Vascular and Neuronal Protection in the Developing Retina: Potential Therapeutic Targets for Retinopathy of Prematurity


Abstract
:1. Introduction
2. Vasculature in the Retina
2.1. Normal Development of the Retinal Vasculature
2.2. Pathogenesis of Retinopathy of Prematurity
2.2.1. Vascular Endothelial Growth Factor (VEGF)
2.2.2. Insulin-Like Growth Factor-1 (IGF-1)
2.2.3. Erythropoietin (Epo)
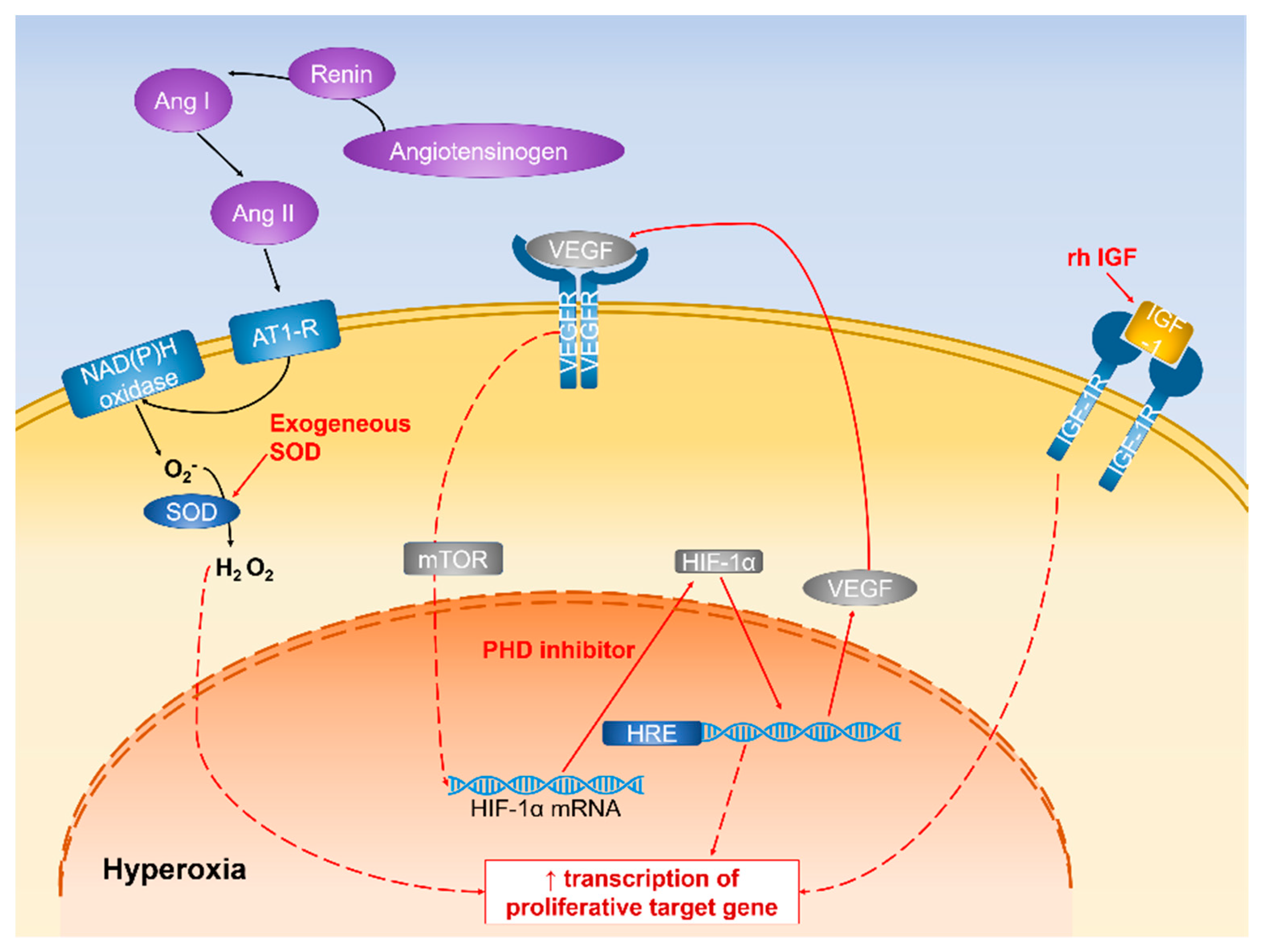
2.2.4. Hypoxia-Inducible Factor-1 (HIF-1)
2.2.5. Nitric Oxide (NO)
2.2.6. Adenosine
2.2.7. β-Adrenergic Receptor (β-AR)
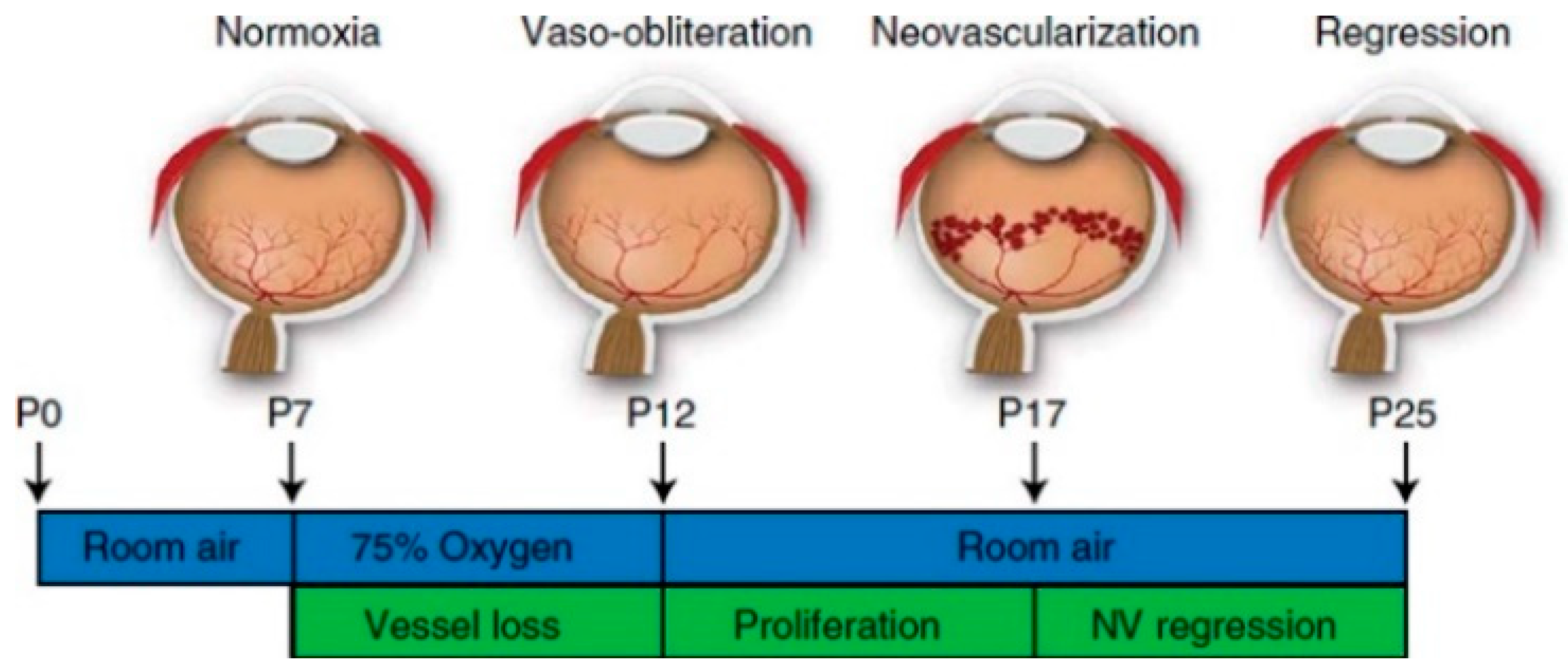
3. Animal Models for ROP—Oxygen-Induced Retinopathy (OIR)
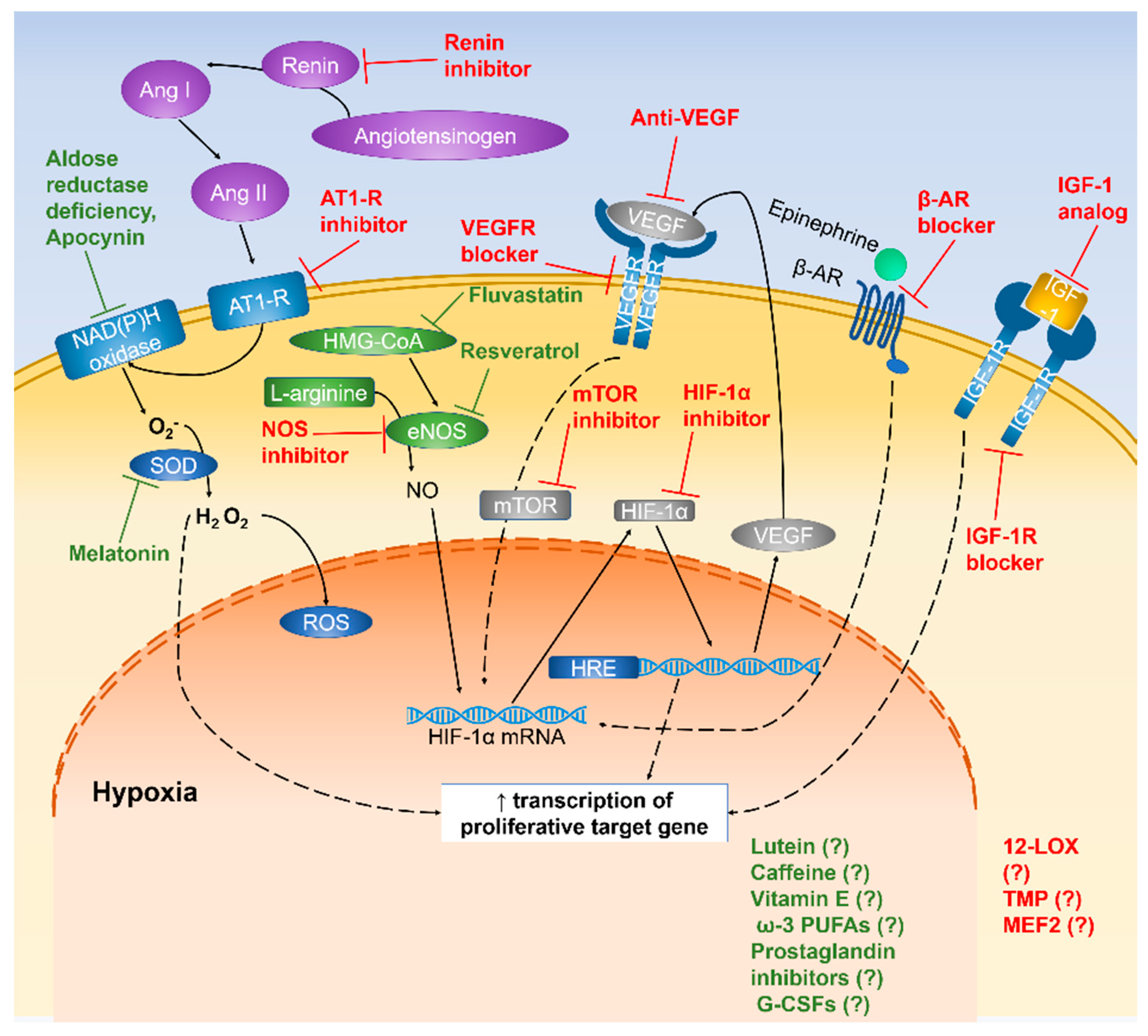
4. Vascular Protection in the ROP
4.1. Growth Factors
4.1.1. Anti-VEGF
4.1.2. IGF-Binding Protein (IGFBP)
4.2. Transcription Factors
4.2.1. Regulation of HIF-1α Expression
4.2.2. Inhibitory Effect of NOS Expression
4.2.3. Blockage of β-ARs
4.3. Anti-Angiogenesis
4.3.1. Steroid Agents
4.3.2. Other Angiogenic Inhibitors
5. Neuroprotective Agents in ROP
5.1. Antioxidants
5.1.1. Nutritional Antioxidants
Lutein
Caffeine
Omega-3 Long-Chain Polyunsaturated Fatty Acids (ω-3 PUFAs)
Resveratrol
Vitamin E
5.1.2. Endogenous Antioxidants
Suppression of Aldose Reductase
Superoxide Dismutase (SOD)
Statin
Melatonin
Apocynin
5.2. Anti-Inflammatory Agents
5.2.1. Prostaglandin Inhibitors
5.2.2. Granulocyte Colony-Stimulating Factor (G-CSF)
5.3. Others
Inhibition of Renin-Angiotensin System (RAS)
6. Stem Cell Therapy in ROP
7. Current Treatments in ROP
8. Conclusions and Future Perspectives
Funding
Conflicts of Interest
Abbreviations
| 5′ N | 5′ nucleotidase |
| AD | Alzheimer’s disease |
| AMD | Age-related macular degeneration |
| AT1-R | Angiotensin II type-1 receptor |
| β-AR | β-adrenergic receptor |
| BRB | Blood-retinal barrier |
| COX | Cyclooxygenase |
| DHA | Docosahexaenoic |
| DR | Diabetic retinopathy |
| EPA | Eicosapentaenoic |
| eEPC | Early endothelial progenitor cells |
| Epo | Erythropoietin |
| FDA | Food and Drug Administration |
| G-CSF | Granulocyte colony-stimulating factor |
| HIF | Hypoxia-inducible factor |
| HRE | Hypoxia response element |
| HSPG | Heparin sulphate proteoglycans |
| IGF-1 | Insulin-like growth factor-1 |
| KDR | Kinase insert domain-containing receptor |
| KLT-1 | Kms-related tyrosine kinase 1 |
| mTOR | Mammalian target of rapamycin |
| NOS | Nitric oxide synthetase |
| NRP | Neurophilin |
| ω-3 PUFAs | Omega-3 long-chain polyunsaturated fatty acids |
| OIR | Oxygen-induced retinopathy |
| OPC | Outgrowth endothelial cells |
| PD | Parkinson’s disease |
| PHD | Prolyl hydroxylase |
| RAS | Renin-angiotensin system |
| RGC | Retinal ganglion cells |
| ROP | Retinopathy of prematurity |
| ROS | Reactive oxygen species |
| SC | Stem cell |
| SRPK1 | Serine arginine protein kinase 1 |
| SRSF1 | Serine-rich splicing factor-1 |
| STOP-ROP | Supplemental Therapeutic Oxygen for Prethreshold Retinopathy Of Prematurity |
| VEGF | Vascular endothelial growth factor |
References
- Terry, T.L. Extreme prematurity and fibroblastic overgrowth of persistent vascular sheath behind each crystalline lens: I. Preliminary report. Am. J. Ophthalmol. 1942, 25, 203–204. [Google Scholar] [CrossRef]
- Jain, V.; Langham, M.C.; Wehrli, F.W. MRI estimation of global brain oxygen consumption rate. Br. J. Pharmacol. 2010, 30, 1987. [Google Scholar]
- Anderson, B.; Saltzman, H.A. Retinal Oxygen Utilization Measured by Hyperbaric Blackout. Arch. Ophthalmol. 1964, 72, 792–795. [Google Scholar] [CrossRef] [PubMed]
- Anderson, B., Jr. Ocular effects of changes in oxygen and carbon dioxide tension. Trans. Am. Ophthalmol. Soc. 1968, 66, 423. [Google Scholar] [PubMed]
- Nag, T.; Wadhwa, S. Morphological and Neurochemical Development of the Human Neural Retina. Neuroembryol. Aging 2006, 4, 19–30. [Google Scholar] [CrossRef]
- Weidman, T.A. Fine Structure of the Developing Retina. Int. Ophthalmol. Clin. 1975, 15, 65–84. [Google Scholar] [CrossRef] [PubMed]
- Van Cruchten, S.; Vrolyk, V.; Lepage, M.-F.P.; Baudon, M.; Voute, H.; Schoofs, S.; Haruna, J.; Benoit-Biancamano, M.-O.; Ruot, B.; Allegaert, K.; et al. Pre- and Postnatal Development of the Eye: A Species Comparison. Birth Defects Res. 2017, 109, 1540–1567. [Google Scholar] [CrossRef] [PubMed]
- Scott, A.; Fruttiger, M. Oxygen-induced retinopathy: A model for vascular pathology in the retina. Eye 2010, 24, 416–421. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Lai, C.H.; Lo, A.C. Therapeutic strategies for retinopathy of prematurity. Hong Kong J. Ophthalmol. 2015, 19, 8–15. [Google Scholar]
- Group, S.-R.M.S. Supplemental therapeutic oxygen for prethreshold retinopathy of prematurity (STOP-ROP), a randomized, controlled trial. I: Primary outcomes. Pediatrics 2000, 105, 295–310. [Google Scholar]
- Chen, J.; Smith, L.E. Retinopathy of prematurity. Angiogenesis 2007, 10, 133–140. [Google Scholar] [CrossRef]
- Hellström, A.; LSmith, E.; Dammann, O. Retinopathy of prematurity. Lancet 2013, 382, 1445–1457. [Google Scholar] [CrossRef] [Green Version]
- Sapieha, P.; Joyal, J.-S.; Rivera, J.C.; Kermorvant-Duchemin, E.; Sennlaub, F.; Hardy, P.; Lachapelle, P.; Chemtob, S. Retinopathy of prematurity: Understanding ischemic retinal vasculopathies at an extreme of life. J. Clin. Investig. 2010, 120, 3022–3032. [Google Scholar] [CrossRef]
- Tin, W.; Gupta, S. Optimum oxygen therapy in preterm babies. Arch. Dis. Child. Fetal Neonatal Ed. 2007, 92, F143–F147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Connor, K.M.; Krah, N.M.; Dennison, R.J.; Aderman, C.M.; Chen, J.; Guerin, K.I.; Sapieha, P.; Stahl, A.; Willett, K.L.; Smith, L.E.H. Quantification of oxygen-induced retinopathy in the mouse: A model of vessel loss, vessel regrowth and pathological angiogenesis. Nat. Protoc. 2009, 4, 1565–1573. [Google Scholar] [CrossRef] [PubMed]
- Alon, T.; Hemo, I.; Itin, A.; Pe’Er, J.; Stone, J.; Keshet, E. Vascular endothelial growth factor acts as a survival factor for newly formed retinal vessels and has implications for retinopathy of prematurity. Nat. Med. 1995, 1, 1024–1028. [Google Scholar] [CrossRef] [PubMed]
- Mintz-Hittner, H.A.; Kennedy, K.A.; Chuang, A.Z. Efficacy of intravitreal bevacizumab for stage 3+ retinopathy of prematurity. N. Engl. J. Med. 2011, 364, 603–615. [Google Scholar] [CrossRef] [PubMed]
- Sato, T.; Wada, K.; Arahori, H.; Kuno, N.; Imoto, K.; Iwahashi-Shima, C.; Kusaka, S. Serum Concentrations of Bevacizumab (Avastin) and Vascular Endothelial Growth Factor in Infants with Retinopathy of Prematurity. Am. J. Ophthalmol. 2012, 153, 327–333. [Google Scholar] [CrossRef]
- Wu, W.-C.; Shih, C.-P.; Lien, R.; Wang, N.-K.; Chen, Y.-P.; Chao, A.-N.; Chen, K.-J.; Chen, T.-L.; Hwang, Y.-S.; Lai, C.-C. Serum vascular endothelial growth factor after bevacizumab or ranibizumab treatment for retinopathy of prematurity. Retina 2017, 37, 1–701. [Google Scholar] [CrossRef]
- Chung, E.J.; Kim, J.H.; Ahn, H.S.; Koh, H.J. Combination of laser photocoagulation and intravitreal bevacizumab (Avastin®) for aggressive zone I retinopathy of prematurity. Graefe’s Arch. Clin. Exp. Ophthalmol. 2007, 245, 1727–1730. [Google Scholar] [CrossRef]
- Kim, J.; Kim, S.J.; Chang, Y.S.; Park, W.S. Combined intravitreal bevacizumab injection and zone I sparing laser photocoagulation in patients with zone I retinopathy of prematurity. Retina 2014, 34, 77–82. [Google Scholar] [CrossRef] [PubMed]
- Martínez-Castellanos, M.A.; Schwartz, S.; Hernández-Rojas, M.L.; Kon-Jara, V.A.; García-Aguirre, G.; Guerrero-Naranjo, J.L.; Chan, R.V.P.; Quiroz-Mercado, H. Long-term effect of antiangiogenic therapy for retinopathy of prematurity up to 5 Years of Follow-up. Retina 2013, 33, 329–338. [Google Scholar] [CrossRef] [PubMed]
- Morin, J.; Luu, T.M.; Superstein, R.; Ospina, L.H.; Lefebvre, F.; Simard, M.-N.; Shah, V.; Shah, P.S.; Kelly, E.N.; The Canadian Neonatal Network and the Canadian Neonatal Follow-Up Network Investigators. Neurodevelopmental Outcomes Following Bevacizumab Injections for Retinopathy of Prematurity. Pediatrics 2016, 137, e20153218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Castellanos, M.A.M.; Schwartz, S.; García-Aguirre, G.; Quiroz-Mercado, H. Short-term outcome after intravitreal ranibizumab injections for the treatment of retinopathy of prematurity. Br. J. Ophthalmol. 2013, 97, 816–819. [Google Scholar] [CrossRef] [PubMed]
- Mota, Á.; Carneiro, Â.; Breda, J.; Rosas, V.; Magalhães, A.; Silva, R.; Falcão-Reis, F. Combination of Intravitreal Ranibizumab and Laser Photocoagulation for Aggressive Posterior Retinopathy of Prematurity. Case Rep. Ophthalmol. 2012, 3, 136–141. [Google Scholar] [CrossRef] [PubMed]
- Tokunaga, C.C.; Mitton, K.; Dailey, W.; Massoll, C.; Roumayah, K.; Guzmán, E.; Tarabishy, N.; Cheng, M.; Drenser, K.A. Effects of Anti-VEGF Treatment on the Recovery of the Developing Retina Following Oxygen-Induced Retinopathy. Investig. Opthalmology Vis. Sci. 2014, 55, 1884–1892. [Google Scholar] [CrossRef] [Green Version]
- Jiang, Y.; Wang, H.; Culp, D.; Yang, Z.; Fotheringham, L.; Flannery, J.; Hammond, S.; Kafri, T.; Hartnett, M.E. Targeting Müller Cell–Derived VEGF164 to Reduce Intravitreal Neovascularization in the Rat Model of Retinopathy of Prematurity. Investig. Opthalmol. Vis. Sci. 2014, 55, 824–831. [Google Scholar] [CrossRef]
- McLeod, D.S.; Taomoto, M.; Cao, J.; Zhu, Z.; Witte, L.; Lutty, G.A. Localization of VEGF receptor-2 (KDR/Flk-1) and effects of blocking it in oxygen-induced retinopathy. Investig. Ophthalmol. Vis. Sci. 2002, 43, 474–482. [Google Scholar]
- Gammons, M.V.; Dick, A.D.; Harper, S.J.; Bates, D.O. SRPK1 inhibition modulates VEGF splicing to reduce pathological neovascularization in a rat model of retinopathy of prematurity. Investig. Ophthalmol. Vis. Sci. 2013, 54, 5797–5806. [Google Scholar] [CrossRef]
- Yagasaki, R.; Nakahara, T.; Ushikubo, H.; Mori, A.; Sakamoto, K.; Ishii, K. Anti-angiogenic Effects of Mammalian Target of Rapamycin Inhibitors in a Mouse Model of Oxygen-Induced Retinopathy. Boil. Pharm. Bull. 2014, 37, 1838–1842. [Google Scholar] [CrossRef] [Green Version]
- Vanhaesebrouck, S.; Daniëls, H.; Moons, L.; Vanhole, C.; Carmeliet, P.; De Zegher, F. Oxygen-Induced Retinopathy in Mice: Amplification by Neonatal IGF-I Deficit and Attenuation by IGF-I Administration. Pediatr. Res. 2009, 65, 307–310. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Löfqvist, C.; Chen, J.; Connor, K.M.; Smith, A.C.H.; Aderman, C.M.; Liu, N.; Pintar, J.E.; Ludwig, T.; Hellström, A.; Smith, L.E.H. IGFBP3 suppresses retinopathy through suppression of oxygen-induced vessel loss and promotion of vascular regrowth. Proc. Natl. Acad. Sci. USA 2007, 104, 10589–10594. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kielczewski, J.L.; Hu, P.; Shaw, L.C.; Calzi, S.L.; Mames, R.N.; Gardiner, T.A.; McFarland, E.; Chan-Ling, T.; Grant, M.B. Novel Protective Properties of IGFBP-3 Result in Enhanced Pericyte Ensheathment, Reduced Microglial Activation, Increased Microglial Apoptosis, and Neuronal Protection after Ischemic Retinal Injury. Am. J. Pathol. 2011, 178, 1517–1528. [Google Scholar] [CrossRef] [PubMed]
- Smith, L.E.H.; Shen, W.; Perruzzi, C.; Soker, S.; Kinose, F.; Xu, X.; Robinson, G.; Driver, S.; Bischoff, J.; Zhang, B.; et al. Regulation of vascular endothelial growth factor-dependent retinal neovascularization by insulin-like growth factor-1 receptor. Nat. Med. 1999, 5, 1390–1395. [Google Scholar] [CrossRef] [PubMed]
- Sears, J.E.; Hoppe, G.; Ebrahem, Q.; Anand-Apte, B. Prolyl hydroxylase inhibition during hyperoxia prevents oxygen-induced retinopathy. Proc. Natl. Acad. Sci. USA 2008, 105, 19898–19903. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duan, L.-J.; Takeda, K.; Fong, G.-H. Prolyl Hydroxylase Domain Protein 2 (PHD2) Mediates Oxygen-Induced Retinopathy in Neonatal Mice. Am. J. Pathol. 2011, 178, 1881–1890. [Google Scholar] [CrossRef] [PubMed]
- Brafman, A.; Mett, I.; Shafir, M.; Gottlieb, H.; Damari, G.; Gozlan-Kelner, S.; Vishnevskia-Dai, V.; Skaliter, R.; Einat, P.; Faerman, A.; et al. Inhibition of Oxygen-Induced Retinopathy in RTP801-Deficient Mice. Investig. Opthalmol. Vis. Sci. 2004, 45, 3796–3805. [Google Scholar] [CrossRef]
- Beauchamp, M.H.; Sennlaub, F.; Speranza, G.; Gobeil, F.; Checchin, D.; Kermorvant-Duchemin, E.; Abran, D.; Hardy, P.; Lachapelle, P.; Varma, D.R.; et al. Redox-dependent effects of nitric oxide on microvascular integrity in oxygen-induced retinopathy. Free. Radic. Boil. Med. 2004, 37, 1885–1894. [Google Scholar] [CrossRef]
- Brooks, S.E.; Gu, X.; Samuel, S.; Marcus, D.M.; Bartoli, M.; Huang, P.L.; Caldwell, R.B. Reduced severity of oxygen-induced retinopathy in eNOS-deficient mice. Investig. Ophthalmol. Vis. Sci. 2001, 42, 222–228. [Google Scholar]
- Zhang, Q.; Zhang, J.; Guan, Y.; Zhang, S.; Zhu, C.; Xu, G.-T.; Wang, L. Suppression of retinal neovascularization by the iNOS inhibitor aminoguanidine in mice of oxygen-induced retinopathy. Graefe’s Arch. Clin. Exp. Ophthalmol. 2009, 247, 919–927. [Google Scholar] [CrossRef]
- Ristori, C.; Filippi, L.; Dal Monte, M.; Martini, D.; Cammalleri, M.; Fortunato, P.; la Marca, G.; Fiorini, P.; Bagnoli, P. Role of the adrenergic system in a mouse model of oxygen-induced retinopathy: Antiangiogenic effects of β-adrenoreceptor blockade. Investig. Ophthalmol. Vis. Sci. 2011, 52, 155–170. [Google Scholar] [CrossRef] [PubMed]
- Monte, M.D.; Casini, G.; La Marca, G.; Isacchi, B.; Filippi, L.; Bagnoli, P. Eye drop propranolol administration promotes the recovery of oxygen-induced retinopathy in mice. Exp. Eye Res. 2013, 111, 27–35. [Google Scholar] [CrossRef] [PubMed]
- Martini, D.; Monte, M.D.; Ristori, C.; Cupisti, E.; Mei, S.; Fiorini, P.; Filippi, L.; Bagnoli, P. Antiangiogenic effects of β2-adrenergic receptor blockade in a mouse model of oxygen-induced retinopathy. J. Neurochem. 2011, 119, 1317–1329. [Google Scholar] [CrossRef] [PubMed]
- Rotschild, T.; Nandgaonkar, B.N.; Yu, K.; Higgins, R.D. Dexamethasone Reduces Oxygen Induced Retinopathy in a Mouse Model. Pediatr. Res. 1999, 46, 94–100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Penn, J.S.; Rajaratnam, V.S.; Collier, R.J.; Clark, A.F. The effect of an angiostatic steroid on neovascularization in a rat model of retinopathy of prematurity. Investig. Ophthalmol. Vis. Sci. 2001, 42, 283–290. [Google Scholar]
- Kim, J.H.; Yu, Y.S.; Shin, J.Y.; Lee, H.-Y.; Kim, K.-W. Deguelin inhibits retinal neovascularization by down-regulation of HIF-1α in oxygen-induced retinopathy. J. Cell. Mol. Med. 2008, 12, 2407–2415. [Google Scholar] [CrossRef]
- DeNiro, M.; Al-Halafi, A.; Al-Mohanna, F.H.; AlSmadi, O.; Al-Mohanna, F.A. Pleiotropic effects of YC-1 selectively inhibit pathological retinal neovascularization and promote physiological revascularization in a mouse model of oxygen-induced retinopathy. Mol. Pharmacol. 2010, 77, 348–367. [Google Scholar] [CrossRef]
- Park, S.W.; Kim, J.H.; Kim, K.E.; Jeong, M.H.; Park, H.; Park, B.; Suh, Y.G.; Park, W.J.; Kim, J.H. Beta-lapachone inhibits pathological retinal neovascularization in oxygen-induced retinopathy via regulation of HIF-1α. J. Cell. Mol. Med. 2014, 18, 875–884. [Google Scholar] [CrossRef]
- Pan, H.; Nguyen, N.-Q.-N.; Yoshida, H.; Bentzien, F.; Shaw, L.C.; Rentier-Delrue, F.; Martial, J.A.; Weiner, R.; Struman, I.; Grant, M.B. Molecular targeting of antiangiogenic factor 16K hPRL inhibits oxygen-induced retinopathy in mice. Investig. Opthalmol. Vis. Sci. 2004, 45, 2413–2419. [Google Scholar] [CrossRef]
- Al-Shabrawey, M.; Mussell, R.; Kahook, K.; Tawfik, A.; Eladl, M.; Sarthy, V.; Nussbaum, J.; El-Marakby, A.; Park, S.Y.; Gurel, Z. Increased expression and activity of 12-lipoxygenase in oxygen-induced ischemic retinopathy and proliferative diabetic retinopathy: Implications in retinal neovascularization. Diabetes 2011, 60, 614–624. [Google Scholar] [CrossRef]
- Liang, X.; Zhou, H.; Ding, Y.; Li, J.; Yang, C.; Luo, Y.; Li, S.; Sun, G.; Liao, X.; Min, W. TMP Prevents Retinal Neovascularization and Imparts Neuroprotection in an Oxygen-Induced Retinopathy Model. Investig. Opthalmol. Vis. Sci. 2012, 53, 2157–2169. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.X.; Sima, J.; Shao, C.; Fant, J.; Chen, Y.; Rohrer, B.; Gao, G.; Ma, J.-X. Plasminogen kringle 5 reduces vascular leakage in the retina in rat models of oxygen-induced retinopathy and diabetes. Diabetologia 2004, 47, 124–131. [Google Scholar] [CrossRef] [Green Version]
- Xu, Z.; Gong, J.; Maiti, D.; Vong, L.; Wu, L.; Schwarz, J.J.; Duh, E.J. MEF2C Ablation in Endothelial Cells Reduces Retinal Vessel Loss and Suppresses Pathologic Retinal Neovascularization in Oxygen-Induced Retinopathy. Am. J. Pathol. 2012, 180, 2548–2560. [Google Scholar] [CrossRef] [Green Version]
- Ferrara, N.; Davis-Smyth, T. The Biology of Vascular Endothelial Growth Factor. Endocr. Rev. 1997, 18, 4–25. [Google Scholar] [CrossRef]
- Ferrara, N.; Gerber, H.P. Vascular endothelial growth factor molecular and biological aspects. In Advances in Organ Biology; Elsevier: Amsterdam, The Netherlands, 1999; pp. 25–57. [Google Scholar]
- Cavallaro, G.; Filippi, L.; Bagnoli, P.; La Marca, G.; Cristofori, G.; Raffaeli, G.; Padrini, L.; Araimo, G.; Fumagalli, M.; Groppo, M.; et al. The pathophysiology of retinopathy of prematurity: An update of previous and recent knowledge. Acta Ophthalmol. 2014, 92, 2–20. [Google Scholar] [CrossRef] [PubMed]
- Abbracchio, M.; Brambilla, R.; Ceruti, S.; Kim, H.; Von Lubitz, D.; Jacobson, K.; Cattabeni, F. G-protein-dependent activation of phospholipase C by adenosine A3 receptors in rat brain. Pharmacol. Res. 1995, 31, 168. [Google Scholar] [CrossRef]
- Bellik, L.; Vinci, M.C.; Filippi, S.; Ledda, F.; Parenti, A. Intracellular pathways triggered by the selective FLT-1-agonist placental growth factor in vascular smooth muscle cells exposed to hypoxia. Br. J. Pharmacol. 2005, 146, 568–575. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Terman, B.I.; Dougher-Vermazen, M.; Carrion, M.E.; Dimitrov, D.; Armellino, D.C.; Gospodarowicz, D.; Böhlen, P. Identification of the KDR tyrosine kinase as a receptor for vascular endothelial cell growth factor. Biochem. Biophys. Res. Commun. 1992, 187, 1579–1586. [Google Scholar] [CrossRef]
- Quinn, T.P.; Peters, K.G.; De Vries, C.; Ferrara, N.; Williams, L.T. Fetal liver kinase 1 is a receptor for vascular endothelial growth factor and is selectively expressed in vascular endothelium. Proc. Natl. Acad. Sci. USA 1993, 90, 7533–7537. [Google Scholar] [CrossRef] [PubMed]
- Waltenberger, J.; Claesson-Welsh, L.; Siegbahn, A.; Shibuya, M.; Heldin, C.H. Different signal transduction properties of KDR and Flt1, two receptors for vascular endothelial growth factor. J. Boil. Chem. 1994, 269, 26988–26995. [Google Scholar]
- Bernatchez, P.N.; Soker, S.; Sirois, M.G. Vascular Endothelial Growth Factor Effect on Endothelial Cell Proliferation, Migration, and Platelet-activating Factor Synthesis Is Flk-1-dependent. J. Boil. Chem. 1999, 274, 31047–31054. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaur, C.; Foulds, W.; Ling, E. Blood–retinal barrier in hypoxic ischaemic conditions: Basic concepts, clinical features and management. Prog. Retin. Eye Res. 2008, 27, 622–647. [Google Scholar] [CrossRef] [PubMed]
- Provis, J. Development of the Primate Retinal Vasculature. Prog. Retin. Eye Res. 2001, 20, 799–821. [Google Scholar] [CrossRef]
- Gariano, R.F.; Gardner, T.W. Retinal angiogenesis in development and disease. Nature 2005, 438, 960–966. [Google Scholar] [CrossRef] [PubMed]
- Stone, J.; Maslim, J. Mechanisms of retinal angiogenesis. Prog. Retin. Eye Res. 1997, 16, 157–181. [Google Scholar] [CrossRef]
- Hellström, A. IGF-I Is Critical for Normal Vascularization of the Human Retina. J. Clin. Endocrinol. Metab. 2002, 87, 3413–3416. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hellström, A.; Perruzzi, C.; Ju, M.; Engström, E.; Hård, A.-L.; Liu, J.-L.; Albertsson-Wikland, K.; Carlsson, B.; Niklasson, A.; Sjödell, L.; et al. Low IGF-I suppresses VEGF-survival signaling in retinal endothelial cells: Direct correlation with clinical retinopathy of prematurity. Proc. Natl. Acad. Sci. USA 2001, 98, 5804–5808. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamaji, R.; Okada, T.; Moriya, M.; Naito, M.; Tsuruo, T.; Miyatake, K.; Nakano, Y. Brain Capillary Endothelial Cells Express two forms of Erythropoietin Receptor mRNA. JBIC J. Boil. Inorg. Chem. 1996, 239, 494–500. [Google Scholar] [CrossRef] [PubMed]
- Watts, K.D.; McColley, S.A. Elevated vascular endothelial growth factor is correlated with elevated erythropoietin in stable, young cystic fibrosis patients. Pediatr. Pulmonol. 2011, 46, 683–687. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Connor, K.M.; Aderman, C.M.; Smith, L.E. Erythropoietin deficiency decreases vascular stability in mice. J. Clin. Investig. 2008, 118, 526–533. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hartnett, M.E. Pathophysiology and mechanisms of severe retinopathy of prematurity. Ophthalmology 2015, 122, 200–210. [Google Scholar] [CrossRef] [PubMed]
- Rusai, K.; Vannay, A.; Szebeni, B.; Borgulya, G.; Fekete, A.; Vásárhelyi, B.; Tulassay, T.; Szabó, A.J. Endothelial nitric oxide synthase gene T−786C and 27-bp repeat gene polymorphisms in retinopathy of prematurity. Mol. Vis. 2008, 14, 286–290. [Google Scholar] [PubMed]
- Yanamandra, K.; Napper, D.; Pramanik, A.; Bocchini, J.A.; Dhanireddy, R. Endothelial Nitric Oxide Synthase genotypes in the etiology of retinopathy of prematurity in premature infants. Ophthalmic Genet. 2010, 31, 173–177. [Google Scholar] [CrossRef] [PubMed]
- Hashiguchi, A.; Yano, S.; Morioka, M.; Hamada, J.; Ushio, Y.; Takeuchi, Y.; Fukunaga, K. Up-Regulation of Endothelial Nitric Oxide Synthase via Phosphatidylinositol 3-Kinase Pathway Contributes to Ischemic Tolerance in the CA1 Subfield of Gerbil Hippocampus. Br. J. Pharmacol. 2004, 24, 271–279. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haskó, G.; Cronstein, B.N. Regulation of Inflammation by Adenosine. Front. Immunol. 2013, 4, 85. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Madeira, M.H.; Boia, R.; Elvas, F.; Martins, T.; Cunha, R.A.; Ambrósio, A.F.; Santiago, A.R.; Information, P.E.K.F.C. Selective A2A receptor antagonist prevents microglia-mediated neuroinflammation and protects retinal ganglion cells from high intraocular pressure–induced transient ischemic injury. Transl. Res. 2016, 169, 112–128. [Google Scholar] [CrossRef]
- Cerri, S.; Levandis, G.; Ambrosi, G.; Montepeloso, E.; Antoninetti, G.F.; Franco, R.; Lanciego, J.L.; Baqi, Y.; Müller, C.E.; Pinna, A. Neuroprotective potential of adenosine A2A and cannabinoid CB1 receptor antagonists in an animal model of Parkinson disease. J. Neuropathol. Exp. Neurol. 2014, 73, 414–424. [Google Scholar] [CrossRef]
- Gyoneva, S.; Shapiro, L.; Lazo, C.; Garnier-Amblard, E.; Smith, Y.; Miller, G.W.; Traynelis, S.F. Adenosine A2A receptor antagonism reverses inflammation-induced impairment of microglial process extension in a model of Parkinson’s disease. Neurobiol. Dis. 2014, 67, 191–202. [Google Scholar] [CrossRef]
- Canas, P.M.; Porciúncula, L.O.; Cunha, G.M.A.; Silva, C.G.; Machado, N.J.; Oliveira, J.M.A.; Oliveira, C.R.; Cunha, R.A. Adenosine A2A Receptor Blockade Prevents Synaptotoxicity and Memory Dysfunction Caused by β-Amyloid Peptides via p38 Mitogen-Activated Protein Kinase Pathway. J. Neurosci. 2009, 29, 14741–14751. [Google Scholar] [CrossRef]
- Chen, J.F.; Huang, Z.; Ma, J.; Zhu, J.; Moratalla, R.; Standaert, D.; Moskowitz, M.A.; Fink, J.S.; Schwarzschild, M.A. A2A adenosine receptor deficiency attenuates brain injury induced by transient focal ischemia in mice. J. Neurosci. 1999, 19, 9192–9200. [Google Scholar] [CrossRef]
- Santiago, A.R.; Baptista, F.I.; Santos, P.F.; Cristóvão, G.; Ambrósio, A.F.; Cunha, R.A.; Gomes, C.A. Role of microglia adenosine A2A receptors in retinal and brain neurodegenerative diseases. Mediat. Inflamm. 2014, 2014, 465694. [Google Scholar] [CrossRef]
- Fischer, S.; Sharma, H.; Karliczek, G.; Schaper, W. Expression of vascular permeability factor/vascular endothelial growth factor in pig cerebral microvascular endothelial cells and its upregulation by adenosine. Mol. Brain Res. 1995, 28, 141–148. [Google Scholar] [CrossRef]
- Takagi, H.; King, G.L.; Ferrara, N.; Aiello, L.P. Hypoxia regulates vascular endothelial growth factor receptor KDR/Flk gene expression through adenosine A2 receptors in retinal capillary endothelial cells. Investig. Ophthalmol. Vis. Sci. 1996, 37, 1311–1321. [Google Scholar]
- Grant, M.B.; Davis, M.I.; Caballero, S.; Feoktistov, I.; Biaggioni, I.; Belardinelli, L. Proliferation, migration, and ERK activation in human retinal endothelial cells through A2B adenosine receptor stimulation. Investig. Ophthalmol. Vis. Sci. 2001, 42, 2068–2073. [Google Scholar]
- De Hoz, R.; Gallego, B.I.; Ramírez, A.I.; Rojas, B.; Salazar, J.J.; Valiente-Soriano, F.J.; Avilés-Trigueros, M.; Villegas-Perez, M.P.; Vidal-Sanz, M.; Triviño, A. Rod-like microglia are restricted to eyes with laser-induced ocular hypertension but absent from the microglial changes in the contralateral untreated eye. PLoS ONE 2013, 8, e83733. [Google Scholar] [CrossRef] [PubMed]
- Taylor, S.; Calder, C.J.; Albon, J.; Erichsen, J.T.; Boulton, M.E.; Morgan, J.E. Involvement of the CD200 receptor complex in microglia activation in experimental glaucoma. Exp. Eye Res. 2011, 92, 338–343. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, C.P.; Sharma, S.; Steinle, J.J. Age-related changes in sympathetic neurotransmission in rat retina and choroid. Exp. Eye Res. 2007, 84, 75–81. [Google Scholar] [CrossRef]
- Steinle, J.J.; Smith, P.G. Role of adrenergic receptors in vascular remodelling of the rat choroid. Br. J. Pharmacol. 2002, 136, 730–734. [Google Scholar] [CrossRef] [PubMed]
- Guimarães, S.; Moura, D. Vascular adrenoceptors: An update. Pharmacol. Rev. 2001, 53, 319–356. [Google Scholar]
- Smith, L.; Wesolowski, E.; McLellan, A.; Kostyk, S.K.; D’Amato, R.; Sullivan, R.; D’Amore, P.A. Oxygen-induced retinopathy in the mouse. Investig. Ophthalmol. Vis. Sci. 1994, 35, 101–111. [Google Scholar]
- Grossniklaus, H.E.; Kang, S.J.; Berglin, L. Animal Models of Choroidal and Retinal Neovascularization. Prog. Retin. Eye Res. 2010, 29, 500–519. [Google Scholar] [CrossRef] [PubMed]
- Penn, J.S.; Tolman, B.L.; Lowery, L.A. Variable oxygen exposure causes preretinal neovascularization in the newborn rat. Investig. Ophthalmol. Vis. Sci. 1993, 34, 576–585. [Google Scholar]
- Rapisarda, A.; Uranchimeg, B.; Sordet, O.; Pommier, Y.; Shoemaker, R.H.; Melillo, G. Topoisomerase I-mediated inhibition of hypoxia-inducible factor 1: Mechanism and therapeutic implications. Cancer Res. 2004, 64, 1475–1482. [Google Scholar] [CrossRef] [PubMed]
- Yu, T.; Tang, B.; Sun, X. Development of Inhibitors Targeting Hypoxia-Inducible Factor 1 and 2 for Cancer Therapy. Yonsei Med. J. 2017, 58, 489–496. [Google Scholar] [CrossRef] [PubMed]
- Miwa, Y.; Hoshino, Y.; Shoda, C.; Jiang, X.; Tsubota, K.; Kurihara, T. Pharmacological HIF inhibition prevents retinal neovascularization with improved visual function in a murine oxygen-induced retinopathy model. Neurochem. Int. 2019, 128, 21–31. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Joyal, J.-S.; Hatton, C.J.; Juan, A.M.; Pei, D.T.; Hurst, C.G.; Xu, D.; Stahl, A.; Hellström, A.; Smith, L.E.H. Propranolol Inhibition of β-Adrenergic Receptor Does Not Suppress Pathologic Neovascularization in Oxygen-Induced Retinopathy. Investig. Opthalmol. Vis. Sci. 2012, 53, 2968–2977. [Google Scholar] [CrossRef] [PubMed]
- Yun, J.-H.; Koh, Y.J.; Jeong, H.-S.; Lee, D.-H.; Lee, E.H.; Cho, C.-H. Propranolol increases vascular permeability through pericyte apoptosis and exacerbates oxygen-induced retinopathy. Biochem. Biophys. Res. Commun. 2018, 503, 2792–2799. [Google Scholar] [CrossRef] [PubMed]
- Parupia, M.H.; Dhanireddy, R. Association of Postnatal Dexamethasone Use and Fungal Sepsis in the Development of Severe Retinopathy of Prematurity and Progression to Laser Therapy in Extremely Low-Birth-Weight Infants. J. Perinatol. 2001, 21, 242–247. [Google Scholar] [CrossRef] [Green Version]
- Pisani, F.; Cammalleri, M.; Dal Monte, M.; Locri, F.; Mola, M.G.; Nicchia, G.P.; Frigeri, A.; Bagnoli, P.; Svelto, M. Potential role of the methylation of VEGF gene promoter in response to hypoxia in oxygen-induced retinopathy: Beneficial effect of the absence of AQP4. J. Cell. Mol. Med. 2018, 22, 613–627. [Google Scholar] [CrossRef]
- Kolibabka, M.; Dietrich, N.; Klein, T.; Hammes, H.-P. Anti-angiogenic effects of the DPP-4 inhibitor linagliptin via inhibition of VEGFR signalling in the mouse model of oxygen-induced retinopathy. Diabetologia 2018, 61, 2412–2421. [Google Scholar] [CrossRef] [Green Version]
- Vähätupa, M.; Cordova, Z.M.; Barker, H.; Aittomäki, S.; Uusitalo, H.; Järvinen, T.A.; Pesu, M.; Uusitalo-Järvinen, H. Furin deficiency in myeloid cells leads to attenuated revascularization in a mouse-model of oxygen-induced retinopathy. Exp. Eye Res. 2018, 166, 160–167. [Google Scholar] [CrossRef] [PubMed]
- Lu, C.-S.; Lee, Y.-N.; Wang, S.-W.; Wu, Y.-J.; Su, C.-H.; Hsieh, C.-L.; Tien, T.Y.; Wang, B.-J.; Chen, M.-C.; Chen, C.-W.; et al. KC21 Peptide Inhibits Angiogenesis and Attenuates Hypoxia-Induced Retinopathy. J. Cardiovasc. Transl. Res. 2019, 12, 366–377. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, F.; Leblanc, M.E.; Wang, W.; Liang, D.; Chen, P.; Chou, T.-H.; Tian, H.; Li, W. Anti-secretogranin III therapy of oxygen-induced retinopathy with optimal safety. Angiogenesis 2019, 22, 369–382. [Google Scholar] [CrossRef] [PubMed]
- Geng, W.; Qin, F.; Ren, J.; Xiao, S.; Wang, A. Mini-peptide RPL41 attenuated retinal neovascularization by inducing degradation of ATF4 in oxygen-induced retinopathy mice. Exp. Cell Res. 2018, 369, 243–250. [Google Scholar] [CrossRef] [PubMed]
- Iizuka, N.; Morita, A.; Kawano, C.; Mori, A.; Sakamoto, K.; Kuroyama, M.; Ishii, K.; Nakahara, T. Anti-angiogenic effects of valproic acid in a mouse model of oxygen-induced retinopathy. J. Pharmacol. Sci. 2018, 138, 203–208. [Google Scholar] [CrossRef] [PubMed]
- Lalkovičová, M.; Danielisová, V. Neuroprotection and antioxidants. Neural Regen. Res. 2016, 11, 865. [Google Scholar] [CrossRef] [PubMed]
- Sohn, E.H.; Van Dijk, H.W.; Jiao, C.; Kok, P.H.B.; Jeong, W.; Demirkaya, N.; Garmager, A.; Wit, F.; Kucukevcilioglu, M.; Van Velthoven, M.E.J.; et al. Retinal neurodegeneration may precede microvascular changes characteristic of diabetic retinopathy in diabetes mellitus. Proc. Natl. Acad. Sci. USA 2016, 113, E2655–E2664. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Hu, Y.; Zhou, T.; Zhou, K.K.; Mott, R.; Wu, M.; Boulton, M.; Lyons, T.J.; Gao, G.; Ma, J.-X. Activation of the Wnt Pathway Plays a Pathogenic Role in Diabetic Retinopathy in Humans and Animal Models. Am. J. Pathol. 2009, 175, 2676–2685. [Google Scholar] [CrossRef] [Green Version]
- Simó, R.; Carrasco, E.; García-Ramírez, M.; Hernández, C. Angiogenic and antiangiogenic factors in proliferative diabetic retinopathy. Curr. Diabetes Rev. 2006, 2, 71–98. [Google Scholar] [CrossRef]
- Vessey, K.; Wilkinson-Berka, J.; Fletcher, E. Characterization of retinal function and glial cell response in a mouse model of oxygen-induced retinopathy. J. Comp. Neurol. 2011, 519, 506–527. [Google Scholar] [CrossRef]
- Obrosova, I.G. Increased Sorbitol Pathway Activity Generates Oxidative Stress in Tissue Sites for Diabetic Complications. Antioxid. Redox Signal. 2005, 7, 1543–1552. [Google Scholar] [CrossRef] [PubMed]
- Meneses, P.; Hajjar, K.; Berns, K.; Duvoisin, R. Recombinant angiostatin prevents retinal neovascularization in a murine proliferative retinopathy model. Gene Ther. 2001, 8, 646. [Google Scholar] [CrossRef] [PubMed]
- Kermorvant-Duchemin, E.; Sapieha, P.; Sirinyan, M.; Beauchamp, M.; Checchin, D.; Hardy, P.; Sennlaub, F.; Lachapelle, P.; Chemtob, S. Understanding ischemic retinopathies: Emerging concepts from oxygen-induced retinopathy. Doc. Ophthalmol. 2010, 120, 51–60. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Tsao, R.; Zhang, S.; Dong, Z.; Yang, R.; Gong, J.; Pei, Y. Antioxidant activity, mutagenicity/anti-mutagenicity, and clastogenicity/anti-clastogenicity of lutein from marigold flowers. Food Chem. Toxicol. 2006, 44, 1522–1529. [Google Scholar] [CrossRef] [PubMed]
- Maoka, T.; Tokuda, H.; Suzuki, N.; Kato, H.; Etoh, H. Anti-Oxidative, Anti-Tumor-Promoting, and Anti-Carcinogensis Activities of Nitroastaxanthin and Nitrolutein, the Reaction Products of Astaxanthin and Lutein with Peroxynitrite. Mar. Drugs 2012, 10, 1391–1399. [Google Scholar] [CrossRef] [PubMed]
- Li, S.-Y.; Fu, Z.-J.; Ma, H.; Jang, W.-C.; So, K.-F.; Wong, D.; Lo, A.C.Y. Effect of Lutein on Retinal Neurons and Oxidative Stress in a Model of Acute Retinal Ischemia/Reperfusion. Investig. Opthalmol. Vis. Sci. 2009, 50, 836–843. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, S.-Y.; Yang, D.; Fu, Z.J.; Woo, T.; Wong, D.; Lo, A.C.Y. Lutein enhances survival and reduces neuronal damage in a mouse model of ischemic stroke. Neurobiol. Dis. 2012, 45, 624–632. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, M.; Ozawa, Y.; Kurihara, T.; Noda, K.; Imamura, Y.; Kobayashi, S.; Ishida, S.; Tsubota, K. Neuroprotective Effect of an Antioxidant, Lutein, during Retinal Inflammation. Investig. Opthalmol. Vis. Sci. 2009, 50, 1433–1439. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alves-Rodrigues, A.; Shao, A. The science behind lutein. Toxicol. Lett. 2004, 150, 57–83. [Google Scholar] [CrossRef] [PubMed]
- Snodderly, D.M. Evidence for protection against age-related macular degeneration by carotenoids and antioxidant vitamins. Am. J. Clin. Nutr. 1995, 62, 1448S–1461S. [Google Scholar] [CrossRef]
- Bone, R.A.; Landrum, J.T.; Friedes, L.M.; Gomez, C.M.; Kilburn, M.D.; Menendez, E.; Vidal, I.; Wang, W. Distribution of Lutein and Zeaxanthin Stereoisomers in the Human Retina. Exp. Eye Res. 1997, 64, 211–218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sommerburg, O.; Keunen, J.E.E.; Bird, A.C.; Van Kuijk, F.J.G.M. Fruits and vegetables that are sources for lutein and zeaxanthin: The macular pigment in human eyes. Br. J. Ophthalmol. 1998, 82, 907–910. [Google Scholar] [CrossRef] [PubMed]
- Fung, F.K.C.; Wong, D.; Chan, H.H.L.; Lo, A.C.Y.; Li, S.-Y.; Fu, Z.J. Anti-Inflammatory Effects of Lutein in Retinal Ischemic/Hypoxic Injury: In Vivo and In Vitro Studies. Investig. Opthalmol. Vis. Sci. 2012, 53, 5976–5984. [Google Scholar] [Green Version]
- Li, S.-Y.; Lo, A.C.Y. Lutein Protects RGC-5 Cells Against Hypoxia and Oxidative Stress. Int. J. Mol. Sci. 2010, 11, 2109–2117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Woo, T.T.; Li, S.-Y.; Lai, W.W.; Wong, D.; Lo, A.C. Neuroprotective effects of lutein in a rat model of retinal detachment. Graefe’s Archive for Clin. Exp. Ophthalmol. 2013, 251, 41–51. [Google Scholar] [CrossRef]
- Zhang, C.; Wang, Z.; Zhao, J.; Li, Q.; Huang, C.; Zhu, L.; Lu, D. Neuroprotective effect of lutein on NMDA-induced retinal ganglion cell injury in rat retina. Cell. Mol. Neurobiol. 2016, 36, 531–540. [Google Scholar] [CrossRef]
- Fu, Z.; Meng, S.S.; Burnim, S.B.; Smith, L.E.; Lo, A.C. Lutein facilitates physiological revascularization in a mouse model of retinopathy of prematurity. Clin. Exp. Ophthalmol. 2017, 45, 529–538. [Google Scholar] [CrossRef]
- Shi, X.; Dalal, N.; Jain, A. Antioxidant behaviour of caffeine: Efficient scavenging of hydroxyl radicals. Food Chem. Toxicol. 1991, 29, 1–6. [Google Scholar] [CrossRef]
- Barcelos, R.P.; Souza, M.A.; Amaral, G.P.; Stefanello, S.T.; Bresciani, G.; Fighera, M.R.; Soares, F.A.A.; Barbosa, N.V. Caffeine supplementation modulates oxidative stress markers in the liver of trained rats. Life Sci. 2014, 96, 40–45. [Google Scholar] [CrossRef] [Green Version]
- Devasagayam, T.; Kamat, J.; Mohan, H.; Kesavan, P. Caffeine as an antioxidant: Inhibition of lipid peroxidation induced by reactive oxygen species. Biochim. Biophys. Acta (BBA) Biomembr. 1996, 1282, 63–70. [Google Scholar] [CrossRef]
- Chavez-Valdez, R.; Wills-Karp, M.; Ahlawat, R.; Cristofalo, E.A.; Nathan, A.; Gauda, E.B. Caffeine Modulates TNF-α Production by Cord Blood Monocytes: The Role of Adenosine Receptors. Pediatr. Res. 2009, 65, 203–208. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Li, G.; Hu, J.-L.; Fu, X.-H.; Zeng, Y.-J.; Zhou, Y.-G.; Xiong, G.; Yang, N.; Dai, S.-S.; He, F.-T. Chronic or high dose acute caffeine treatment protects mice against oleic acid-induced acute lung injury via an adenosine A2A receptor-independent mechanism. Eur. J. Pharmacol. 2011, 654, 295–303. [Google Scholar] [CrossRef] [PubMed]
- Dall’Lgna, O.P.; Porciúncula, L.O.; Souza, D.O.; Cunha, R.A.; Lara, D.R. Neuroprotection by caffeine and adenosine A2A receptor blockade of β-amyloid neurotoxicity. Br. J. Pharmacol. 2003, 138, 1207–1209. [Google Scholar] [CrossRef] [PubMed]
- Endesfelder, S.; Zaak, I.; Weichelt, U.; Bührer, C.; Schmitz, T. Caffeine protects neuronal cells against injury caused by hyperoxia in the immature brain. Free. Radic. Boil. Med. 2014, 67, 221–234. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Zhou, R.; Li, B.; Li, H.; Wang, Y.; Gu, X.; Tang, L.; Wang, C.; Zhong, D.; Ge, Y.; et al. Caffeine preferentially protects against oxygen-induced retinopathy. FASEB J. 2017, 31, 3334–3348. [Google Scholar] [CrossRef] [PubMed]
- Barberger-Gateau, P.; Letenneur, L.; Deschamps, V.; Pérès, K.; Dartigues, J.-F.; Renaud, S. Fish, meat, and risk of dementia: Cohort study. BMJ 2002, 325, 932–933. [Google Scholar] [CrossRef]
- Morris, M.C.; Evans, D.A.; Bienias, J.L.; Tangney, C.C.; Bennett, D.A.; Wilson, R.S.; Aggarwal, N.; Schneider, J. Consumption of Fish and n-3 Fatty Acids and Risk of Incident Alzheimer Disease. Arch. Neurol. 2003, 60, 940–946. [Google Scholar] [CrossRef] [PubMed]
- Calon, F.; Cole, G. Neuroprotective action of omega-3 polyunsaturated fatty acids against neurodegenerative diseases: Evidence from animal studies. Prostaglandins Leukot. Essent. Fat. Acids 2007, 77, 287–293. [Google Scholar] [CrossRef] [PubMed]
- Calon, F.; Lim, G.P.; Yang, F.; Morihara, T.; Teter, B.; Ubeda, O.; Rostaing, P.; Triller, A.; Salem, N.; Ashe, K.H.; et al. Docosahexaenoic Acid Protects from Dendritic Pathology in an Alzheimer’s Disease Mouse Model. Neuron 2004, 43, 633–645. [Google Scholar] [CrossRef]
- Shimazawa, M.; Nakajima, Y.; Mashima, Y.; Hara, H. Docosahexaenoic acid (DHA) has neuroprotective effects against oxidative stress in retinal ganglion cells. Brain Res. 2009, 1251, 269–275. [Google Scholar] [CrossRef]
- Sapieha, P.; Stahl, A.; Chen, J.; Seaward, M.R.; Willett, K.L.; Krah, N.M.; Dennison, R.J.; Connor, K.M.; Aderman, C.M.; Liclican, E.; et al. 5-Lipoxygenase Metabolite 4-HDHA Is a Mediator of the Antiangiogenic Effect of ω-3 Polyunsaturated Fatty Acids. Sci. Transl. Med. 2011, 3, 69ra12. [Google Scholar] [CrossRef] [PubMed]
- Stahl, A.; Sapieha, P.; Connor, K.M.; SanGiovanni, J.P.; Chen, J.; Aderman, C.M.; Willett, K.L.; Krah, N.M.; Dennison, R.J.; Seaward, M.R.; et al. PPARγ mediates a direct anti-angiogenic effect of ω3-PUFAs in proliferative retinopathy. Circ. Res. 2010, 107, 495–500. [Google Scholar] [CrossRef] [PubMed]
- Beharry, K.D.; Cai, C.L.; Siddiqui, F.; Chowdhury, S.; D’Agrosa, C.; Valencia, G.B.; Aranda, J.V. Comparative Effects of Coenzyme Q10 or n-3 Polyunsaturated Fatty Acid Supplementation on Retinal Angiogenesis in a Rat Model of Oxygen-Induced Retinopathy. Antioxidants 2018, 7, 160. [Google Scholar] [CrossRef] [PubMed]
- De La Lastra, C.A.; Villegas, I. Resveratrol as an antioxidant and pro-oxidant agent: Mechanisms and clinical implications. Biochem. Soc. Trans. 2007, 35, 1156–1160. [Google Scholar] [CrossRef] [PubMed]
- Zini, R.; Morin, C.; Bertelli, A.A.; Tillement, J.P. Effects of resveratrol on the rat brain respiratory chain. Drugs Under Exp. Clin. Res. 1999, 25, 87–97. [Google Scholar]
- Sinha, K.; Chaudhary, G.; Gupta, Y.K. Protective effect of resveratrol against oxidative stress in middle cerebral artery occlusion model of stroke in rats. Life Sci. 2002, 71, 655–665. [Google Scholar] [CrossRef]
- Martín, A.R.; Villegas, I.; La Casa, C.; de la Lastra, C.A. Resveratrol, a polyphenol found in grapes, suppresses oxidative damage and stimulates apoptosis during early colonic inflammation in rats. Biochem. Pharmacol. 2004, 67, 1399–1410. [Google Scholar] [PubMed]
- Martín, A.R.; Villegas, I.; Sánchez-Hidalgo, M.; De La Lastra, C.A. The effects of resveratrol, a phytoalexin derived from red wines, on chronic inflammation induced in an experimentally induced colitis model. Br. J. Pharmacol. 2006, 147, 873–885. [Google Scholar] [CrossRef] [Green Version]
- Jin, F.; Wu, Q.; Lu, Y.-F.; Gong, Q.-H.; Shi, J.-S. Neuroprotective effect of resveratrol on 6-OHDA-induced Parkinson’s disease in rats. Eur. J. Pharmacol. 2008, 600, 78–82. [Google Scholar] [CrossRef]
- Ates, O.; Cayli, S.; Altinoz, E.; Gurses, I.; Yucel, N.; Sener, M.; Kocak, A.; Yologlu, S. Neuroprotection by resveratrol against traumatic brain injury in rats. Mol. Cell. Biochem. 2007, 294, 137–144. [Google Scholar] [CrossRef]
- Lopez, M.S.; Dempsey, R.J.; Vemuganti, R. Resveratrol neuroprotection in stroke and traumatic CNS injury. Neurochem. Int. 2015, 89, 75–82. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, F.; Gong, Q.; Dong, H.; Shi, J. Resveratrol, a neuroprotective supplement for Alzheimer’s disease. Curr. Pharm. Des. 2012, 18, 27–33. [Google Scholar] [CrossRef] [PubMed]
- Bastianetto, S.; Ménard, C.; Quirion, R. Neuroprotective action of resveratrol. Biochim. Biophys. Acta (BBA) Mol. Basis Dis. 2015, 1852, 1195–1201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lançon, A.; Frazzi, R.; Latruffe, N. Anti-Oxidant, Anti-Inflammatory and Anti-Angiogenic Properties of Resveratrol in Ocular Diseases. Molecules 2016, 21, 304. [Google Scholar] [CrossRef] [PubMed]
- Kim, W.T.; Suh, E.S. Retinal Protective Effects of Resveratrol via Modulation of Nitric Oxide Synthase on Oxygen-induced Retinopathy. Korean J. Ophthalmol. 2010, 24, 108–118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, W.; Jiang, D. Effect of resveratrol on Bcl-2 and VEGF expression in oxygen-induced retinopathy of prematurity. J. Pediatr. Ophthalmol. Strabismus 2012, 49, 230–235. [Google Scholar] [CrossRef] [PubMed]
- Akama, K.T.; Albanese, C.; Pestell, R.G.; Van Eldik, L.J. Amyloid β-peptide stimulates nitric oxide production in astrocytes through an NFκB-dependent mechanism. Proc. Natl. Acad. Sci. USA 1998, 95, 5795–5800. [Google Scholar] [CrossRef]
- Sano, M.; Ernesto, C.; Thomas, R.G.; Klauber, M.R.; Schafer, K.; Grundman, M.; Woodbury, P.; Growdon, J.; Cotman, C.W.; Pfeiffer, E. A controlled trial of selegiline, alpha-tocopherol, or both as treatment for Alzheimer’s disease. N. Engl. J. Med. 1997, 336, 1216–1222. [Google Scholar] [CrossRef]
- Penn, J.S.; Tolman, B.L.; Bullard, L.E. Effect of a Water-Soluble Vitamin E Analog, Trolox C, on Retinal Vascular Development in an Animal Model of Retinopathy of Prematurity. Free. Radic. Boil. Med. 1997, 22, 977–984. [Google Scholar] [CrossRef]
- Penn, J.S.; Thum, L.A.; Naash, M.I. Oxygen-induced retinopathy in the rat. Vitamins C and E as potential therapies. Investig. Ophthalmol. Vis. Sci. 1992, 33, 1836–1845. [Google Scholar]
- Johnson, L.; Quinn, G.E.; Abbasi, S.; Otis, C.; Goldstein, D.; Sacks, L.; Porat, R.; Fong, E.; Delivoria-Papadopoulos, M.; Peckham, G.; et al. Effect of sustained pharmacologic vitamin E levels on incidence and severity of retinopathy of prematurity: A controlled clinical trial. J. Pediatr. 1989, 114, 827–838. [Google Scholar] [CrossRef]
- Raju, T.N.; Langenberg, P.; Bhutani, V.; Quinn, G.E. Vitamin E prophylaxis to reduce retinopathy of prematurity: A reappraisal of published trials. J. Pediatr. 1997, 131, 844–850. [Google Scholar] [CrossRef]
- Finer, N.N.; Schindler, R.F.; Grant, G.; Hill, G.B.; Peters, K. Effect of intramuscular vitamin E on frequency and severity of retrolental fibroplasia. A controlled trial. Lancet 1982, 1, 1087–1091. [Google Scholar] [CrossRef]
- Phelps, D.L.; Rosenbaum, A.L.; Isenberg, S.J.; Leake, R.D.; Dorey, F.J. Tocopherol efficacy and safety for preventing retinopathy of prematurity: A randomized, controlled, double-masked trial. Pediatrics 1987, 79, 489–500. [Google Scholar] [PubMed]
- Obrosova, I.G.; Pacher, P.; Szabó, C.; Zsengeller, Z.; Hirooka, H.; Stevens, M.J.; Yorek, M.A. Aldose Reductase Inhibition Counteracts Oxidative-Nitrosative Stress and Poly(ADP-Ribose) Polymerase Activation in Tissue Sites for Diabetes Complications. Diabetes 2005, 54, 234–242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chung, S.S.; Ho, E.C.; Lam, K.S. Contribution of Polyol Pathway to Diabetes-Induced Oxidative Stress. J. Am. Soc. Nephrol. 2003, 14, 233–236. [Google Scholar] [CrossRef]
- Lee, A.Y.W.; Chung, S.S.M. Contributions of polyol pathway to oxidative stress in diabetic cataract. FASEB J. 1999, 13, 23–30. [Google Scholar] [CrossRef] [Green Version]
- Lou, M.F.; Dickerson, J.E.; Garadi, R.; York, B.M. Glutathione depletion in the lens of galactosemic and diabetic rats. Exp. Eye Res. 1988, 46, 517–530. [Google Scholar] [CrossRef]
- Song, Z.; Fu, D.T.; Chan, Y.-S.; Leung, S.; Chung, S.S.; Chung, S.K. Transgenic mice overexpressing aldose reductase in Schwann cells show more severe nerve conduction velocity deficit and oxidative stress under hyperglycemic stress. Mol. Cell. Neurosci. 2003, 23, 638–647. [Google Scholar] [CrossRef]
- Cheung, A.K.; Lo, A.C.; So, K.F.; Chung, S.S.; Chung, S.K.; Lo, A.C.Y. Gene deletion and pharmacological inhibition of aldose reductase protect against retinal ischemic injury. Exp. Eye Res. 2007, 85, 608–616. [Google Scholar] [CrossRef]
- Obrosova, I.G.; Minchenko, A.G.; Vasupuram, R.; White, L.; Abatan, O.I.; Kumagai, A.K.; Frank, R.N.; Stevens, M.J.; Minchenko, O. Aldose Reductase Inhibitor Fidarestat Prevents Retinal Oxidative Stress and Vascular Endothelial Growth Factor Overexpression in Streptozotocin-Diabetic Rats. Diabetes 2003, 52, 864–871. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheung, A.K.; Fung, M.K.; Lo, A.C.; Lam, T.T.; So, K.F.; Chung, S.S.; Chung, S.K. Aldose reductase deficiency prevents diabetes-induced blood-retinal barrier breakdown, apoptosis, and glial reactivation in the retina of db/db mice. Diabetes 2005, 54, 3119–3125. [Google Scholar] [CrossRef] [PubMed]
- Thomas, T.; Rauscher, F.; Sanders, R.; Veltman, J.; Watkins, J.B. Effects of Aldose Reductase Inhibitors on Antioxidant Defense in Rat and Rabbit Liver. Toxicol. Sci. 2000, 53, 145–149. [Google Scholar] [CrossRef] [PubMed]
- Fu, Z.J.; Li, S.-Y.; Kociok, N.; Wong, D.; Chung, S.K.; Lo, A.C.Y. Aldose Reductase Deficiency Reduced Vascular Changes in Neonatal Mouse Retina in Oxygen-Induced Retinopathy. Investig. Opthalmol. Vis. Sci. 2012, 53, 5698–5712. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fu, Z.; Nian, S.; Li, S.-Y.; Wong, D.; Chung, S.K.; Lo, A.C.Y. Deficiency of aldose reductase attenuates inner retinal neuronal changes in a mouse model of retinopathy of prematurity. Graefe’s Arch. Clin. Exp. Ophthalmol. 2015, 253, 1503–1513. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, K.; Rajapakse, N.; Horiguchi, T.; Payne, R.; Busija, D.W. Neuroprotection against hypoxia-ischemia in neonatal rat brain by novel superoxide dismutase mimetics. Neurosci. Lett. 2003, 346, 41–44. [Google Scholar] [CrossRef]
- Spierer, A.; Rabinowitz, R.; Pri-Chen, S.; Rosner, M. An increase in superoxide dismutase ameliorates oxygen-induced retinopathy in transgenic mice. Eye 2005, 19, 86. [Google Scholar] [CrossRef]
- Huang, H.F.; Guo, F.; Cao, Y.Z.; Shi, W.; Xia, Q. Neuroprotection by Manganese Superoxide Dismutase (M n SOD) Mimics: Antioxidant Effect and Oxidative Stress Regulation in Acute Experimental Stroke. CNS Neurosci. Ther. 2012, 18, 811–818. [Google Scholar] [CrossRef]
- Niesman, M.R.; Johnson, K.A.; Penn, J.S. Therapeutic Effect of Liposomal Superoxide Dismutase in an Animal Model of Retinopathy of Prematurity. Neurochem. Res. 1997, 22, 597–605. [Google Scholar] [CrossRef]
- Dohare, P.; Hyzinski-García, M.C.; Vipani, A.; Bowens, N.H.; Nalwalk, J.W.; Feustel, P.J.; Keller, R.W., Jr.; Jourd’Heuil, D.; Mongin, A.A. The neuroprotective properties of the superoxide dismutase mimetic tempol correlate with its ability to reduce pathological glutamate release in a rodent model of stroke. Free. Radic. Boil. Med. 2014, 77, 168–182. [Google Scholar] [CrossRef] [Green Version]
- Paraskevas, K.I.; Tzovaras, A.S.; Briana, D.D.; Mikhailidis, D.P. Emerging indications for statins: A pluripotent family of agents with several potential applications. Curr. Pharm. Des. 2007, 13, 3622–3636. [Google Scholar] [CrossRef] [PubMed]
- Bonetti, P.; Lerman, L.; Napoli, C. Statin effects beyond lipid lowering—Are they clinically relevant? Eur. Hear. J. 2003, 24, 225–248. [Google Scholar] [CrossRef]
- Wood, W.G.; Eckert, G.P.; Igbavboa, U.; Müller, W.E. Statins and neuroprotection: a prescription to move the field forward. Ann. N. Y. Acad. Sci. 2010, 1199, 69–76. [Google Scholar] [CrossRef] [PubMed]
- Sigurdsson, G.; Haraldsdottir, S.O.; Melberg, T.H.; Tikkanen, M.J.; Miettinen, T.E.; Kristianson, K.J. Simvastatin compared to fluvastatin in the reduction of serum lipids and apolipoproteins in patients with ischaemic heart disease and moderate hypercholesterolaemia. Acta Cardiol. 1998, 53, 7–14. [Google Scholar] [PubMed]
- Johnson-Anuna, L.N.; Eckert, G.P.; Keller, J.H.; Igbavboa, U.; Franke, C.; Fechner, T.; Schubert-Zsilavecz, M.; Karas, M.; Müller, W.E.; Wood, W.G. Chronic administration of statins alters multiple gene expression patterns in mouse cerebral cortex. J. Pharmacol. Exp. Ther. 2005, 312, 786–793. [Google Scholar] [CrossRef]
- Franke, C.; Nöldner, M.; Abdel-Kader, R.; Johnson-Anuna, L.N.; Wood, W.G.; Müller, W.E.; Eckert, G.P. Bcl-2 upregulation and neuroprotection in guinea pig brain following chronic simvastatin treatment. Neurobiol. Dis. 2007, 25, 438–445. [Google Scholar] [CrossRef] [PubMed]
- Bartoli, M.; Al-Shabrawey, M.; Labazi, M.; Behzadian, M.A.; Istanboli, M.; El-Remessy, A.B.; Caldwell, R.W.; Marcus, D.M.; Caldwell, R.B. HMG-CoA reductase inhibitors (statin) prevents retinal neovascularization in a model of oxygen-induced retinopathy. Investig. Ophthalmol. Vis. Sci. 2009, 50, 4934–4940. [Google Scholar] [CrossRef]
- Lee, M.-Y.; Kuan, Y.-H.; Chen, H.-Y.; Chen, T.-Y.; Chen, S.-T.; Huang, C.-C.; Yang, I.-P.; Hsu, Y.-S.; Wu, T.-S.; Lee, E.-J. Intravenous administration of melatonin reduces the intracerebral cellular inflammatory response following transient focal cerebral ischemia in rats. J. Pineal Res. 2007, 42, 297–309. [Google Scholar] [CrossRef]
- Chen, T.-Y.; Lee, M.-Y.; Chen, H.-Y.; Kuo, Y.-L.; Lin, S.-C.; Wu, T.-S.; Lee, E.-J. Melatonin attenuates the postischemic increase in blood-brain barrier permeability and decreases hemorrhagic transformation of tissue-plasminogen activator therapy following ischemic stroke in mice. J. Pineal Res. 2006, 40, 242–250. [Google Scholar] [CrossRef]
- Kondoh, T.; Uneyama, H.; Nishino, H.; Torii, K. Melatonin reduces cerebral edema formation caused by transient forebrain ischemia in rats. Life Sci. 2002, 72, 583–590. [Google Scholar] [CrossRef]
- Watson, N.; Diamandis, T.; Gonzales-Portillo, C.; Reyes, S.; Borlongan, C.V. Melatonin as an Antioxidant for Stroke Neuroprotection. Cell Transplant. 2016, 25, 883–891. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alghamdi, B.S. The neuroprotective role of melatonin in neurological disorders. J. Neurosci. Res. 2018, 96, 1136–1149. [Google Scholar] [CrossRef] [PubMed]
- Alonso-Alconada, D.; Álvarez, A.; Arteaga, O.; Martínez-Ibargüen, A.; Hilario, E. Neuroprotective Effect of Melatonin: A Novel Therapy against Perinatal Hypoxia-Ischemia. Int. J. Mol. Sci. 2013, 14, 9379–9395. [Google Scholar] [CrossRef] [Green Version]
- Tomás-Zapico, C.; Coto-Montes, A.; Tomás-Zapico, C.; Coto-Montes, A.; Tomás-Zapico, C.; Coto-Montes, A. A proposed mechanism to explain the stimulatory effect of melatonin on antioxidative enzymes. J. Pineal Res. 2005, 39, 99–104. [Google Scholar] [CrossRef]
- Kaur, C.; Sivakumar, V.; Robinson, R.; Foulds, W.S.; Luu, C.D.; Ling, E.A. Neuroprotective effect of melatonin against hypoxia-induced retinal ganglion cell death in neonatal rats. J. Pineal Res. 2013, 54, 190–206. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Lu, X.; Hu, Y.; Yang, B.; Tsui, C.-K.; Yu, S.; Lu, L.; Liang, X. Melatonin attenuated retinal neovascularization and neuroglial dysfunction by inhibition of HIF-1α-VEGF pathway in oxygen-induced retinopathy mice. J. Pineal Res. 2018, 64, e12473. [Google Scholar] [CrossRef] [PubMed]
- Simonyi, A. The neuroprotective effects of apocynin. Front. Biosci. 2012, 4, 2183. [Google Scholar] [CrossRef]
- Jackman, K.; Miller, A.; De Silva, T.; Crack, P.J.; Drummond, G.; Sobey, C. Reduction of cerebral infarct volume by apocynin requires pretreatment and is absent in Nox2-deficient mice. Br. J. Pharmacol. 2009, 156, 680–688. [Google Scholar] [CrossRef] [Green Version]
- Chen, H.; Song, Y.S.; Chan, P.H. Inhibition of NADPH oxidase is neuroprotective after ischemia-reperfusion. Br. J. Pharmacol. 2009, 29, 1262–1272. [Google Scholar] [CrossRef]
- Kelly, K.A.; Li, X.; Tan, Z.; Vangilder, R.L.; Rosen, C.L.; Huber, J.D. NOX2 inhibition with apocynin worsens stroke outcome in aged rats. Brain Res. 2009, 1292, 165–172. [Google Scholar] [CrossRef] [Green Version]
- Impellizzeri, D.; Mazzon, E.; Esposito, E.; Paterniti, I.; Bramanti, P.; Cuzzocrea, S. Effect of Apocynin, an inhibitor of NADPH oxidase, in the inflammatory process induced by an experimental model of spinal cord injury. Free Radic. Res. 2011, 45, 221–236. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Pallares, J.; Parga, J.A.; Muñoz, A.; Rey, P.; Guerra, M.J.; Labandeira-Garcia, J.L. Mechanism of 6-hydroxydopamine neurotoxicity: The role of NADPH oxidase and microglial activation in 6-hydroxydopamine-induced degeneration of dopaminergic neurons. J. Neurochem. 2007, 103, 145–156. [Google Scholar] [CrossRef] [PubMed]
- Rey, P.; Lopez-Real, A.; Sánchez-Iglesias, S.; Muñoz, A.; Soto-Otero, R.; Labandeira-Garcia, J. Angiotensin type-1-receptor antagonists reduce 6-hydroxydopamine toxicity for dopaminergic neurons. Neurobiol. Aging 2007, 28, 555–567. [Google Scholar] [CrossRef] [PubMed]
- Al-Shabrawey, M.; Bartoli, M.; El-Remessy, A.B.; Platt, D.H.; Matragoon, S.; Behzadian, M.A.; Caldwell, R.W.; Caldwell, R.B. Inhibition of NAD(P)H Oxidase Activity Blocks Vascular Endothelial Growth Factor Overexpression and Neovascularization during Ischemic Retinopathy. Am. J. Pathol. 2005, 167, 599–607. [Google Scholar] [CrossRef] [Green Version]
- Saito, Y.; Uppal, A.; Byfield, G.; Budd, S.; Hartnett, M.E. Activated NAD(P)H Oxidase from Supplemental Oxygen Induces Neovascularization Independent of VEGF in Retinopathy of Prematurity Model. Investig. Opthalmol. Vis. Sci. 2008, 49, 1591–1598. [Google Scholar] [CrossRef] [PubMed]
- Johnston, P.G.; Gillam-Krakauer, M.; Fuller, M.P.; Reese, J. Evidence-Based Use of Indomethacin and Ibuprofen in the Neonatal Intensive Care Unit. Clin. Perinatol. 2012, 39, 111–136. [Google Scholar] [CrossRef] [Green Version]
- Parikh, P.; Juul, S.E. Neuroprotective Strategies in Neonatal Brain Injury. J. Pediatr. 2018, 192, 22–32. [Google Scholar] [CrossRef]
- Tutak, E.; Satar, M.; Zorludemir, S.; Erdogan, S.; Yapıcıoğlu, H.; Narlı, N. Neuroprotective Effects of Indomethacin and Aminoguanidine in the Newborn Rats with Hypoxic-Ischemic Cerebral Injury. Neurochem. Res. 2005, 30, 937–942. [Google Scholar] [CrossRef]
- Lambat, Z.; Conrad, N.; Anoopkumar-Dukie, S.; Walker, R.B.; Daya, S. An Investigation into the Neuroprotective Properties of Ibuprofen. Metab. Brain Dis. 2000, 15, 249–256. [Google Scholar] [CrossRef]
- Iwata, Y.; Nicole, O.; Zurakowski, D.; Okamura, T.; Jonas, R.A. Ibuprofen for neuroprotection after cerebral ischemia. J. Thorac. Cardiovasc. Surg. 2010, 139, 489–493. [Google Scholar] [CrossRef] [Green Version]
- Świątkiewicz, M.; Zaremba, M.; Joniec, I.; Członkowski, A.; Kurkowska-Jastrzębska, I. Potential neuroprotective effect of ibuprofen, insights from the mice model of Parkinson’s disease. Pharmacol. Rep. 2013, 65, 1227–1236. [Google Scholar] [CrossRef]
- Nandgaonkar, B.N.; Rotschild, T.; Yu, K.; Higgins, R.D. Indomethacin Improves Oxygen-Induced Retinopathy in the Mouse. Pediatr. Res. 1999, 46, 184–188. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharma, J.; Barr, S.M.; Geng, Y.; Yun, Y.; Higgins, R.D. Ibuprofen improves oxygen-induced retinopathy in a mouse model. Curr. Eye Res. 2003, 27, 309–314. [Google Scholar] [CrossRef] [PubMed]
- Solaroglu, I. Neuroprotective Effect of Granulocyte-Colony Stimulating Factor. Front. Biosci. 2007, 12, 712. [Google Scholar] [CrossRef]
- Schäbitz, W.-R.; Kollmar, R.; Schwaninger, M.; Juettler, E.; Bardutzky, J.; Schölzke, M.N.; Sommer, C.; Schwab, S. Neuroprotective Effect of Granulocyte Colony–Stimulating Factor After Focal Cerebral Ischemia. Stroke 2003, 34, 745–751. [Google Scholar]
- Komine-Kobayashi, M.; Zhang, N.; Liu, M.; Tanaka, R.; Hara, H.; Osaka, A.; Mochizuki, H.; Mizuno, Y.; Urabe, T. Neuroprotective effect of recombinant human granulocyte colony-stimulating factor in transient focal ischemia of mice. J. Cereb. Blood Flow Metab. 2006, 26, 402–413. [Google Scholar] [CrossRef] [PubMed]
- Meuer, K.; Pitzer, C.; Teismann, P.; Krüger, C.; Göricke, B.; Laage, R.; Lingor, P.; Peters, K.; Schlachetzki, J.C.; Kobayashi, K. Granulocyte-colony stimulating factor is neuroprotective in a model of Parkinson’s disease. J. Neurochem. 2006, 97, 675–686. [Google Scholar] [CrossRef] [PubMed]
- Yanqing, Z.; Yu-Min, L.; Jian, Q.; Bao-Guo, X.; Chuan-Zhen, L. Fibronectin and neuroprotective effect of granulocyte colony-stimulating factor in focal cerebral ischemia. Brain Res. 2006, 1098, 161–169. [Google Scholar] [CrossRef]
- Hartung, T. Anti-inflammatory effects of granulocyte colony-stimulating factor. Curr. Opin. Hematol. 1998, 5, 221–225. [Google Scholar] [CrossRef]
- Kojima, H.; Otani, A.; Oishi, A.; Makiyama, Y.; Nakagawa, S.; Yoshimura, N. Granulocyte colony-stimulating factor attenuates oxidative stress–induced apoptosis in vascular endothelial cells and exhibits functional and morphologic protective effect in oxygen-induced retinopathy. Blood 2011, 117, 1091–1100. [Google Scholar] [CrossRef]
- Wilkinson-Berka, J.L.; Tan, G.; Binger, K.J.; Sutton, L.; McMaster, K.; Deliyanti, D.; Perera, G.; Campbell, D.J.; Miller, A.G.; Wilkinson-Berka, J.; et al. Aliskiren reduces vascular pathology in diabetic retinopathy and oxygen-induced retinopathy in the transgenic (mRen-2)27 rat. Diabetologia 2011, 54, 2724–2735. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Downie, L.E.; Pianta, M.J.; Vingrys, A.J.; Wilkinson-Berka, J.L.; Fletcher, E.L.; Wilkinson-Berka, J.L.; Wilkinson-Berka, J.L. AT1 receptor inhibition prevents astrocyte degeneration and restores vascular growth in oxygen-induced retinopathy. Glia 2008, 56, 1076–1090. [Google Scholar] [CrossRef] [PubMed]
- Downie, L.E.; Hatzopoulos, K.M.; Pianta, M.J.; Vingrys, A.J.; Wilkinson-Berka, J.L.; Kalloniatis, M.; Fletcher, E.L.; Wilkinson-Berka, J.L.; Wilkinson-Berka, J.L. Angiotensin type-1 receptor inhibition is neuroprotective to amacrine cells in a rat model of retinopathy of prematurity. J. Comp. Neurol. 2010, 518, 41–63. [Google Scholar] [CrossRef] [PubMed]
- Biehl, J.K.; Russell, B. Introduction to stem cell therapy. J. Cardiovasc. Nurs. 2009, 24, 98–103. [Google Scholar] [CrossRef] [PubMed]
- Lai, A.; Fu, Z.; Lo, A. Stem cell therapy for retinopathy of prematurity. Anat. Physiol. 2013. [Google Scholar] [CrossRef]
- Machalińska, A.; Modrzejewska, M.; Kotowski, M.; Dziedziejko, V.; Kucia, M.; Kawa, M.; Safranow, K.; Baśkiewicz-Masiuk, M.; Modrzejewska, A.; Karczewicz, D. Circulating stem cell populations in preterm infants: Implications for the development of retinopathy of prematurity. Arch. Ophthalmol. 2010, 128, 1311–1319. [Google Scholar] [CrossRef] [PubMed]
- Paczkowska, E.; Kucia, M.; Koziarska, D.; Halasa, M.; Safranow, K.; Masiuk, M.; Karbicka, A.; Nowik, M.; Nowacki, P.; Ratajczak, M.Z.; et al. Clinical Evidence That Very Small Embryonic-Like Stem Cells Are Mobilized into Peripheral Blood in Patients After Stroke. Stroke 2009, 40, 1237–1244. [Google Scholar] [CrossRef]
- Ratajczak, M.; Machalinski, B.; Wojakowski, W.; Ratajczak, J.; Kucia, M. A hypothesis for an embryonic origin of pluripotent Oct-4+ stem cells in adult bone marrow and other tissues. Leukemia 2007, 21, 860. [Google Scholar] [CrossRef]
- Ritter, M.R.; Banin, E.; Moreno, S.K.; Aguilar, E.; Dorrell, M.I.; Friedlander, M. Myeloid progenitors differentiate into microglia and promote vascular repair in a model of ischemic retinopathy. J. Clin. Investig. 2006, 116, 3266–3276. [Google Scholar] [CrossRef]
- Medina, R.J.; O’Neill, C.L.; Humphreys, M.W.; Gardiner, T.A.; Stitt, A.W. Outgrowth Endothelial Cells: Characterization and Their Potential for Reversing Ischemic Retinopathy. Investig. Opthalmol. Vis. Sci. 2010, 51, 5906–5913. [Google Scholar] [CrossRef]
- Noueihed, B.; Rivera, J.C.; Chemtob, S. AB028. Mesenchymal stem cells repair retinal vascular damage in retinopathy of prematurity mouse model. Ann. Eye Sci. 2018, 3, AB028. [Google Scholar] [CrossRef]
- Moisseiev, E.; Anderson, J.D.; Oltjen, S.; Goswami, M.; Zawadzki, R.J.; Nolta, J.A.; Park, S.S. Protective Effect of Intravitreal Administration of Exosomes Derived from Mesenchymal Stem Cells on Retinal Ischemia. Curr. Eye Res. 2017, 42, 1358–1367. [Google Scholar] [CrossRef] [PubMed]
- Ma, Q.; Liu, F.; Shi, M.; Sun, C.; Tan, Z.; Chang, X.; Li, Q.; Feng, Z. Bone marrow mesenchymal stem cells modified by angiogenin-1 promotes tissue repair in mice with oxygen-induced retinopathy of prematurity by promoting retinal stem cell proliferation and differentiation. J. Cell. Physiol. 2019, 234, 21027–21038. [Google Scholar] [CrossRef] [PubMed]
- Mendel, T.A.; Clabough, E.B.D.; Kao, D.S.; Demidova-Rice, T.N.; Durham, J.T.; Zotter, B.C.; Seaman, S.A.; Cronk, S.M.; Rakoczy, E.P.; Katz, A.J.; et al. Correction: Pericytes Derived from Adipose-Derived Stem Cells Protect against Retinal Vasculopathy. PLoS ONE 2013, 8, 65691. [Google Scholar] [CrossRef]
- Dogra, M.R.; Katoch, D. An Update on Retinopathy of Prematurity (ROP). Indian J. Pediatr. 2017, 84, 930–936. [Google Scholar] [CrossRef] [PubMed]
- Broxterman, E.C.; Hug, D.A. Retinopathy of Prematurity: A Review of Current Screening Guidelines and Treatment Options. Mo. Med. 2016, 113, 187–190. [Google Scholar] [PubMed]
- Cryotherapy for Retinopathy of Prematurity Cooperative Group. Multicenter trial of cryotherapy for retinopathy of prematurity: Preliminary results. Pediatrics 1988, 81, 697–706. [Google Scholar]
- Elsas, F.; Collins, M.; Jones, J.; Kimble, J.; Kline, L.; Witherspoon, D.; Roth, A.; Demorest, B.; Gilbert, W.; Plotsky, D. Multicenter trial of cryotherapy for retinopathy of prematurity: Ophthalmological outcomes at 10 years. Arch. Ophthalmol. 2001, 119, 1110–1118. [Google Scholar]
- Palmer, E.; Hardy, R.; Dobson, V.; Phelps, D.; Quinn, G.; Summers, C.; Krom, C.; Tung, B. 15-year outcomes following threshold retinopathy of prematurity: Final results from the multicenter trial of cryotherapy for retinopathy of prematurity. Arch. Ophthalmol. 2005, 123, 311–318. [Google Scholar] [PubMed]
- Clark, D.; Mandal, K. Treatment of retinopathy of prematurity. Early Hum. Dev. 2008, 84, 95–99. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Pathogenic Agents in ROP | Phase 1 in ROP Development | Phase 2 in ROP Development | Relevant Vascular Protective Agents in ROP Development (Phase of ROP) | Intervention | Animal Model | Beneficial Effect | Adverse Effect | Reference |
|---|---|---|---|---|---|---|---|---|
| VEGF | ↓ | ↑ | VEGF (Phase 1) | Intraocular injection | Rat OIR model |
| / | [16] |
| Bevacizumab (Phase 2) | Intravitreal injection | (Clinical study) |
|
| [17,18,19,20,21,22,23] | |||
| Ranibizumab (Phase 2) | Intravitreal injection | (Clinical study) | / | [19,24,25] | ||||
| Aflibercept (Phase 2) | Intravitreal injection | Mouse OIR model | / | [26] | ||||
| VEGFA shRNA (Phase 2) | Subretinal injection | Rat OIR model |
| / | [27] | |||
| Anti-KDR (Phase 2) | Surgical implantation | Dog OIR model |
| / | [28] | |||
| SRPIN340 (Phase 2) | Intraocular injection | Rat OIR model |
| / | [29] | |||
| Rapamycin (Phase 2) | Subcutaneous injection | Mouse OIR model |
| / | [30] | |||
| IGF-1 | ↓ | ↑ | rhIGF-1 (Phase 1) | Intraperitoneal injection | Mouse OIR model |
| / | [31] |
| IGFBP3 (Phase 1 and 2) | Knockout mouse | Mouse OIR model |
| / | [32,33] | |||
| Jb3 (Phase 2) | Subcutaneous injection | Mouse OIR model |
| / | [34] | |||
| Epo | ↓ | ↑ | / | / | / |
| / | / |
| HIF-1 | ↓ | ↑ | DMOG (Phase 1) | Intraperitoneal injection | Mouse OIR model |
| / | [35] |
| PHD2 (Phase 1 and 2) | Knockout mouse | Mouse OIR model | / | [36] | ||||
| RTP801 (Phase 1 and 2) | Knockout mouse | Mouse OIR model |
| / | [37] | |||
| NO | ↓ | ↑ | l-NA (Phase 2) | Intraperitoneal injection | Rat OIR model |
| / | [38] |
| l-NNA (Phase 2) | Intraperitoneal injection | Mouse OIR model | / | [39] | ||||
| AG (Phase 2) | Intravitreal injection | Mouse OIR model | / | [40] | ||||
| Adenosine | ↓ | ↑ | / | / | / | / | / | / |
| β-AR | ? | ↑ | Propranolol (Phase 2) | Subcutaneous injection | Mouse OIR model |
| / | [41] |
| Topical administration | Mouse OIR model | / | [42] | |||||
| Atenolol (Phase 2) | Subcutaneous injection | Mouse OIR model |
| / | [43] | |||
| ICI 118,551 (Phase 2) | / | |||||||
| SR59230A (Phase 2) | / | |||||||
| Other angiogenic agents | ? | ↑ | Dexamethasone (Phase 2) | Subcutaneous injection | Mouse OIR model |
|
| [44] |
| Anecortave acetate (Phase 2) | Intravitreal injection | Rat OIR model | [45] | |||||
| Degulin (Phase 2) | Intravitreal injection | Mouse OIR model |
| / | [46] | |||
| YC-1 (Phase 2) | Intravitreal injection | Mouse OIR model | / | [47] | ||||
| β-lapachone (Phase 2) | Intravitreal injection | Mouse OIR model | / | [48] | ||||
| 16K HPRL (Phase 2) | Intravitreal injection | Mouse OIR model |
| / | [49] | |||
| 12-LOX (Phase 2) | Intraperitoneal injection | Mouse OIR model | / | [50] | ||||
| TMP (Phase 2) | Intraperitoneal injection | Mouse OIR model |
| / | [51] | |||
| K5 (Phase 2) | Intravitreal injection | Rat OIR model | / | [52] | ||||
| MEF2C (Phase 1 and 2) | Knockout mouse | Mouse OIR model | / | [53] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tsang, J.K.W.; Liu, J.; Lo, A.C.Y. Vascular and Neuronal Protection in the Developing Retina: Potential Therapeutic Targets for Retinopathy of Prematurity. Int. J. Mol. Sci. 2019, 20, 4321. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20174321
Tsang JKW, Liu J, Lo ACY. Vascular and Neuronal Protection in the Developing Retina: Potential Therapeutic Targets for Retinopathy of Prematurity. International Journal of Molecular Sciences. 2019; 20(17):4321. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20174321
Chicago/Turabian StyleTsang, Jessica K. W., Jin Liu, and Amy C. Y. Lo. 2019. "Vascular and Neuronal Protection in the Developing Retina: Potential Therapeutic Targets for Retinopathy of Prematurity" International Journal of Molecular Sciences 20, no. 17: 4321. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20174321