; Steward, N.; Bhutani, N.; Gersbach, C.A.; Guilak, F. Step-wise chondrogenesis of human induced pluripotent stem cells and purification via a reporter allele generated by CRISPR-Cas9 genome editing. Stem Cells 2019, 37, 65–76. [Google Scholar] [CrossRef] [PubMed]
- Diederichs, S.; Tuan, R.S. Functional comparison of human-induced pluripotent stem cell-derived mesenchymal cells and bone marrow-derived mesenchymal stromal cells from the same donor. Stem Cells Dev. 2014, 23, 1594–1610. [Google Scholar] [CrossRef] [PubMed]
- Koyama, N.; Miura, M.; Nakao, K.; Kondo, E.; Fujii, T.; Taura, D.; Kanamoto, N.; Sone, M.; Yasoda, A.; Arai, H.; et al. Human induced pluripotent stem cells differentiated into chondrogenic lineage via generation of mesenchymal progenitor cells. Stem Cells Dev. 2013, 22, 102–113. [Google Scholar] [CrossRef] [PubMed]
- Nejadnik, H.; Diecke, S.; Lenkov, O.D.; Chapelin, F.; Donig, J.; Tong, X.; Derugin, N.; Chan, R.C.; Gaur, A.; Yang, F.; et al. Improved approach for chondrogenic differentiation of human induced pluripotent stem cells. Stem Cell Rev. 2015, 11, 242–253. [Google Scholar] [CrossRef] [PubMed]
- Hynes, K.; Menicanin, D.; Mrozik, K.; Gronthos, S.; Bartold, P.M. Generation of functional mesenchymal stem cells from different induced pluripotent stem cell lines. Stem Cells Dev. 2014, 23, 1084–1096. [Google Scholar] [CrossRef]
- Frobel, J.; Hemeda, H.; Lenz, M.; Abagnale, G.; Joussen, S.; Denecke, B.; Saric, T.; Zenke, M.; Wagner, W. Epigenetic rejuvenation of mesenchymal stromal cells derived from induced pluripotent stem cells. Stem Cell Rep. 2014, 3, 414–422. [Google Scholar] [CrossRef] [PubMed]
- Bieback, K.; Kern, S.; Kluter, H.; Eichler, H. Critical parameters for the isolation of mesenchymal stem cells from umbilical cord blood. Stem Cells 2004, 22, 625–634. [Google Scholar] [CrossRef]
- Brady, K.; Dickinson, S.C.; Guillot, P.V.; Polak, J.; Blom, A.W.; Kafienah, W.; Hollander, A.P. Human fetal and adult bone marrow-derived mesenchymal stem cells use different signaling pathways for the initiation of chondrogenesis. Stem Cells Dev. 2014, 23, 541–554. [Google Scholar] [CrossRef]
- Kern, S.; Eichler, H.; Stoeve, J.; Kluter, H.; Bieback, K. Comparative analysis of mesenchymal stem cells from bone marrow, umbilical cord blood, or adipose tissue. Stem Cells 2006, 24, 1294–1301. [Google Scholar] [CrossRef]
- Andrzejewska, A.; Lukomska, B.; Janowski, M. Concise review: Mesenchymal stem cells: From roots to boost. Stem Cells 2019, 37, 855–864. [Google Scholar] [CrossRef]
- Davidson, D. The function and evolution of Msx genes: Pointers and paradoxes. Trends Genet. 1995, 11, 405–411. [Google Scholar] [CrossRef]
- Spitzhorn, L.S.; Megges, M.; Wruck, W.; Rahman, M.S.; Otte, J.; Degistirici, O.; Meisel, R.; Sorg, R.V.; Oreffo, R.O.C.; Adjaye, J. Human iPSC-derived MSCs (iMSCs) from aged individuals acquire a rejuvenation signature. Stem Cell Res. Ther. 2019, 10, 100. [Google Scholar] [CrossRef] [PubMed]
- Thomas, P.D.; Campbell, M.J.; Kejariwal, A.; Mi, H.; Karlak, B.; Daverman, R.; Diemer, K.; Muruganujan, A.; Narechania, A. PANTHER: A library of protein families and subfamilies indexed by function. Genome Res. 2003, 13, 2129–2141. [Google Scholar] [CrossRef] [PubMed]
- Howe, A.K.; Aplin, A.E.; Juliano, R.L. Anchorage-dependent ERK signaling-mechanisms and consequences. Current Opin. Genet. Dev. 2002, 12, 30–35. [Google Scholar] [CrossRef]
- Umeda, K.; Zhao, J.; Simmons, P.; Stanley, E.; Elefanty, A.; Nakayama, N. Human chondrogenic paraxial mesoderm, directed specification and prospective isolation from pluripotent stem cells. Sci. Rep. 2012, 2, 455. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alvarez, R.; Lee, H.L.; Wang, C.Y.; Hong, C. Characterization of the osteogenic potential of mesenchymal stem cells from human periodontal ligament based on cell surface markers. Int. J. Oral Sci. 2015, 7, 213–219. [Google Scholar] [CrossRef] [PubMed]
- Pinho, S.; Lacombe, J.; Hanoun, M.; Mizoguchi, T.; Bruns, I.; Kunisaki, Y.; Frenette, P.S. PDGFRα and CD51 mark human nestin+ sphere-forming mesenchymal stem cells capable of hematopoietic progenitor cell expansion. J. Exp. Med. 2013, 210, 1351–1367. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Ghazanfari, R.; Zacharaki, D.; Ditzel, N.; Isern, J.; Ekblom, M.; Mendez-Ferrer, S.; Kassem, M.; Scheding, S. Low/negative expression of PDGFR-α identifies the candidate primary mesenchymal stromal cells in adult human bone marrow. Stem Cell Rep. 2014, 3, 965–974. [Google Scholar] [CrossRef]
- Phillips, M.D.; Kuznetsov, S.A.; Cherman, N.; Park, K.; Chen, K.G.; McClendon, B.N.; Hamilton, R.S.; McKay, R.D.; Chenoweth, J.G.; Mallon, B.S.; et al. Directed differentiation of human induced pluripotent stem cells toward bone and cartilage: In vitro versus in vivo assays. Stem Cells Transl. Med. 2014, 3, 867–878. [Google Scholar] [CrossRef]
- Chan, C.K.F.; Gulati, G.S.; Sinha, R.; Tompkins, J.V.; Lopez, M.; Carter, A.C.; Ransom, R.C.; Reinisch, A.; Wearda, T.; Murphy, M.; et al. Identification of the human skeletal stem cell. Cell 2018, 175, 43–56.e21. [Google Scholar] [CrossRef]
- Casey, R.C.; Burleson, K.M.; Skubitz, K.M.; Pambuccian, S.E.; Oegema, T.R., Jr.; Ruff, L.E.; Skubitz, A.P. β1-integrins regulate the formation and adhesion of ovarian carcinoma multicellular spheroids. Am. J. Pathol. 2001, 159, 2071–2080. [Google Scholar] [CrossRef]
- Ghosh, S.; Laha, M.; Mondal, S.; Sengupta, S.; Kaplan, D.L. In vitro model of mesenchymal condensation during chondrogenic development. Biomaterials 2009, 30, 6530–6540. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, I.G.; Ko, J.; Lee, H.R.; Do, S.H.; Park, K. Mesenchymal cells condensation-inducible mesh scaffolds for cartilage tissue engineering. Biomaterials 2016, 85, 18–29. [Google Scholar] [CrossRef] [PubMed]
- Tavella, S.; Raffo, P.; Tacchetti, C.; Cancedda, R.; Castagnola, P. N-CAM and N-cadherin expression during in vitro chondrogenesis. Exp. Cell Res. 1994, 215, 354–362. [Google Scholar] [CrossRef] [PubMed]
- Hall, B.K.; Miyake, T. All for one and one for all: Condensations and the initiation of skeletal development. Bioessays 2000, 22, 138–147. [Google Scholar] [CrossRef]
- Miyamoto, S.; Teramoto, H.; Gutkind, J.S.; Yamada, K.M. Integrins can collaborate with growth factors for phosphorylation of receptor tyrosine kinases and MAP kinase activation: Roles of integrin aggregation and occupancy of receptors. J. Cell Biol. 1996, 135, 1633–1642. [Google Scholar] [CrossRef]
- Hynes, R.O. The extracellular matrix: Not just pretty fibrils. Science 2009, 326, 1216–1219. [Google Scholar] [CrossRef] [PubMed]
- Geiger, B.; Yamada, K.M. Molecular architecture and function of matrix adhesions. Cold Spring Harbor Perspect. Biol. 2011, 3, a005033. [Google Scholar] [CrossRef]
- Alexopoulos, L.G.; Youn, I.; Bonaldo, P.; Guilak, F. Developmental and osteoarthritic changes in Col6a1-knockout mice: Biomechanics of type VI collagen in the cartilage pericellular matrix. Arthr. Rheum. 2009, 60, 771–779. [Google Scholar] [CrossRef]
- Izu, Y.; Ansorge, H.L.; Zhang, G.; Soslowsky, L.J.; Bonaldo, P.; Chu, M.L.; Birk, D.E. Dysfunctional tendon collagen fibrillogenesis in collagen VI null mice. Matrix Biol. 2011, 30, 53–61. [Google Scholar] [CrossRef] [Green Version]
- Cescon, M.; Gattazzo, F.; Chen, P.; Bonaldo, P. Collagen VI at a glance. J. Cell Sci. 2015, 128, 3525–3531. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gubbiotti, M.A.; Vallet, S.D.; Ricard-Blum, S.; Iozzo, R.V. Decorin interacting network: A comprehensive analysis of decorin-binding partners and their versatile functions. Matrix Biol. 2016, 55, 7–21. [Google Scholar] [CrossRef] [PubMed]
- Iwasaki, S.; Hosaka, Y.; Iwasaki, T.; Yamamoto, K.; Nagayasu, A.; Ueda, H.; Kokai, Y.; Takehana, K. The modulation of collagen fibril assembly and its structure by decorin: An electron microscopic study. Arch. Histol. Cytol. 2008, 71, 37–44. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mohammadzadeh, N.; Lunde, I.G.; Andenaes, K.; Strand, M.E.; Aronsen, J.M.; Skrbic, B.; Marstein, H.S.; Bandlien, C.; Nygard, S.; Gorham, J.; et al. The extracellular matrix proteoglycan lumican improves survival and counteracts cardiac dilatation and failure in mice subjected to pressure overload. Sci. Rep. 2019, 9, 9206. [Google Scholar] [CrossRef] [PubMed]
- Wiberg, C.; Klatt, A.R.; Wagener, R.; Paulsson, M.; Bateman, J.F.; Heinegard, D.; Morgelin, M. Complexes of matrilin-1 and biglycan or decorin connect collagen VI microfibrils to both collagen II and aggrecan. J. Biol. Chem. 2003, 278, 37698–37704. [Google Scholar] [CrossRef] [PubMed]
- Seidler, D.G.; Schaefer, L.; Robenek, H.; Iozzo, R.V.; Kresse, H.; Schonherr, E. A physiologic three-dimensional cell culture system to investigate the role of decorin in matrix organisation and cell survival. Biochem. Biophys. Res. Commun. 2005, 332, 1162–1170. [Google Scholar] [CrossRef] [PubMed]
- Moazedi-Fuerst, F.C.; Gruber, G.; Stradner, M.H.; Guidolin, D.; Jones, J.C.; Bodo, K.; Wagner, K.; Peischler, D.; Krischan, V.; Weber, J.; et al. Effect of Laminin-A4 inhibition on cluster formation of human osteoarthritic chondrocytes. J. Orthop. Res. 2016, 34, 419–426. [Google Scholar] [CrossRef] [PubMed]
- Aplin, A.E.; Stewart, S.A.; Assoian, R.K.; Juliano, R.L. Integrin-mediated adhesion regulates ERK nuclear translocation and phosphorylation of Elk-1. J. Cell Biol. 2001, 153, 273–282. [Google Scholar] [CrossRef] [PubMed]
- Mebratu, Y.; Tesfaigzi, Y. How ERK1/2 activation controls cell proliferation and cell death: Is subcellular localization the answer? Cell Cycle 2009, 8, 1168–1175. [Google Scholar] [CrossRef] [PubMed]
- Sart, S.; Tsai, A.C.; Li, Y.; Ma, T. Three-dimensional aggregates of mesenchymal stem cells: Cellular mechanisms, biological properties, and applications. Tissue Eng. Part B Rev. 2014, 20, 365–380. [Google Scholar] [CrossRef]
- Robinson, E.E.; Zazzali, K.M.; Corbett, S.A.; Foty, R.A. α5β1 integrin mediates strong tissue cohesion. J. Cell Sci. 2003, 116, 377–386. [Google Scholar] [CrossRef] [PubMed]
- Rockey, D.C.; Weymouth, N.; Shi, Z. Smooth muscle alpha actin (Acta2) and myofibroblast function during hepatic wound healing. PloS ONE 2013, 8, e77166. [Google Scholar] [CrossRef] [PubMed]
- Kinner, B.; Zaleskas, J.M.; Spector, M. Regulation of smooth muscle actin expression and contraction in adult human mesenchymal stem cells. Exp. Cell Res. 2002, 278, 72–83. [Google Scholar] [CrossRef] [PubMed]
- Sancho, A.; Vandersmissen, I.; Craps, S.; Luttun, A.; Groll, J. A new strategy to measure intercellular adhesion forces in mature cell-cell contacts. Sci. Rep. 2017, 7, 46152. [Google Scholar] [CrossRef] [PubMed]
- Foty, R.A. Measurement of intercellular cohesion by tissue surface tensiometry. Methods Mol. Biol. 2015, 1189, 237–254. [Google Scholar] [PubMed]
- Butler, C.M.; Foty, R.A. Measurement of aggregate cohesion by tissue surface tensiometry. J. Vis. Exp. 2011, 50, 2739. [Google Scholar] [CrossRef] [PubMed]
- Medvedev, S.P.; Grigor’eva, E.V.; Shevchenko, A.I.; Malakhova, A.A.; Dementyeva, E.V.; Shilov, A.A.; Pokushalov, E.A.; Zaidman, A.M.; Aleksandrova, M.A.; Plotnikov, E.Y.; et al. Human induced pluripotent stem cells derived from fetal neural stem cells successfully undergo directed differentiation into cartilage. Stem Cells Dev. 2011, 20, 1099–1112. [Google Scholar] [CrossRef]
- Nasu, A.; Ikeya, M.; Yamamoto, T.; Watanabe, A.; Jin, Y.; Matsumoto, Y.; Hayakawa, K.; Amano, N.; Sato, S.; Osafune, K.; et al. Genetically matched human iPS cells reveal that propensity for cartilage and bone differentiation differs with clones, not cell type of origin. PloS ONE 2013, 8, e53771. [Google Scholar] [CrossRef]
- Villa-Diaz, L.G.; Brown, S.E.; Liu, Y.; Ross, A.M.; Lahann, J.; Parent, J.M.; Krebsbach, P.H. Derivation of mesenchymal stem cells from human induced pluripotent stem cells cultured on synthetic substrates. Stem Cells 2012, 30, 1174–1181. [Google Scholar] [CrossRef]
- Nam, Y.; Rim, Y.A.; Jung, S.M.; Ju, J.H. Cord blood cell-derived iPSCs as a new candidate for chondrogenic differentiation and cartilage regeneration. Stem Cell Res. Ther. 2017, 8, 16. [Google Scholar] [CrossRef]
- Li, Y.; Liu, T.; Van Halm-Lutterodt, N.; Chen, J.; Su, Q.; Hai, Y. Reprogramming of blood cells into induced pluripotent stem cells as a new cell source for cartilage repair. Stem Cell Res. Ther. 2016, 7, 31. [Google Scholar] [CrossRef] [PubMed]
- Wu, L.; Bluguermann, C.; Kyupelyan, L.; Latour, B.; Gonzalez, S.; Shah, S.; Galic, Z.; Ge, S.; Zhu, Y.; Petrigliano, F.A.; et al. Human developmental chondrogenesis as a basis for engineering chondrocytes from pluripotent stem cells. Stem Cell Rep. 2013, 1, 575–589. [Google Scholar] [CrossRef] [PubMed]
- Dickhut, A.; Gottwald, E.; Steck, E.; Heisel, C.; Richter, W. Chondrogenesis of mesenchymal stem cells in gel-like biomaterials in vitro and in vivo. Front. Biosci. 2008, 13, 4517–4528. [Google Scholar] [CrossRef] [PubMed]
- Hinz, B. The extracellular matrix and transforming growth factor-β1: Tale of a strained relationship. Matrix Biol. 2015, 47, 54–65. [Google Scholar] [CrossRef] [PubMed]
- Hesse, E.; Freudenberg, U.; Niemietz, T.; Greth, C.; Weisser, M.; Hagmann, S.; Binner, M.; Werner, C.; Richter, W. Peptide-functionalized starPEG/heparin hydrogels direct mitogenicity, cell morphology and cartilage matrix distribution in vitro and in vivo. J. Tissue Eng. Regen. Med. 2018, 12, 229–239. [Google Scholar] [CrossRef] [PubMed]
- Almeida, H.V.; Liu, Y.; Cunniffe, G.M.; Mulhall, K.J.; Matsiko, A.; Buckley, C.T.; O’Brien, F.J.; Kelly, D.J. Controlled release of transforming growth factor-beta3 from cartilage-extra-cellular-matrix-derived scaffolds to promote chondrogenesis of human-joint-tissue-derived stem cells. Acta Biomater. 2014, 10, 4400–4409. [Google Scholar] [CrossRef]
- Rowland, C.R.; Lennon, D.P.; Caplan, A.I.; Guilak, F. The effects of crosslinking of scaffolds engineered from cartilage ECM on the chondrogenic differentiation of MSCs. Biomaterials 2013, 34, 5802–5812. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pei, M.; Li, J.T.; Shoukry, M.; Zhang, Y. A review of decellularized stem cell matrix: A novel cell expansion system for cartilage tissue engineering. Eur. Cells Mater. 2011, 22, 333–343. [Google Scholar] [CrossRef]
- Thakkar, S.; Ghebes, C.A.; Ahmed, M.; Kelder, C.; van Blitterswijk, C.A.; Saris, D.; Fernandes, H.A.; Moroni, L. Mesenchymal stromal cell-derived extracellular matrix influences gene expression of chondrocytes. Biofabrication 2013, 5, 025003. [Google Scholar] [CrossRef]
- Torocsik, B.; Szeberenyi, J. Anisomycin uses multiple mechanisms to stimulate mitogen-activated protein kinases and gene expression and to inhibit neuronal differentiation in PC12 phaeochromocytoma cells. Eur. J. Neurosci. 2000, 12, 527–532. [Google Scholar] [CrossRef]
- Hong, S.S.; Qian, H.; Zhao, P.; Bazzy-Asaad, A.; Xia, Y. Anisomycin protects cortical neurons from prolonged hypoxia with differential regulation of p38 and ERK. Brain Res. 2007, 1149, 76–86. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jimenez-Sainz, M.C.; Fast, B.; Mayor, F., Jr.; Aragay, A.M. Signaling pathways for monocyte chemoattractant protein 1-mediated extracellular signal-regulated kinase activation. Mol. Pharmacol. 2003, 64, 773–782. [Google Scholar] [CrossRef] [PubMed]
- Ronkina, N.; Menon, M.B.; Schwermann, J.; Arthur, J.S.; Legault, H.; Telliez, J.B.; Kayyali, U.S.; Nebreda, A.R.; Kotlyarov, A.; Gaestel, M. Stress induced gene expression: A direct role for MAPKAP kinases in transcriptional activation of immediate early genes. Nucleic Acids Res. 2011, 39, 2503–2518. [Google Scholar] [CrossRef] [PubMed]
- Ueda, Y.; Hirai, S.; Osada, S.; Suzuki, A.; Mizuno, K.; Ohno, S. Protein kinase C activates the MEK-ERK pathway in a manner independent of Ras and dependent on Raf. J. Biol. Chem. 1996, 271, 23512–23519. [Google Scholar] [CrossRef] [PubMed]
- Wilson, L.S.; Baillie, G.S.; Pritchard, L.M.; Umana, B.; Terrin, A.; Zaccolo, M.; Houslay, M.D.; Maurice, D.H. A phosphodiesterase 3B-based signaling complex integrates exchange protein activated by cAMP 1 and phosphatidylinositol 3-kinase signals in human arterial endothelial cells. J. Biol. Chem. 2011, 286, 16285–16296. [Google Scholar] [CrossRef] [PubMed]
- Dominici, M.; Le Blanc, K.; Mueller, I.; Slaper-Cortenbach, I.; Marini, F.; Krause, D.; Deans, R.; Keating, A.; Prockop, D.; Horwitz, E. Minimal criteria for defining multipotent mesenchymal stromal cells. The International Society for Cellular Therapy position statement. Cytotherapy 2006, 8, 315–317. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Vodyanik, M.A.; Smuga-Otto, K.; Antosiewicz-Bourget, J.; Frane, J.L.; Tian, S.; Nie, J.; Jonsdottir, G.A.; Ruotti, V.; Stewart, R.; et al. Induced pluripotent stem cell lines derived from human somatic cells. Science 2007, 318, 1917–1920. [Google Scholar] [CrossRef]
- Scholtes, S.; Kramer, E.; Weisser, M.; Roth, W.; Luginbuhl, R.; Grossner, T.; Richter, W. Global chondrocyte gene expression after a single anabolic loading period: Time evolution and re-inducibility of mechano-responses. J. Cell. Physiol. 2018, 233, 699–711. [Google Scholar] [CrossRef]
- Saeed, A.I.; Sharov, V.; White, J.; Li, J.; Liang, W.; Bhagabati, N.; Braisted, J.; Klapa, M.; Currier, T.; Thiagarajan, M.; et al. TM4: a free, open-source system for microarray data management and analysis. BioTechniques 2003, 34, 374–378. [Google Scholar] [CrossRef]
- Tusher, V.G.; Tibshirani, R.; Chu, G. Significance analysis of microarrays applied to the ionizing radiation response. Proc. Natl. Acad. Sci. USA 2001, 98, 5116–5121. [Google Scholar] [CrossRef] [Green Version]
- Ashburner, M.; Ball, C.A.; Blake, J.A.; Botstein, D.; Butler, H.; Cherry, J.M.; Davis, A.P.; Dolinski, K.; Dwight, S.S.; Eppig, J.T.; et al. Gene ontology: Tool for the unification of biology. The gene ontology consortium. Nat. Genet. 2000, 25, 25–29. [Google Scholar] [CrossRef] [PubMed]
- Krase, A.; Abedian, R.; Steck, E.; Hurschler, C.; Richter, W. BMP activation and Wnt-signalling affect biochemistry and functional biomechanical properties of cartilage tissue engineering constructs. Osteoarthr. Cartil. 2014, 22, 284–292. [Google Scholar] [CrossRef] [PubMed]
- Vogel, J.P.; Szalay, K.; Geiger, F.; Kramer, M.; Richter, W.; Kasten, P. Platelet-rich plasma improves expansion of human mesenchymal stem cells and retains differentiation capacity and in vivo bone formation in calcium phosphate ceramics. Platelets 2006, 17, 462–469. [Google Scholar] [CrossRef] [PubMed]
- Schneider, C.A.; Rasband, W.S.; Eliceiri, K.W. NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 2012, 9, 671–675. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene Symbol | iMPC | MSC | Fold Difference | Gene Symbol | iMPC | MSC | Fold Difference |
|---|---|---|---|---|---|---|---|
| CD10 (MME) | 160 | b | 1.7 | CD166 (ALCAM) | 2174 | 937 | 2.3 |
| CD14 | 87 | 171 | 2.0 | CD340 (ERBB2) | 292 | 237 | 1.2 |
| CD29 (ITGB1) | 2986 | 2510 | 1.2 | ANXA5 | 3396 | 3520 | 1.0 |
| CD44 | 2947 | 1695 | 1.7 | COL1A1 | 4127 | 3117 | 1.3 |
| CD49E (ITGA5) | 1064 | 1383 | 1.3 | MSX1 | 3016 | 511 | 5.9 |
| CD73 (NT5E) | 1420 | 2207 | −1.6 | PDGFRA | 266 | 2782 | −10.5 |
| CD90 (THY1) | 630 | 1009 | −1.6 | PDGFRB | 813 | 2739 | −3.4 |
| CD105 (ENG) | 266 | 1824 | −6.9 | PPARG | 196 | 240 | −1.2 |
| CD106 (VCAM1) | b | 1748 | −19.4 | RUNX2 | 139 | 131 | 1.0 |
| VIM | 11725 | 11777 | 1.0 |
| Gene Symbol | iMPC | MSC | Fold Difference | Gene Name |
|---|---|---|---|---|
| LRRC32 | 131 | 993 | −7.6 | Leucine-rich repeat-containing protein |
| GAS6 | 364 | 2283 | −6.3 | Growth arrest-specific protein 6 |
| ADAMTS1 | 294 | 1838 | −6.3 | A disintegrin and MMP with thrombosponding motifs 1 |
| VASN | 460 | 2516 | −5.5 | Vasorin |
| ISLR | b | 417 | −5.1 | Ig superfamily containg leucine-rich protein |
| SEPP1 | b | 415 | −4.8 | Selenoprotein P |
| EGFLAM | b | 365 | −4.4 | Pikachurin |
| COL8A1 1 | 929 | 3854 | −4.1 | Collagen type VIIIα1 |
| CLEC3B 1 | b | 336 | −3.9 | Tetranectin |
| LTBP2 1 | 425 | 1402 | −3.3 | Latent TGFβ-binding protein 2 |
| CLEC11A1 1 | 158 | 495 | −3.1 | C-type lectin dmoain family 11 member A |
| EFEMP2 1 | 876 | 2657 | −3.0 | EGF-containing fibulin-like ECM protein 2 |
| SCUBE3 | b | 226 | −2.6 | Signal peptide CUB and EGF-like DCP3 |
| LAMB2 | 317 | 794 | −2.5 | Laminin subunit beta-2 |
| ADAMTS5 | b | 199 | −2.4 | A disintegrin and MMP with thrombospondin motifs 5 |
| LAMA3 | b | 196 | −2.3 | Laminin subunit alpha 3 |
| AGRN | 284 | 126 | 2.3 | Agrin |
| FBLN2 1 | 904 | 177 | 5.1 | Fibulin-2 |
| Pathway | Number of Genes | p-Value |
|---|---|---|
| Integrin signaling pathway | 24 | 0.0015 |
| Opioid proenkephalin pathway | 9 | 0.0023 |
| Inflammation mediated by chemokine and cytokine signaling | 21 | 0.0149 |
| Plasminogen activating cascade | 2 | 0.0158 |
| Endothelin signaling pathway | 7 | 0.0202 |
| De novo purine biosynthesis | 5 | 0.0209 |
| Pyridoxal phosphate salvage pathway | 2 | 0.0482 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Buchert, J.; Diederichs, S.; Kreuser, U.; Merle, C.; Richter, W. The Role of Extracellular Matrix Expression, ERK1/2 Signaling and Cell Cohesiveness for Cartilage Yield from iPSCs. Int. J. Mol. Sci. 2019, 20, 4295. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20174295
Buchert J, Diederichs S, Kreuser U, Merle C, Richter W. The Role of Extracellular Matrix Expression, ERK1/2 Signaling and Cell Cohesiveness for Cartilage Yield from iPSCs. International Journal of Molecular Sciences. 2019; 20(17):4295. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20174295
Chicago/Turabian StyleBuchert, Justyna, Solvig Diederichs, Ursula Kreuser, Christian Merle, and Wiltrud Richter. 2019. "The Role of Extracellular Matrix Expression, ERK1/2 Signaling and Cell Cohesiveness for Cartilage Yield from iPSCs" International Journal of Molecular Sciences 20, no. 17: 4295. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20174295