The Role of Protein Misfolding and Tau Oligomers (TauOs) in Alzheimer′s Disease (AD)

Abstract
:
1. Introduction
1.1. Protein Misfolding
1.2. Pathological Changes in Alzheimer’s Disease
2. Materials and Methods
Literature Search and Data Extraction
3. Tau Protein
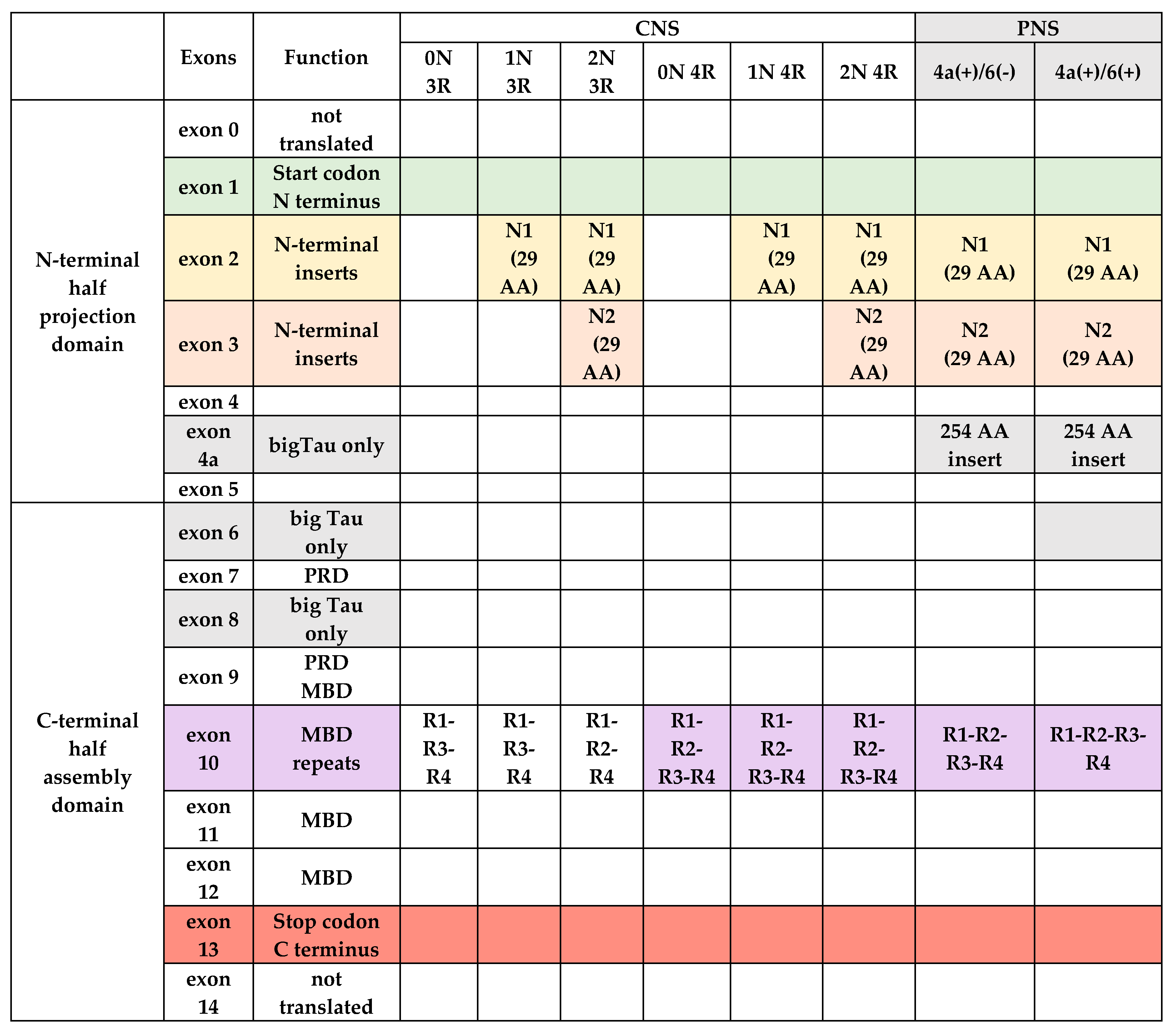
3.1. Tau Protein Gene and Its Transcription Products
3.2. Post-Translational Modifications of Tau
3.3. Tau Protein Structure
3.4. Biological Functions of Tau
3.5. Aggregation and Deposition of Misfolded Tau Protein
4. Toxic Activity of Tau and TauOs in AD
4.1. Tau Protein Toxicity Related to Its Post-Translational Modifications and Missorting
4.2. Relationship between Toxic AβOs and TauOs
4.3. Influence of Tau on Synaptic Transmission
4.4. Toxic Tau Aggregates
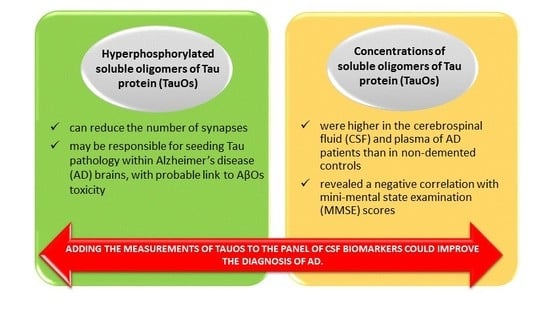
5. Diagnostic Possibilities of TauOs Determination
6. Conclusions
Funding
Conflicts of Interest
Abbreviations
| AA | amino acids |
| NFTs | neurofibrillary tangles |
| NDs | neurodegenerative diseases |
| BSE | bovine spongiform encephalopathy |
| CJD | Creutzfeldt-Jakob disease |
| AD | Alzheimer’s disease |
| HD | Huntington’s disease |
| PD | Parkinson’s disease |
| Aβ | amyloid beta |
| AβOs | oligomers of amyloid β peptide |
| LTP | long-term potentiation |
| LTD | enhanced long-term synaptic depression |
| LMW | low molecular weight |
| HMW | high molecular weight |
| MT | Microtubule |
| MAP | microtubule-associated proteins |
| PRD | proline-rich domain |
| MBD | microtubule binding domain |
| PNS | peripheral nervous system |
| PSP | progressive supranuclear palsy |
| CBD | cortico-basal degeneration |
| CNS | central nervous system |
| PHFs | paired helical filaments |
| hTau | hyperphosphorylated Tau |
| MARK | microtubule affinity regulating kinase |
| Cdk5 | cycline-dependent kinase 5 |
| GSK3b | glycogen synthase kinase 3b |
| NMDAR | N-methyl-d-aspartate receptor |
| AMPAR | α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor |
| IDP | intrinsically disordered protein |
| NGF | nerve growth factor |
| ROS | reactive oxygen species |
| IGF-1 | insulin-like growth factor-1 |
| IRS-1 | Insulin receptor substrate 1 |
| PTEN | phosphatase and tensin homologue on chromosome 10 |
| TauOs | oligomers of Tau protein |
| SF | straight filaments |
| PSD-95 | post synaptic density 95 protein |
| MMSE | Mini Mental State Examination test |
| MRI | magnetic resonance imaging |
| PET | positron emission tomography |
| CSF | cerebrospinal fluid |
| NFL | neurofilament light protein |
| scFvs | single chain antibody fragments |
| MMPs | matrix metalloproteinases |
| MCI | mild cognitive impairment |
| TTR | transthyretin |
| ATTRwt | wild-type amyloidogenic transthyretin |
| ATTRv | variant amyloidogenic transthyretin |
| TTR-FAP | transthyretin-related familial amyloid polyneuropathy |
References
- Alberts, B.; Johnson, A.; Lewis, J.; Raff, M.; Roberts, K.; Walter, P. Protein Function. In Molecular Biology of the Cell, 4th ed.; Garland Science: New York, NY, USA, 2002. [Google Scholar]
- Anfinsen, D.C.B. The Formation and Stabilization of Protein Structure. Biochem. J. 1972, 128, 737–749. [Google Scholar] [CrossRef] [PubMed]
- Taylor, J.P.; Hardy, J.; Fischbeck, K.H. Toxic proteins in neurodegenerative disease. Science 2002, 296, 1991–1995. [Google Scholar] [CrossRef] [PubMed]
- Moreno-Gonzalez, I.; Soto, C. Misfolded protein aggregates: Mechanisms, structures and potential for disease transmission. Semin. Cell Dev. Biol. 2011, 22, 482–487. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ingram, V.M. Gene mutations in human haemoglobin: The chemical difference between normal and sickle cell haemoglobin. Nature 1957, 180, 326–328. [Google Scholar] [CrossRef] [PubMed]
- Gibson, J.S.; Ellory, J.C. Membrane transport in sickle cell disease. Blood Cells Mol. Dis. 2002, 28, 303–314. [Google Scholar] [CrossRef]
- Chamcheu, J.C.; Siddiqui, I.A.; Syed, D.N.; Adhami, V.M.; Liovic, M.; Mukhtar, H. Keratin gene mutations in disorders of human skin and its appendages. Arch. Biochem. Biophys. 2011, 508, 123–137. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koike, H.; Katsuno, M. Ultrastructure in Transthyretin Amyloidosis: From Pathophysiology to Therapeutic Insights. Biomedicines 2019, 7, 11. [Google Scholar] [CrossRef]
- Kumar, V.; Sami, N.; Kashav, T.; Islam, A.; Ahmad, F.; Hassan, M.I. Protein aggregation and neurodegenerative diseases: From theory to therapy. Eur. J. Med. Chem. 2016, 124, 1105–1120. [Google Scholar] [CrossRef]
- Dovidchenko, N.V.; Leonova, E.I.; Galzitskaya, O.V. Mechanisms of amyloid fibril formation. Biochemistry 2014, 79, 1515–1527. [Google Scholar] [CrossRef]
- Toyama, B.H.; Weissman, J.S. Amyloid structure: Conformational diversity and consequences. Ann. Rev. Biochem. 2011, 80, 557–585. [Google Scholar] [CrossRef]
- Prusiner, S.B. Biology and genetics of prions causing neurodegeneration. Annu. Rev. Genet. 2013, 47, 601–623. [Google Scholar] [CrossRef] [PubMed]
- Baumann, F.; Tolnay, M.; Brabeck, C.; Pahnke, J.; Kloz, U.; Niemann, H.H.; Heikenwalder, M.; Rülicke, T.; Bürkle, A.; Aguzzi, A. Lethal recessive myelin toxicity of prion protein lacking its central domain. Embo J. 2007, 26, 538547. [Google Scholar] [CrossRef] [PubMed]
- Soto, C.; Estrada, L.; Castilla, J. Amyloids, prions and the inherent infectious nature of misfolded protein aggregates. Trends Biochem. Sci. 2007, 31, 150–155. [Google Scholar] [CrossRef] [PubMed]
- Dobson, C.M. The structural basis of protein folding and its links with human disease. Philos. Trans. R Soc. Lond. B Biol. Sci. 2001, 356, 133–145. [Google Scholar] [CrossRef] [PubMed]
- Knowles, T.P.; Vendruscolo, M.; Dobson, C.M. The amyloid state and its association with protein misfolding diseases. Nat. Rev. Mol. Cell Biol. 2014, 15, 384–396. [Google Scholar] [CrossRef]
- MacLea, K.S. What makes a prion: Infectious proteins from animals to yeast. Int. Rev. Cell Mol. Biol. 2017, 329, 227–276. [Google Scholar] [CrossRef] [PubMed]
- Alzheimer, A. Uber eine eigenartige Erkrankung der Hirnrinde. Allgemeine Zeitschrift fur Psychiatrie und psychisch-gerichtliche Medizin 1907, 64, 146–148. [Google Scholar]
- Terry, R.; Hansen, L.; Masliah, E. Structural basis of the cognitive alterations in Alzheimer disease. In Alzheimer Disease; Terry, R., Katzman, R., Eds.; Raven: New York, NY, USA, 1994; pp. 179–196. [Google Scholar]
- Mroczko, B.; Groblewska, M.; Litman-Zawadzka, A.; Kornhuber, J.; Lewczuk, P. Amyloid β oligomers (AβOs) in Alzheimer’s disease. J. Neural Transm. 2018, 125, 177–191. [Google Scholar] [CrossRef] [PubMed]
- Lansbury, P.T. Evolution of amyloid: What normal protein folding may tell us about fibrillogenesis and disease. Proc. Natl. Acad. Sci. USA 1999, 96, 3342–3344. [Google Scholar] [CrossRef] [Green Version]
- Kayed, R.; Lasagna-Reeves, C.A. Molecular mechanisms of amyloid oligomers toxicity. J. Alzheimers Dis. 2013, 33, S67–S78. [Google Scholar] [CrossRef]
- Scheff, S.W.; Price, D.A.; Schmitt, F.A.; DeKosky, S.T.; Mufson, E.J. Synaptic alterations in CA1 in mild Alzheimer disease and mild cognitive impairment. Neurology 2007, 68, 1501–1508. [Google Scholar] [CrossRef] [PubMed]
- Shankar, G.M.; Li, S.; Mehta, T.H.; Garcia-Munoz, A.; Shepardson, N.E.; Smith, I.; Brett, F.M.; Farrell, M.A.; Rowan, M.J.; Lemere, C.A.; et al. Amyloid-beta protein dimers isolated directly from Alzheimer’s brains impair synaptic plasticity and memory. Nat. Med. 2008, 14, 837–842. [Google Scholar] [CrossRef] [PubMed]
- Condello, C.; Stöehr, J. Aβ propagation and strains: Implications for the phenotypic diversity in Alzheimer’s disease. Neurobiol. Dis. 2018, 109, 191–200. [Google Scholar] [CrossRef] [PubMed]
- Mroczko, B.; Groblewska, M.; Litman-Zawadzka, A.; Kornhuber, J.; Lewczuk, P. Cellular Receptors of Amyloid β Oligomers (AβOs) in Alzheimer’s Disease. Int. J. Mol. Sci. 2018, 19, 1884. [Google Scholar] [CrossRef]
- Chételat, G. Alzheimer disease: Aβ-independent processes-rethinking preclinical AD. Nat. Rev. Neurol. 2013, 9, 123–124. [Google Scholar] [CrossRef] [PubMed]
- Chételat, G. Reply: The amyloid cascade is not the only pathway to AD. Nat. Rev. Neurol. 2013, 9, 356. [Google Scholar] [CrossRef] [PubMed]
- Vishnu, V.Y. Can tauopathy shake the amyloid cascade hypothesis? Nat. Rev. Neurol. 2013, 9, 356. [Google Scholar] [CrossRef]
- Mudher, A.; Lovestone, S. Alzheimer’s disease-do tauists and baptists finally shake hands? Trends Neurosci. 2002, 25, 22–26. [Google Scholar] [CrossRef]
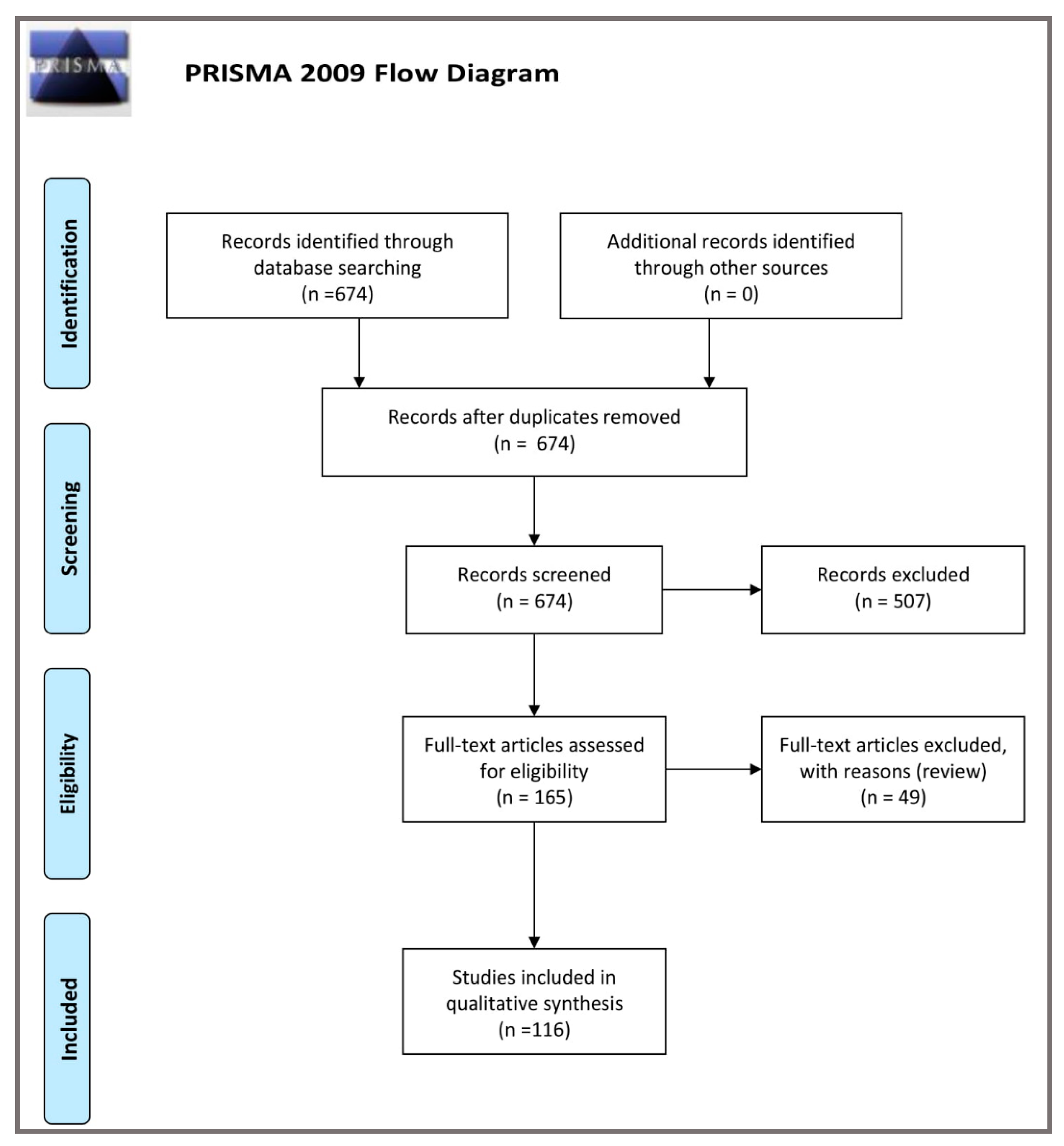
- Moher, D.; Liberati, A.; Tetzlaff, J.; Altman, D.G.; The PRISMA Group. Preferred Reporting Items for Systematic Reviews and Meta-Analyses: The PRISMA Statement. PLoS Med 2009, 6, e1000097. [Google Scholar] [CrossRef]
- Trojanowski, J.Q.; Schuck, T.; Schmidt, M.L.; Lee, V.M. Distribution of tau proteins in the normal human central and peripheral nervous system. J. Histochem. Cytochem. 1989, 37, 209–215. [Google Scholar] [CrossRef]
- Avila, J.; de Barreda, E.G.; Pallas-Bazarra, N.; Hernandez, F. Tau and neuron aging. Aging Dis. 2013, 4, 23–28. [Google Scholar] [PubMed]
- Guo, T.; Noble, W.; Hanger, D.P. Roles of tau protein in health and disease. Acta. Neuropathol. 2017, 133, 665–704. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- León-Espinosa, G.; García, E.; García-Escudero, V.; Hernández, F.; Defelipe, J.; Avila, J. Changes in Tau phosphorylation in hibernating rodents. J. Neurosci. Res. 2013, 91, 954–962. [Google Scholar] [CrossRef] [PubMed]
- Hernández, F.; Avila, J. Tauopathies. Cell Mol. Life Sci. 2007, 64, 2219–2233. [Google Scholar] [CrossRef] [PubMed]
- Goedert, M.; Spillantini, M.G.; Crowther, R.A. Cloning of a big tau microtubule-associated protein characteristic of the peripheral nervous system. Proc. Natl. Acad. Sci. USA 1992, 89, 1983–1987. [Google Scholar] [CrossRef] [PubMed]
- Boutajangout, A.; Authelet, M.; Blanchard, V.; Touchet, N.; Tremp, G.; Pradier, L.; Brion, J.P. Characterisation of cytoskeletal abnormalities in mice transgenic for wild-type human tau and familial Alzheimer’s disease mutants of APP and presenilin-1. Neurobiol. Dis. 2004, 15, 47–60. [Google Scholar] [CrossRef] [PubMed]
- Martin, L.; Latypova, X.; Terro, F. Post-translational modifications of tau protein: Implications for Alzheimer’s disease. Neurochem. Int. 2011, 58, 458–471. [Google Scholar] [CrossRef] [PubMed]
- Caffrey, T.M.; Joachim, C.; Wade-Martins, R. Haplotype-specific expression of the N-terminal exons 2 and 3 at the human MAPT locus. Neurobiol. Aging 2008, 29, 1923–1929. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arendt, T.; Stieler, J.T.; Holzer, M. Tau and tauopathies. Brain Res. Bull. 2016, 126, 238–292. [Google Scholar] [CrossRef]
- Jones, E.L.; Margallo-Lana, M.; Prasher, V.P.; Ballard, C.G. The extended tau haplotype and the age of onset of dementia in Down syndrome. Dement. Geriatr. Cogn. Disord. 2008, 26, 199–202. [Google Scholar] [CrossRef]
- Wang, J.Z.; Liu, F. Microtubule-associated protein tau in development, degeneration and protection of neurons. Prog. Neurobiol. 2008, 85, 148–175. [Google Scholar] [CrossRef] [PubMed]
- Goedert, M.; Jakes, R. Expression of separate isoforms of human tau protein: Correlation with the tau pattern in brain and effects on tubulin polymerization. Embo J. 1990, 9, 4225–4230. [Google Scholar] [CrossRef] [PubMed]
- Spires-Jones, T.L.; Stoothoff, W.H.; de Calignon, A.; Jones, P.B.; Hyman, B.T. Tau pathophysiology in neurodegeneration: A tangled issue. Trends Neurosci. 2009, 32, 150–159. [Google Scholar] [CrossRef] [PubMed]
- Stoothoff, W.; Jones, P.B.; Spires-Jones, T.L.; Joyner, D.; Chhabra, E.; Bercury, K.; Fan, Z.; Xie, H.; Bacskai, B.; Edd, J.; et al. Differential effect of three-repeat and four-repeat tau on mitochondrial axonal transport. J. Neurochem. 2009, 111, 417–427. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goedert, M.; Spillantini, M.G.; Jakes, R.; Rutherford, D.; Crowther, R.A. Multiple isoforms of human microtubule-associated protein tau: Sequences and localization in neurofibrillary tangles of Alzheimer’s disease. Neuron 1989, 3, 519–526. [Google Scholar] [CrossRef]
- Knowles, R.; LeClerc, N.; Kosik, K.S. Organization of actin and microtubules during process formation in tau-expressing Sf9 cells. Cell Motil. Cytoskeleton. 1994, 28, 256–264. [Google Scholar] [CrossRef] [PubMed]
- McMillan, P.; Korvatska, E.; Poorkaj, P.; Evstafjeva, Z.; Robinson, L.; Greenup, L.; Leverenz, J.; Schellenberg, G.D.; D’Souza, I. Tau isoform regulation is region- and cell-specific in mouse brain. J. Comp. Neurol. 2008, 511, 788–803. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schweers, O.; Mandelkow, E.M.; Biernat, J.; Mandelkow, E. Oxidation of cysteine-322 in the repeat domain of microtubule-associated protein tau controls the in vitro assembly of paired helical filaments. Proc. Natl. Acad. Sci. USA 1995, 92, 8463–8467. [Google Scholar] [CrossRef]
- Lee, G.; Neve, R.L.; Kosik, K.S. The microtubule binding domain of tau protein. Neuron 1989, 2, 1615–1624. [Google Scholar] [CrossRef]
- Butner, K.A.; Kirschner, M.W. Tau protein binds to microtubules through a flexible array of distributed weak sites. J. Cell Biol. 1991, 115, 717–730. [Google Scholar] [CrossRef]
- Goode, B.L.; Denis, P.E.; Panda, D.; Radeke, M.J.; Miller, H.P.; Wilson, L.; Feinstein, S.C. Functional interactions between the proline-rich and repeat regions of tau enhance microtubule binding and assembly. Mol. Biol. Cell 1997, 8, 353–365. [Google Scholar] [CrossRef] [PubMed]
- Mukrasch, M.D.; Biernat, J.; von Bergen, M.; Griesinger, C.; Mandelkow, E.; Zweckstetter, M. Sites of tau important for aggregation populate beta-structure and bind to microtubules and polyanions. J. Biol. Chem. 2005, 280, 24978–24986. [Google Scholar] [CrossRef] [PubMed]
- Xia, D.; Li, C.; Gotz, J. Pseudophosphorylation of Tau at distinct epitopes or the presence of the P301L mutation targets the microtubule-associated protein Tau to dendritic spines. Biochim. Biophys. Acta 2015, 1852, 913–924. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cardona-Gomez, G.P.; Arango-Davila, C.; Gallego-Gomez, J.C.; Barrera-Ocampo, A.; Pimienta, H.; Garcia-Segura, L.M. Estrogen dissociates Tau and alpha-amino-3-hydroxy-5-methylisoxazole-4-propionic acid receptor subunit in postischemic hippocampus. Neuroreport 2006, 17, 1337–1341. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miyamoto, T.; Stein, L.; Thomas, R.; Djukic, B.; Taneja, P.; Knox, J.; Vossel, K.; Mucke, L. Phosphorylation of tau at Y18, but not tau-fyn binding, is required for tau to modulate NMDA receptor-dependent excitotoxicity in primary neuronal culture. Mol. Neurodegener. 2017, 12, 41. [Google Scholar] [CrossRef] [PubMed]
- Ebneth, A.; Godemann, R.; Stamer, K.; Illenberger, S.; Trinczek, B.; Mandelkow, E.; Mandelkow, E. Overexpression of tau protein inhibits kinesin-dependent trafficking of vesicles, mitochondria, and endoplasmic reticulum: Implications for Alzheimer’s disease. J. Cell Biol. 1998, 143, 777–794. [Google Scholar] [CrossRef] [PubMed]
- Strittmatter, W.J.; Saunders, A.M.; Goedert, M.; Weisgraber, K.H.; Dong, L.M.; Jakes, R.; Huang, D.Y.; Pericak-Vance, M.; Schmechel, D.; Roses, A.D. Isoform-specific interactions of apolipoprotein E with microtubule-associated protein tau: Implications for Alzheimer disease. Proc. Natl. Acad. Sci. USA 1994, 91, 11183–11186. [Google Scholar] [CrossRef]
- Reynolds, C.H.; Garwood, C.J.; Wray, S.; Price, C.; Kellie, S.; Perera, T.; Zvelebil, M.; Yang, A.; Sheppard, P.W.; Varndell, I.M.; et al. Phosphorylation regulates tau interactions with Src homology 3 domains of phosphatidylinositol 3-kinase, phospholipase Cgamma1, Grb2, and Src family kinases. J. Biol. Chem. 2008, 283, 18177–18186. [Google Scholar] [CrossRef]
- Bhaskar, K.; Yen, S.-H.; Lee, G. Disease-related modifications in tau affect the interaction between Fyn and Tau. J. Biol. Chem. 2005, 280, 35119–35125. [Google Scholar] [CrossRef]
- Lee, G. Tau and src family tyrosine kinases. Biochim. Biophys. Acta 2005, 1739, 323–330. [Google Scholar] [CrossRef] [Green Version]
- Qi, H.; Cantrelle, F.-X.; Benhelli-Mokrani, H.; Smet-Nocca, C.; Buée, L.; Lippens, G.; Bonnefoy, E.; Galas, M.-C.; Landrieu, I. Nuclear magnetic resonance spectroscopy characterization of interaction of tau with DNA and its regulation by phosphorylation. Biochemistry 2015, 54, 1525–1533. [Google Scholar] [CrossRef] [PubMed]
- Morris, M.; Maeda, S.; Vossel, K.; Mucke, L. The many faces of tau. Neuron 2011, 70, 410–426. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Mandelkow, E. Tau in physiology and pathology. Nat. Rev. Neurosci. 2015, 17, 22–35. [Google Scholar] [CrossRef]
- Vega, I.E. Increase in tau tyrosine phosphorylation correlates with the formation of tau aggregates. Brain Res. Mol. Brain Res. 2005, 138, 135–144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schneider, A.; Biernat, J.; von Bergen, M.; Mandelkow, E.; Mandelkow, E.M. Phosphorylation that detaches tau protein from microtubules (Ser262, Ser214) also protects it against aggregation into Alzheimer paired helical filaments. Biochemistry 1999, 38, 3549–3558. [Google Scholar] [CrossRef] [PubMed]
- Planel, E. Alterations in glucose metabolism induce hypothermia leading to tau hyperphosphorylation through differential inhibition of kinase and phosphatase activities: Implications for Alzheimer’s disease. J. Neurosci. 2004, 24, 2401–2411. [Google Scholar] [CrossRef]
- Yanagisawa, M.; Planel, E.; Ishiguro, K.; Fujita, S.C. Starvation induces tau hyperphosphorylation in mouse brain: Implications for Alzheimer’s disease. FEBS Lett. 1999, 461, 329–333. [Google Scholar] [CrossRef]
- Sotiropoulos, I.; Catania, C.; Pinto, L.G.; Silva, R.; Pollerberg, G.E.; Takashima, A.; Sousa, N.; Almeida, O.F.X. Stress acts cumulatively to precipitate Alzheimer’s disease-like tau pathology and cognitive deficits. J. Neurosci. 2011, 31, 7840–7847. [Google Scholar] [CrossRef]
- Le Freche, H.; Brouillette, J.; Fernandez-Gomez, F.-J.; Patin, P.; Caillierez, R.; Zommer, N.; Sergeant, N.; Buée-Scherrer, V.; Lebuffe, G.; Blum, D.; et al. Tau phosphorylation and sevoflurane anesthesia. Anesthesiology 2012, 116, 779–787. [Google Scholar] [CrossRef]
- Whittington, R.A.; Bretteville, A.; Dickler, M.F.; Planel, E. Anesthesia and tau pathology. Prog. Neuropsychopharmacol. Biol. Psychiatry 2013, 47, 147–155. [Google Scholar] [CrossRef] [Green Version]
- Andreeva, A.; Howorth, D.; Chothia, C.; Kulesha, E.; Murzin, A.G. SCOP2 prototype: A new approach to protein structure mining. Nucleic Acids Res. 2014, 42, D310–D314. [Google Scholar] [CrossRef] [PubMed]
- Friedhoff, P.; von Bergen, M.; Mandelkow, E.M.; Mandelkow, E. Structure of tau protein and assembly into paired helical filaments. Biochim. Biophys. Acta 2000, 1502, 122–132. [Google Scholar] [CrossRef]
- Avila, J.; Jiménez, J.S.; Sayas, C.L.; Bolós, M.; Zabala, J.C.; Rivas, G.; Hernández, F. Tau Structures. Front. Aging Neurosci. 2016, 8, 262. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jeganathan, S.; von Bergen, M.; Brutlach, H.; Steinhoff, H.-J.; Mandelkow, E. Global hairpin folding of tau in solution. Biochemistry 2006, 45, 2283–2293. [Google Scholar] [CrossRef] [PubMed]
- Al-Bassam, J.; Ozer, R.S.; Safer, D.; Halpain, S.; Milligan, R.A. MAP2 and tau bind longitudinally along the outer ridges of microtubule protofilaments. J. Cell Biol. 2002, 157, 1187–1196. [Google Scholar] [CrossRef]
- Goux, W.J.; Kopplin, L.; Nguyen, A.D.; Leak, K.; Rutkofsky, M.; Shanmuganandam, V.D.; Sharma, D.; Inouye, H.; Kirschner, D.A. The formation of straight and twisted filaments from short tau peptides. J. Biol. Chem. 2004, 279, 26868–26875. [Google Scholar] [CrossRef] [PubMed]
- Zabik, N.L.; Imhof, M.M.; Martic-Milne, S. Structural evaluations of tau protein conformation: Methodologies and approaches. Biochem. Cell Biol. 2017, 95, 338–349. [Google Scholar] [CrossRef]
- Mukrasch, M.D.; Bibow, S.; Korukottu, J.; Jeganathan, S.; Biernat, J.; Griesinger, C.; Mandelkow, E.; Zweckstetter, M. Structural polymorphism of 441-residue tau at single residue resolution. PLoS Biol. 2009, 7, e1000034. [Google Scholar] [CrossRef]
- Andronesi, O.C.; von Bergen, M.; Biernat, J.; Seidel, K.; Griesinger, C.; Mandelkow, E.; Baldus, M. Characterization of Alzheimer’s-like paired helical filaments from the core domain of tau protein using solid-state NMR spectroscopy. J. Am. Chem. Soc. 2008, 130, 5922–5928. [Google Scholar] [CrossRef]
- Wegmann, S.; Medalsy, I.D.; Mandelkow, E.; Muller, D.J. The fuzzy coat of pathological human Tau fibrils is a two-layered polyelectrolyte brush. Proc. Natl. Acad. Sci. USA 2013, 110, E313–E321. [Google Scholar] [CrossRef]
- Mi, K.; Johnson, G.V. The role of tau phosphorylation in the pathogenesis of Alzheimer’s disease. Curr. Alzheimer Res. 2006, 3, 449–463. [Google Scholar] [CrossRef] [PubMed]
- Cuchillo-Ibanez, I.; Seereeram, A.; Byers, H.L.; Leung, K.Y.; Ward, M.A.; Anderton, B.H.; Hanger, D.P. Phosphorylation of tau regulates its axonal transport by controlling its binding to kinesin. FASEB J. 2008, 22, 3186–3195. [Google Scholar] [CrossRef] [PubMed]
- Dixit, R.; Ross, J.L.; Goldman, Y.E.; Holzbaur, E.L. Differential regulation of dynein and kinesin motor proteins by tau. Science 2008, 319, 1086–1089. [Google Scholar] [CrossRef] [PubMed]
- Knops, J.; Kosik, K.S.; Lee, G.; Pardee, J.D.; Cohen-Gould, L.; McConlogue, L. Overexpression of tau in a nonneuronal cell induces long cellular processes. J. Cell Biol. 1991, 114, 725–733. [Google Scholar] [CrossRef] [PubMed]
- Caceres, A.; Kosik, K.S. Inhibition of neurite polarity by tau antisense oligonucleotides in primary cerebellar neurons. Nature 1990, 343, 461–463. [Google Scholar] [CrossRef]
- Lee, G.; Newman, S.T.; Gard, D.L.; Band, H.; Panchamoorthy, G. Tau interacts with src-family non-receptor tyrosine kinases. J. Cell Sci. 1998, 111, 3167–3177. [Google Scholar]
- Westerink, R.H.; Ewing, A.G. The PC12 cell as model for neurosecretion. Acta Physiol. 2008, 192, 273–285. [Google Scholar] [CrossRef]
- Sultan, A.; Nesslany, F.; Violet, M.; Begard, S.; Loyens, A.; Talahari, S. Nuclear tau, a key player in neuronal DNA protection. J. Biol. Chem. 2011, 286, 4566–4575. [Google Scholar] [CrossRef] [PubMed]
- Violet, M.; Delattre, L.; Tardivel, M.; Sultan, A.; Chauderlier, A.; Caillierez, R. A major role for Tau in neuronal DNA and RNA protection in vivo under physiological and hyperthermic conditions. Front. Cell Neurosci. 2014, 8, 84. [Google Scholar] [CrossRef] [Green Version]
- Marciniak, E.; Leboucher, A.; Caron, E.; Ahmed, T.; Tailleux, A.; Dumont, J. Tau deletion promotes brain insulin resistance. J. Exp. Med. 2017, 214, 2257–2269. [Google Scholar] [CrossRef] [PubMed]
- De Felice, F.G.; Lourenco, M.V.; Ferreira, S.T. How does brain insulin resistance develop in Alzheimer’s disease? Alzheimer’s Dement. J. Alzheimer’s Assoc. 2014, 10, S26–S32. [Google Scholar] [CrossRef]
- Tian, H.; Davidowitz, E.; Lopez, P.; Emadi, S.; Moe, J.; Sierks, M. Trimeric tau is toxic to human neuronal cells at low nanomolar concentrations. Int. J. Cell Biol. 2013, 2013, 260787. [Google Scholar] [CrossRef]
- Cowan, C.M.; Bossing, T.; Page, A.; Shepherd, D.; Mudher, A. Soluble hyper-phosphorylated tau causes microtubule breakdown and functionally compromises normal tau in vivo. Acta Neuropathol. 2010, 120, 593–604. [Google Scholar] [CrossRef] [Green Version]
- Zempel, H.; Thies, E.; Mandelkow, E.; Mandelkow, E.M. Abeta oligomers cause localized Ca(2+) elevation, missorting of endogenous Tau into dendrites, Tau phosphorylation, and destruction of microtubules and spines. J. Neurosci. 2010, 30, 11938–11950, [published correction in: J. Neurosci. 2012, 32, 6052]. [Google Scholar] [CrossRef]
- Sahara, N.; Maeda, S.; Murayama, M.; Suzuki, T.; Dohmae, N.; Yen, S.H.; Takashima, A. Assembly of two distinct dimers and higher-order oligomers from full-length tau. Eur. J. Neurosci. 2007, 25, 3020–3029. [Google Scholar] [CrossRef]
- Maeda, S.; Sahara, N.; Saito, Y.; Murayama, M.; Yoshiike, Y.; Kim, H.; Miyasaka, T.; Murayama, S.; Ikai, A.; Takashima, A. Granular tau oligomers as intermediates of tau filaments. Biochemistry 2007, 46, 3856–3861. [Google Scholar] [CrossRef]
- Maeda, S.; Sahara, N.; Saito, Y.; Murayama, S.; Ikai, A.; Takashima, A. Increased levels of granular tau oligomers: An early sign of brain aging and Alzheimer’s disease. Neurosci. Res. 2006, 54, 197–201. [Google Scholar] [CrossRef]
- Cowan, C.M.; Mudher, A. Are tau aggregates toxic or protective in tauopathies? Front. Neurol. 2013, 4, 114. [Google Scholar] [CrossRef]
- Trojanowski, J.Q.; Schmidt, M.L.; Shin, R.W.; Bramblett, G.T.; Rao, D.; Lee, V.M.Y. Altered tau and neurofilament proteins in neurodegenerative diseases: Diagnostic implications for Alzheimer’s disease and Lewy body dementias. Brain Pathol. 1993, 3, 45–54. [Google Scholar] [CrossRef]
- Mirbaha, H.; Holmes, B.B.; Sanders, D.W.; Bieschke, J.; Diamond, M.I. Tau Trimers Are the Minimal Propagation Unit Spontaneously Internalized to Seed Intracellular Aggregation. J. Biol. Chem. 2015, 290, 14893–14903. [Google Scholar] [CrossRef] [Green Version]
- Patterson, K.R.; Remmers, C.; Fu, Y.; Brooker, S.; Kanaan, N.M.; Vana, L.; Ward, S.; Reyes, J.F.; Philibert, K.; Glucksman, M.J.; et al. Characterization of prefibrillar Tau oligomers in vitro and in Alzheimer disease. J. Biol. Chem. 2011, 286, 23063–23076. [Google Scholar] [CrossRef]
- Makrides, V.; Shen, T.E.; Bhatia, R.; Smith, B.L.; Thimm, J.; Lal, R.; Feinstein, S.C. Microtubule-dependent oligomerization of tau. Implications for physiological tau function and tauopathies. J. Biol. Chem. 2003, 278, 33298–33304. [Google Scholar] [CrossRef]
- Lasagna-Reeves, C.A.; Castillo-Carranza, D.L.; Guerrero-Muoz, M.J.; Jackson, G.R.; Kayed, R. Preparation and characterization of neurotoxic tau oligomers. Biochemistry 2010, 49, 10039–10041. [Google Scholar] [CrossRef]
- Henkins, K.M.; Sokolow, S.; Miller, C.A.; Vinters, H.V.; Poon, W.W.; Cornwell, L.B.; Saing, T.; Gylys, K.H. Extensive p-tau pathology and SDS-stable p-tau oligomers in Alzheimer’s cortical synapses. Brain Pathol. 2012, 22, 826–833. [Google Scholar] [CrossRef]
- Soeda, Y.; Yoshikawa, M.; Almeida, O.F.; Sumioka, A.; Maeda, S.; Osada, H.; Kondoh, Y.; Saito, A.; Miyasaka, T.; Kimura, T.; et al. Toxic tau oligomer formation blocked by capping of cysteine residues with 1,2-dihydroxybenzene groups. Nat. Commun. 2015, 6, 10216. [Google Scholar] [CrossRef] [Green Version]
- Kanemaru, K.; Takio, K.; Miura, R.; Titani, K.; Ihara, Y. Fetal-type phosphorylation of the tau in paired helical filaments. J. Neurochem. 1992, 58, 1667–1675. [Google Scholar] [CrossRef]
- Kopke, E.; Tung, Y.C.; Shaikh, S.; Alonso, A.C.; Iqbal, K.; Grundke-Iqbal, I. Microtubule-associated protein tau. Abnormal phosphorylation of a non-paired helical filament pool in Alzheimer disease. J. Biol. Chem. 1993, 268, 24374–24384. [Google Scholar]
- Smith, A.D. Imaging the progression of Alzheimer pathology through the brain. Proc. Natl. Acad. Sci. USA 2002, 99, 4135–4137. [Google Scholar] [CrossRef] [Green Version]
- Zempel, H.; Luedtke, J.; Kumar, Y.; Biernat, J.; Dawson, H.; Mandelkow, E.; Mandelkow, E.M. Amyloid-β oligomers induce synaptic damage via Tau-dependent microtubule severing by TTLL6 and spastin. Embo J. 2013, 32, 2920–2937. [Google Scholar] [CrossRef]
- Gulisano, W.; Maugeri, D.; Baltrons, M.A.; Fà, M.; Amato, A.; Palmeri, A.; D’Adamio, L.; Grassi, C.; Devanand, D.P.; Honig, L.S.; et al. Role of Amyloid-β and Tau Proteins in Alzheimer’s Disease: Confuting the Amyloid Cascade. J. Alzheimers Dis. 2018, 64, S611–S631. [Google Scholar] [CrossRef]
- Evans, D.B.; Rank, K.B.; Bhattacharya, K.; Thomsen, D.R.; Gurney, M.E.; Sharma, S.K. Tau phosphorylation at serine 396 and serine 404 by human recombinant tau protein kinase II inhibits tau’s ability to promote microtubule assembly. J. Biol. Chem. 2000, 275, 24977–24983. [Google Scholar] [CrossRef]
- Mondragon-Rodriguez, S.; Perry, G.; Luna-Munoz, J.; Acevedo-Aquino, M.C.; Williams, S. Phosphorylation of tau protein at sites Ser(396-404) is one of the earliest events in Alzheimer’s disease and Down syndrome. Neuropathol. Appl. Neurobiol. 2014, 40, 121–135. [Google Scholar] [CrossRef]
- Arrasate, M.; Pérez, M.; Valpuesta, J.M.; Avila, J. Role of glycosaminoglycans in determining the helicity of paired helical filaments. Am. J. Pathol. 1997, 151, 1115–1122. [Google Scholar] [CrossRef]
- Huvent, I.; Kamah, A.; Cantrelle, F.X.; Barois, N.; Slomianny, C.; Smet-Nocca, C.; Landrieu, I.; Lippens, G. A functional fragment of Tau forms fibers without the need for an intermolecular cysteine bridge. Biochem. Biophys. Res. Commun. 2014, 445, 299–303. [Google Scholar] [CrossRef]
- Kim, A.C.; Lim, S.; Kim, Y.K. Metal Ion Effects on Aβ and Tau Aggregation. Int. J. Mol. Sci. 2018, 19, 128. [Google Scholar] [CrossRef]
- Zhu, H.L.; Meng, S.R.; Fan, J.B.; Chen, J.; Liang, Y. Fibrillization of human tau is accelerated by exposure to lead via interaction with His-330 and His-362. PLoS ONE 2011, 6, e25020. [Google Scholar] [CrossRef]
- Pittman, A.M.; Fung, H.C.; de Silva, R. Untangling the tau gene association with neurodegenerative disorders. Hum. Mol. Genet. 2006, 15, R188–R195. [Google Scholar] [CrossRef]
- Myers, A.J.; Pittman, A.M.; Zhao, A.S.; Rohrer, K.; Kaleem, M.; Marlowe, L.; Lees, A.; Leung, D.; McKeith, I.G.; Perry, R.H.; et al. The MAPT H1c risk haplotype is associated with increased expression of tau and especially of 4 repeat containing transcripts. Neurobiol. Dis. 2007, 25, 561–570. [Google Scholar] [CrossRef]
- Oddo, S.; Caccamo, A.; Green, K.N.; Liang, K.; Tran, L.; Chen, Y.; Leslie, F.M.; LaFerla, F.M. Chronic nicotine administration exacerbates tau pathology in a transgenic model of Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2005, 102, 3046–3051. [Google Scholar] [CrossRef]
- Ribes, D.; Colomina, M.T.; Vicens, P.; Domingo, J.L. Effects of oral aluminum exposure on behavior and neurogenesis in a transgenic mouse model of Alzheimer’s disease. Exp. Neurol. 2008, 214, 293–300. [Google Scholar] [CrossRef]
- Walton, J.R. Evidence for participation of aluminum in neurofibrillary tangle formation and growth in Alzheimer’s disease. J. Alzheimers Dis. 2010, 22, 65–72. [Google Scholar] [CrossRef]
- Song, M.S.; Rauw, G.; Baker, G.B.; Kar, S. Memantine protects rat cortical cultured neurons against beta-amyloid-induced toxicity by attenuating tau phosphorylation. Eur. J. Neurosci. 2008, 28, 1989–2002. [Google Scholar] [CrossRef]
- Aktas, O.; Ullrich, O.; Infante-Duarte, C.; Nitsch, R.; Zipp, F. Neuronal damage in brain inflammation. Arch. Neurol. 2007, 64, 185–189. [Google Scholar] [CrossRef]
- Yoshiyama, Y.; Higuchi, M.; Zhang, B.; Huang, S.M.; Iwata, N.; Saido, T.C.; Maeda, J.; Suhara, T.; Trojanowski, J.Q.; Lee, V.M. Synaptic loss and microglial activation precede tangles in a P301S tauopathy mouse model. Neuron 2007, 53, 337–351. [Google Scholar] [CrossRef]
- Mateo, I.; Sanchez-Juan, P.; Rodriguez-Rodriguez, E.; Infante, J.; Vázquez-Higuera, J.L.; García-Gorostiaga, I.; Berciano, J.; Combarros, O. Synergistic effect of Heme Oxygenase-1 and Tau genetic variants on Alzheimer’s disease risk. Dement. Geriatr. Cogn. Disord. 2008, 26, 339–342. [Google Scholar] [CrossRef]
- Beach, T.G.; Wilson, J.R.; Sue, L.I.; Newell, A.; Poston, M.; Cisneros, R.; Pandya, Y.; Esh, C.; Connor, D.J.; Sabbagh, M.; et al. Circle of Willis atherosclerosis: Association with Alzheimer’s disease, neurotic plaques and neurofibrillary tangles. Acta Neuropathol. 2007, 113, 13–21. [Google Scholar] [CrossRef]
- Michikawa, M. Role of cholesterol in amyloid cascade: Cholesterol-dependent modulation of tau phosphorylation and mitochondrial function. Acta Neurol. Scand. Suppl. 2006, 185, 21–26. [Google Scholar] [CrossRef]
- Ohm, T.G.; Meske, V. Cholesterol, statins and tau. Acta Neurol. Scand. Suppl. 2006, 185, 93–101. [Google Scholar] [CrossRef]
- Sontag, J.M.; Nunbhakdi-Craig, V.; Montgomery, L.; Arning, E.; Bottiglieri, T.; Sontag, E. Folate deficiency induces in vitro and mouse brain region-specific downregulation of leucine carboxyl methyltransferase-1 and protein phosphatase 2A B(alpha) subunit expression that correlate with enhanced tau phosphorylation. J. Neurosci. 2008, 28, 11477–11487. [Google Scholar] [CrossRef]
- Julien, C.; Tremblay, C.; Phivilay, A.; Berthiaume, L.; Emond, V.; Julien, P.; Calon, F. High-fat diet aggravates amyloid-beta and tau pathologiges in the 3xTg-AD mouse model. Neurobiol. Aging 2010, 31, 1516–1531. [Google Scholar] [CrossRef]
- Rapp, M.A.; Schnaider-Beeri, M.; Grossman, H.T.; Sano, M.; Perl, D.P.; Purohit, D.P.; Gorman, J.M.; Haroutunian, V. Increased hippocampal plaques and tangles in patients with Alzheimer disease with a lifetime history of major depression. Arch. Gen. Psychiatry 2006, 63, 161–167. [Google Scholar] [CrossRef] [PubMed]
- Rapp, M.A.; Schnaider-Beeri, M.; Purohit, D.P.; Perl, D.P.; Haroutunian, V.; Sano, M. Increased neurofibrillary tangles in patients with Alzheimer disease with comorbid depression. Am. J. Geriatr. Psychiatry 2008, 16, 168–174. [Google Scholar] [CrossRef] [PubMed]
- Green, K.N.; Billing, L.M.; Roozendaal, B.; McGaugh, J.L.; LaFerla, F.M. Glucocorticoids increase amyloid-beta and tau pathology in a mouse model of Alzheimer’s disease. J. Neurosci. 2006, 26, 9047–9056. [Google Scholar] [CrossRef] [PubMed]
- Braak, H.; Braak, E. Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol. 1991, 82, 239–259. [Google Scholar] [CrossRef] [PubMed]
- Serrano-Pozo, A.; Frosch, M.P.; Masliah, E.; Hyman, B.T. Neuropathological alterations in Alzheimer disease. Cold Spring Harb. Perspect. Med. 2011, 1, a006189. [Google Scholar] [CrossRef] [PubMed]
- Braak, H.; Braak, E.; Grundke-Iqbal, I.; Iqbal, K. Occurence of neuropil threads in the senile human brains in Alzheimer’s disease: A third location of paired helicial filaments outside of neurofibrillary tangles and neuritic plaques. Neurosci. Lett. 1986, 65, 351–355. [Google Scholar] [CrossRef]
- Perry, G.; Kawai, M.; Tabaton, M.; Onorato, M.; Mulvihill, P.; Richey, P.; Morandi, A.; Connolly, J.A.; Gambetti, P. Neuropil threads of Alzheimer’s disease show a marked alteration of the normal cytoskeleton. J. Neurosci. 1991, 11, 1748–1755. [Google Scholar] [CrossRef]
- Wittmann, C.W.; Wszolek, M.F.; Shulman, J.M.; Salvaterra, P.M.; Lewis, J.; Hutton, M.; Feany, M.B. Tauopathy in Drosophila: Neurodegeneration without neurofibrillary tangles. Science 2001, 293, 711–714. [Google Scholar] [CrossRef] [PubMed]
- Mershin, A.; Pavlopoulos, E.; Fitch, O.; Braden, B.C.; Nanopoulos, D.V.; Skoulakis, E.M. Learning and memory deficits upon TAU accumulation in Drosophila mushroom body neurons. Learn. Mem. 2004, 11, 277–287. [Google Scholar] [CrossRef] [PubMed]
- Ishihara, T.; Zhang, B.; Higuchi, M.; Yoshiyama, Y.; Trojanowski, J.Q.; Lee, V.M. Age-dependent induction of congophilic neurofibrillary tau inclusions in tau transgenic mice. Am. J. Pathol. 2001, 158, 555–562. [Google Scholar] [CrossRef]
- Frandemiche, M.L.; De Seranno, S.; Rush, T.; Borel, E.; Elie, A.; Arnal, I.; Lanté, F.; Buisson, A. Activity-dependent tau protein translocation to excitatory synapse is disrupted by exposure to amyloid-beta oligomers. J. Neurosci. 2014, 34, 6084–6097. [Google Scholar] [CrossRef] [PubMed]
- Ittner, L.M.; Ke, Y.D.; Delerue, F.; Bi, M.; Gladbach, A.; van Eersel, J.; Wölfing, H.; Chieng, B.C.; Christie, M.J.; Napier, I.A.; et al. Dendritic function of tau mediates amyloid-β toxicity in Alzheimer’s disease mouse models. Cell 2010, 142, 387–397. [Google Scholar] [CrossRef] [PubMed]
- Santacruz, K.; Lewis, J.; Spires, T.; Paulson, J.; Kotilinek, L.; Ingelsson, M.; Guimaraes, A.; DeTure, M.; Ramsden, M.; McGowan, E.; et al. Tau suppression in a neurodegenerative mouse model improves memory function. Science 2005, 309, 476–481. [Google Scholar] [CrossRef] [PubMed]
- Van der Jeugd, A. Cognitive defects are reversible in inducible mice expressing pro-aggregant full-length human Tau. Acta Neuropathol. 2012, 123, 787–805. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lasagna-Reeves, C.A.; Castillo-Carranza, D.L.; Sengupta, U.; Clos, A.L.; Jackson, G.R.; Kayed, R. Tau oligomers impair memory and induce synaptic and mitochondrial dysfunction in wild-type mice. Mol. Neurodegener. 2011, 6, 39. [Google Scholar] [CrossRef] [PubMed]
- Flach, K.; Hilbrich, I.; Schiffmann, A.; Gartner, U.; Kruger, M.; Leonhardt, M.; Waschipky, H.; Wick, L.; Arendt, T.; Holzer, M. Tau oligomers impair artificial membrane integrity and cellular viability. J. Biol. Chem. 2012, 287, 43223–43233. [Google Scholar] [CrossRef]
- Mufson, E.J.; Ward, S.; Binder, L. Prefibrillar tau oligomers in mild cognitive impairment and Alzheimer’s Disease. Neurodegener. Dis. 2013, 13, 151–153. [Google Scholar] [CrossRef]
- Violet, M.; Chauderlier, A.; Delattre, L.; Tardivel, M.; Chouala, M.S.; Sultan, A. Prefibrillar Tau oligomers alter the nucleic acid protective function of Tau in hippocampal neurons in vivo. Neurobiol. Dis. 2015, 82, 540–551. [Google Scholar] [CrossRef]
- Mansuroglu, Z.; Benhelli-Mokrani, H.; Marcato, V.; Sultan, A.; Violet, M.; Chauderlier, A.; Delattre, L.; Loyens, A.; Talahari, S.; Bégard, S.; et al. Loss of Tau protein affects the structure, transcription and repair of neuronal pericentromeric heterochromatin. Sci. Rep. 2016, 6, 33047. [Google Scholar] [CrossRef]
- Chin, J.; Scharfman, H.E. Shared cognitive and behavioral impairments in epilepsy and Alzheimer’s disease and potential underlying mechanisms. Epilepsy. Behav. 2013, 26, 343–351. [Google Scholar] [CrossRef]
- Palop, J.J.; Mucke, L. Epilepsy and cognitive impairments in Alzheimer disease. Arch. Neurol. 2009, 66, 435–440. [Google Scholar] [CrossRef] [PubMed]
- DeVos, S.L.; Goncharoff, D.K.; Chen, G.; Kebodeaux, C.S.; Yamada, K.; Stewart, F.R.; Schuler, D.R.; Maloney, S.E.; Wozniak, D.F.; Rigo, F.; et al. Antisense reduction of tau in adult mice protects against seizures. J. Neurosci. 2013, 33, 12887–12897. [Google Scholar] [CrossRef] [PubMed]
- Gheyara, A.L.; Ponnusamy, R.; Djukic, B.; Craft, R.J.; Ho, K.; Guo, W.; Finucane, M.M.; Sanchez, P.E.; Mucke, L. Tau reduction prevents disease in a mouse model of Dravet syndrome. Ann. Neurol. 2014, 76, 443–456. [Google Scholar] [CrossRef] [PubMed]
- Kimura, T.; Whitcomb, D.J.; Jo, J.; Regan, P.; Piers, T.; Heo, S. Microtubule-associated protein tau is essential for long-term depression in the hippocampus. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2013, 369, 20130144. [Google Scholar] [CrossRef] [PubMed]
- Tai, H.C.; Wang, B.Y.; Serrano-Pozo, A.; Frosch, M.P.; Spires-Jones, T.L.; Hyman, B.T. Frequent and symmetric deposition of misfolded tau oligomers within presynaptic and postsynaptic terminals in Alzheimer’s disease. Acta Neuropathol. Commun. 2014, 2, 146. [Google Scholar] [CrossRef] [PubMed]
- Lasagna-Reeves, C.A.; Castillo-Carranza, D.L.; Sengupta, U.; Guerrero-Munoz, M.J.; Kiritoshi, T.; Neugebauer, V.; Jackson, G.R.; Kayed, R. Alzheimer brain-derived tau oligomers propagate pathology from endogenous tau. Sci. Rep. 2012, 2, 700. [Google Scholar] [CrossRef] [PubMed]
- Morozova, O.A.; March, Z.M.; Robinson, A.S.; Colby, D.W. Conformational features of tau fibrils from Alzheimer’s disease brain are faithfully propagated by unmodified recombinant protein. Biochemistry 2013, 52, 6960–6967. [Google Scholar] [CrossRef]
- Gendreau, K.L.; Hall, G.F. Tangles, Toxicity, and Tau Secretion in AD-New Approaches to a Vexing Problem. Front. Neurol. 2013, 4, 160. [Google Scholar] [CrossRef]
- Dubois, B.; Feldman, H.H.; Jacova, C.; Dekosky, S.T.; Barberger-Gateau, P.; Cummings, J.; Delacourte, A.; Galasko, D.; Gauthier, S.; Jicha, G.; et al. Research criteria for the diagnosis of Alzheimer’s disease: Revising the NINCDS-ADRDA criteria. Lancet. Neurol. 2007, 6, 734–746. [Google Scholar] [CrossRef]
- McKhann, G.M.; Knopman, D.S.; Chertkow, H.; Hyman, B.T.; Jack, C.R., Jr.; Kawas, C.H.; Klunk, W.E.; Koroshetz, W.J.; Manly, J.J.; Mayeux, R.; et al. The diagnosis of dementia due to Alzheimer’s disease: Recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement. 2011, 7, 263–269. [Google Scholar] [CrossRef]
- Albert, M.S.; DeKosky, S.T.; Dickson, D.; Dubois, B.; Feldman, H.H.; Fox, N.C.; Gamst, A.; Holtzman, D.M.; Jagust, W.J.; Petersen, R.C.; et al. The diagnosis of mild cognitive impairment due to Alzheimer’s disease: Recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement. 2011, 7, 270–279. [Google Scholar] [CrossRef] [PubMed]
- Olsson, B.; Lautner, R.; Andreasson, U.; Öhrfelt, A.; Portelius, E.; Bjerke, M.; Hölttä, M.; Rosén, C.; Olsson, C.; Strobel, G.; et al. CSF and blood biomarkers for the diagnosis of Alzheimer’s disease: A systematic review and meta-analysis. Lancet Neurol. 2016, 15, 673–684. [Google Scholar] [CrossRef]
- Tian, H.; Davidowitz, E.; Lopez, P.; He, P.; Schulz, P.; Moe, J.; Sierks, M.R. Isolation and characterization of antibody fragments selective for toxic oligomeric tau. Neurobiol. Aging 2015, 36, 1342–1355. [Google Scholar] [CrossRef] [PubMed]
- Sengupta, U.; Portelius, E.; Hansson, O.; Farmer, K.; Castillo-Carranza, D.; Woltjer, R.; Zetterberg, H.; Galasko, D.; Blennow, K.; Kayed, R. Tau oligomers in cerebrospinal fluid in Alzheimer’s disease. Ann. Clin. Transl. Neurol. 2017, 4, 226–235. [Google Scholar] [CrossRef] [PubMed]
- Brkic, M.; Balusu, S.; Wonterghem, E.V.; Gorle, N.; Benilova, I.; Kremer, A.; Hove, I.V.; Moons, L.; Strooper, B.D.; Kanazir, S.; et al. Amyloid oligomers disrupt blood-CSF barrier integrity by activating matrix metalloproteinases. J. Neurosci. 2015, 35, 12766–12778. [Google Scholar] [CrossRef] [PubMed]
- Williams, S.M.; Schulz, P.; Rosenberry, T.L.; Caselli, R.J.; Sierks, M.R. Blood-Based Oligomeric and Other Protein Variant Biomarkers to Facilitate Pre-Symptomatic Diagnosis and Staging of Alzheimer’s Disease. J. Alzheimers Dis. 2017, 58, 23–35. [Google Scholar] [CrossRef] [PubMed]
- Kolarova, M.; Sengupta, U.; Bartos, A.; Ricny, J.; Kayed, R. Tau Oligomers in Sera of Patients with Alzheimer’s Disease and Aged Controls. J. Alzheimers Dis. 2017, 58, 471–478. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
| Species of Tau | Molecular Weight and Size | Toxicity |
|---|---|---|
| Monomer | 60 kDa, 352–441 AA | no |
| Abnormally phosphorylated non-PHF Tau (AD P-Tau) | 67–70 kDa | yes |
| Dimer/trimer | 120–180 kDa, length 22–25 nm | yes, some types trimeric TauOs: |
| Small soluble oligomer (6–8 molecules) (TauOs) | 300-500 kDa | yes, some types |
| TauOs containing 6–8 molecules: | ||
| Granular tau oligomers (36 Tau monomers) (gTauOs) | 1800 kDa, diameter 20–50 nm | yes, some types |
| Straight filaments (SF) | >50 nm length, 10 nm width | not always |
| ||
| Paired helical filaments (PHF) | 10–20 nm width, with 80 nm periodicity, length > 220 nm | probably not toxic |
| ||
| Neurofibrillary tangles (NFT) | NA | probably not toxic |
| ||
| Ghost tangle | NA | probably not toxic |
|
| Physiological Functions of Tau | Pathological Activity of Tau |
|---|---|
| Main distribution in normal healthy neurons in axons [82,83]: Scaffold protein Stabilization and assembly of MTs Regulation of axonal transport Activity through the repeated regions of MBD | Results of post-translational modifications [95,96]: Detachment of tau from microtubules Microtubule disassembly in axons Mislocalization in presynaptic terminals Induction of synaptic dysfunction Reduction in the number of synaptic vesicles in presynaptic terminals Synapse loss |
| Dendritic function associated with N-terminal region of Tau [87]: Detected in dendrites in small amounts Involved in the development of dendrites Important factor for neurite and axonal growth Contribution to synaptic plasticity Neurite outgrowth | Missorting of pathological Tau into dendrites and postsynaptic compartments [96,157]: Induction of postsynaptic dysfunction mediated by Aβ oligomers Synapse loss Dendritic pathological tau [144]: Transport of FYN kinase to the postsynaptic sites. Reduction of dendritic spines and local increase in Ca2+. Enhancement of Aβ toxicity |
| Nuclear function [89,90]: DNA and RNA protection from oxidative stress Regulation of transcriptional activity Maintaining the integrity of genomic DNA and RNA in hyperthermia | Loss of possibility to enter the nucleus by pathologic Tau [93]: Loss of the DNA-protective function of tau DNA damage |
| Neuronal signaling pathways—PRD domain [91]: Regulation of brain insulin pathway signaling | Signaling molecule in the postsynaptic compartment [144,146]: Influence on neuronal activity Disturbed synaptic transmission Loss of LTP in hippocampus Impaired synaptic plasticity Significant reduction of synaptic proteins and dendritic spines Strengthening the excitotoxicity of AβOs by excitatory neurotransmitters NMDA and PSD95 Induction of neuronal hyperexcitability Cognitive deficits in learning/memory performance tests in mice model of tauopathy |
| Formation of aggregates [99,102,112]: Disturbed neuronal function Release of Tau aggregates into extracellular space Uptake of Tau aggregates by other neurons Spread of Tau pathology | |
| Tau knockout [93]: Impaired hippocampal response to insulin and promotion of brain insulin resistance |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mroczko, B.; Groblewska, M.; Litman-Zawadzka, A. The Role of Protein Misfolding and Tau Oligomers (TauOs) in Alzheimer′s Disease (AD). Int. J. Mol. Sci. 2019, 20, 4661. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20194661
Mroczko B, Groblewska M, Litman-Zawadzka A. The Role of Protein Misfolding and Tau Oligomers (TauOs) in Alzheimer′s Disease (AD). International Journal of Molecular Sciences. 2019; 20(19):4661. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20194661
Chicago/Turabian StyleMroczko, Barbara, Magdalena Groblewska, and Ala Litman-Zawadzka. 2019. "The Role of Protein Misfolding and Tau Oligomers (TauOs) in Alzheimer′s Disease (AD)" International Journal of Molecular Sciences 20, no. 19: 4661. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20194661