1. Introduction
The oxidative folding of proteins containing disulfide bridges is a critical process in the pathway leading to native conformations [
1]. A precise understanding of this event is not a merely speculative exercise, but the basis to know the origin of pathological misfoldings that cause severe diseases like Alzheimer’s and Parkinson’s diseases [
2]. It is well known that a correct formation of structural disulfide bonds is crucial for protein integrity and dysregulation of such disulfides may contribute to the pathogenesis of these diseases [
3]. In fact, under appropriate conditions a wide range of disulfide-containing proteins can convert from their normally soluble forms into fibrillar aggregates with all the characteristics of disease-associated amyloid fibrils [
4]. These aggregates are not normally found in biological systems due to mechanisms that inhibit their formation. Furthermore, disulfide bonds have co-evolved with protein sequences to minimize protein misfolding and propensity to form potentially toxic aggregates [
5]; and understanding the nature of such protective mechanisms is a crucial step in the development of strategies to prevent these diseases [
6]. In this context, the discovery of new kinetic properties of structural cysteines in the nascent structure of a protein may be of paramount interest and it represents the main novelty of the present study.
The oxidative folding of ribonuclease (RNase) has been extensively studied in the last century, starting with the historical study by the Nobel Prize winner, Anfinsen [
7]. It is well accepted that, in the presence of glutathione/oxidized glutathione (GSH/GSSG) in a ratio similar to that found in the endoplasmic reticulum, a reduced ribonuclease (rRNase) first forms mixed disulfides with GSSG and then a restricted number of correct and incorrect protein disulfides through a non-random mechanism [
8,
9]. The final disulfide to be formed involves Cys40 and Cys95, although a small fraction involves Cys62-Cys75 [
10,
11]. The possible role of protein disulfide isomerase (PDI) as a catalyst in this process is not supported by mass spectrometry (MS) analyses of the S-S containing peptides, because they are equally populated in the presence or absence of this enzyme [
8]. Furthermore, there is strong evidence for endoplasmic reticulum oxidase (Ero1)-independent mechanisms that lead to the production of the oxidized enzyme [
12]. In the early stage of the folding process, Cys110, present in many proteolytic fragments containing native and incorrect disulfides, appears to be one of the most reactive residues, while Cys26 is the most inert among the eight cysteines. By observing the kinetics of disulfide formation at pH 8.0, approximate values have been calculated for the formation of the first two disulfides (about 115 M
−1 s
−1) [
13], a value more than ten-fold higher than those found in a previous paper [
14] under similar conditions.
Despite these and many other studies that have been made to detail this process, some important particulars remain to be defined. For example, the intrinsic reactivity of each cysteine in the reduced enzyme has been never evaluated as well as no comparison has been made with the reactivity of a free cysteine. Thus, no evidence has been obtained in the past for the possible existence of cysteines hyper-reactive toward GSSG, which may react with this disulfide in a very short time without the assistance of an enzyme. This reaction may be important to drive a correct oxidative folding. The occurrence of non-functional hyper-reactive cysteines was only rarely investigated in the past for other proteins. In the literature, bovine pancreatic trypsin inhibitor (BPTI) is one of the most cited case of hyper-reactive cysteines intended to form disulfides in the native protein [
15]. However, it was recently demonstrated that among its six cysteines, four of them display only about ten times higher reactivity than an unperturbed protein cysteine, while the remaining cysteines are even less reactive [
16]. Conversely, we found that Cys94 in reduced lysozyme [
17] and Cys75 in reduced serum albumin [
16] display thousand times higher reactivity toward GSSG. In the case of lysozyme, glutathionylation of Cys94 causes the instantaneous inhibition of the deleterious aggregation that occurs when lysozyme is in a reduced form [
17], giving a reasonable finality for this phenomenon.
This paper discovers that the rRNase in a molten globule like conformation, representing the early proof of its native conformation, shows one particular cysteine hyper-reactive toward GSSG, which is estimated to be thousand times more reactive than an unperturbed protein cysteine. Our study identifies this particular residue and explores the origin of this phenomenon that is in part due to a low pKa of this cysteine, but mainly to the rapid formation of a productive protein-GSSG complex.
3. Discussion
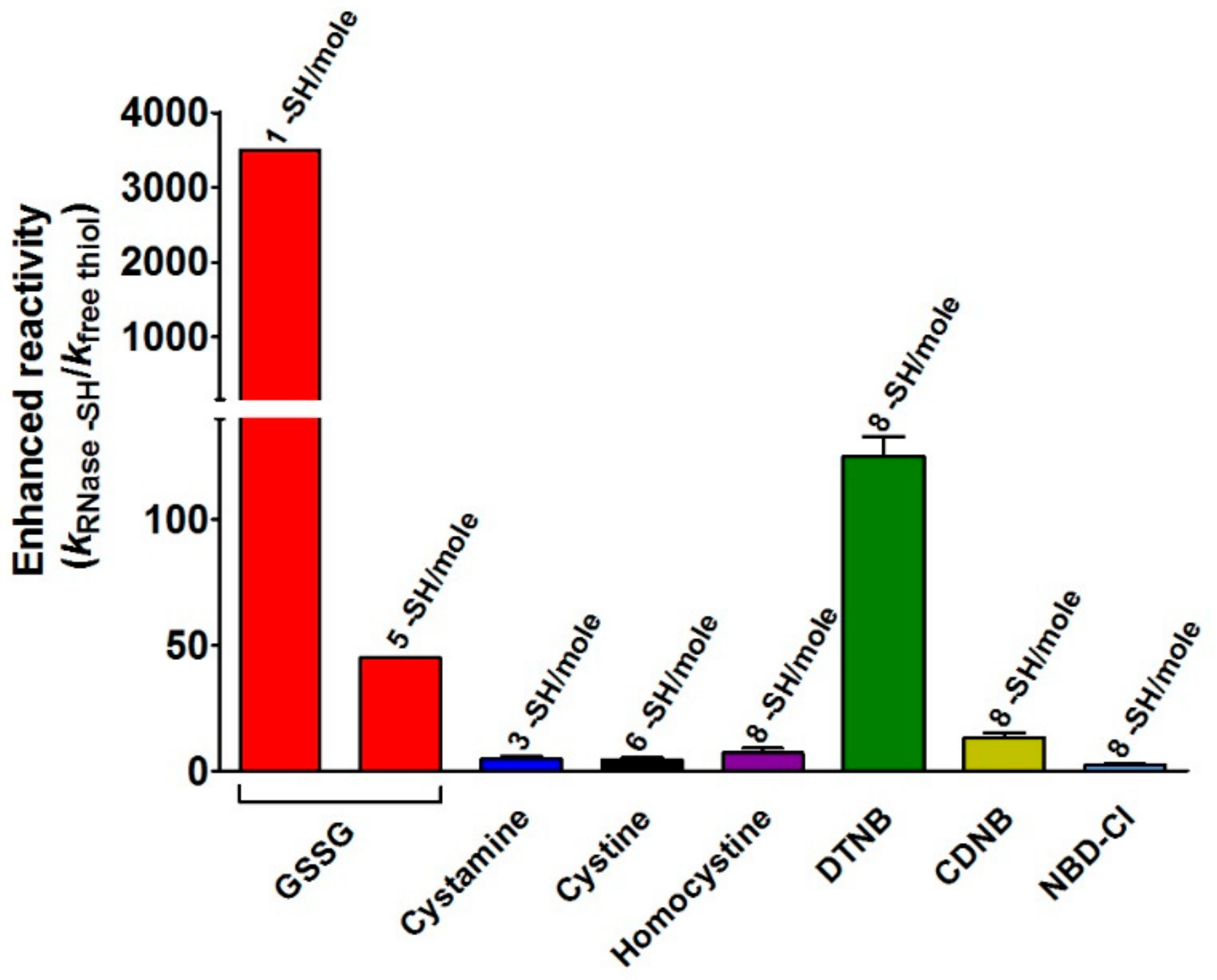
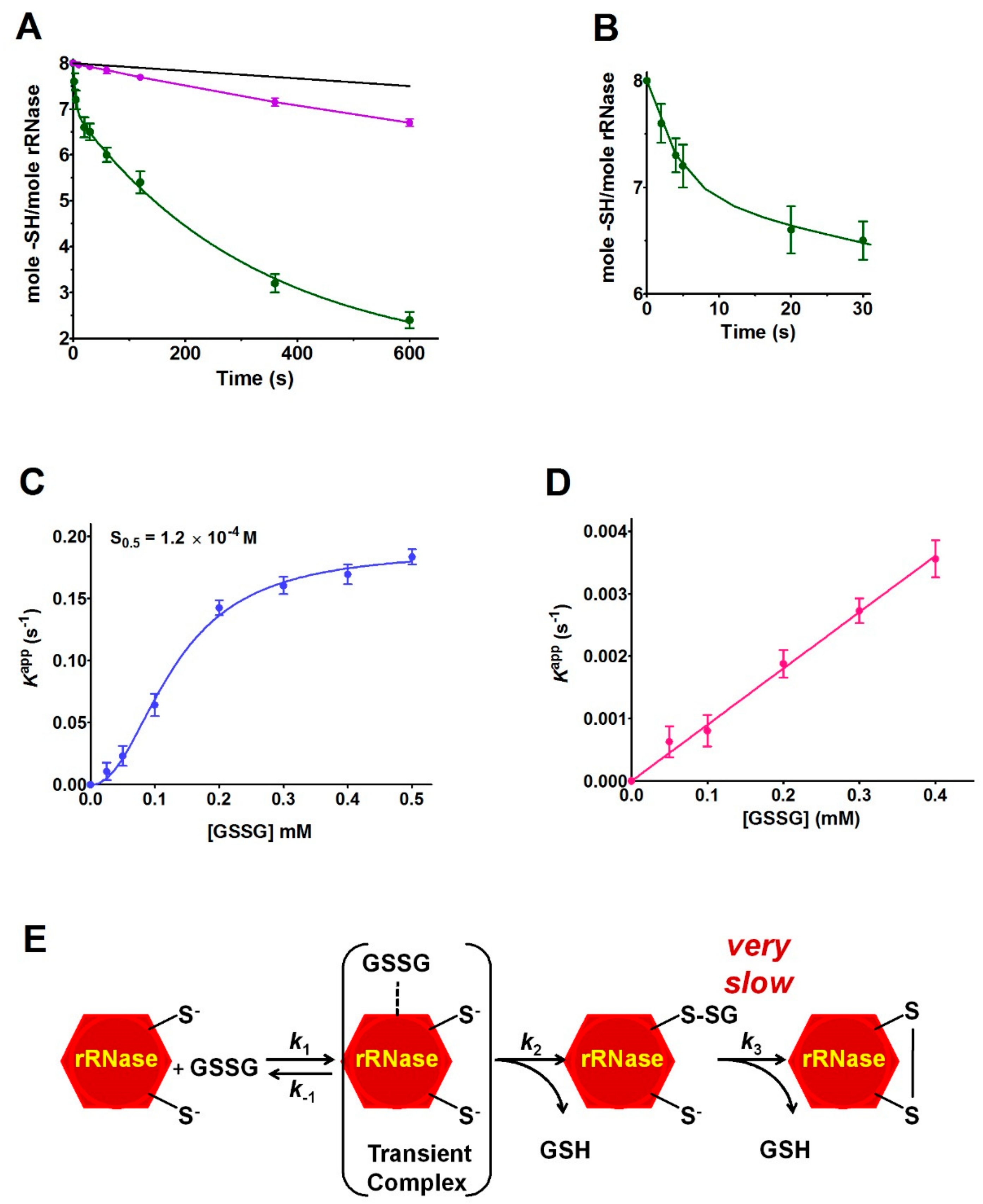
This study discloses for the first time the existence, in the rRNase, of a curious and extraordinary hyper-reactivity of one cysteine toward GSSG and a parallel relevant hyper-reactivity of five other cysteines (3500 and 45 times more reactive when compared to an unperturbed protein cysteine, respectively) (
Table 1,
Figure 2 and
Figure 5).
Hyper-reactivity toward GSSG was previously demonstrated for Cys75 in reduced albumin [
16] and then for Cys94 in reduced lysozyme [
17] (
k > 250 M
−1 s
−1 and
k = 600 M
−1 s
−1, respectively). These values, normalized to that of an unperturbed protein cysteine i.e., 0.2 M
−1 s
−1, fulfill enhanced reactivity > 1250 times and 3000 times for Cys75 and Cys94, respectively.
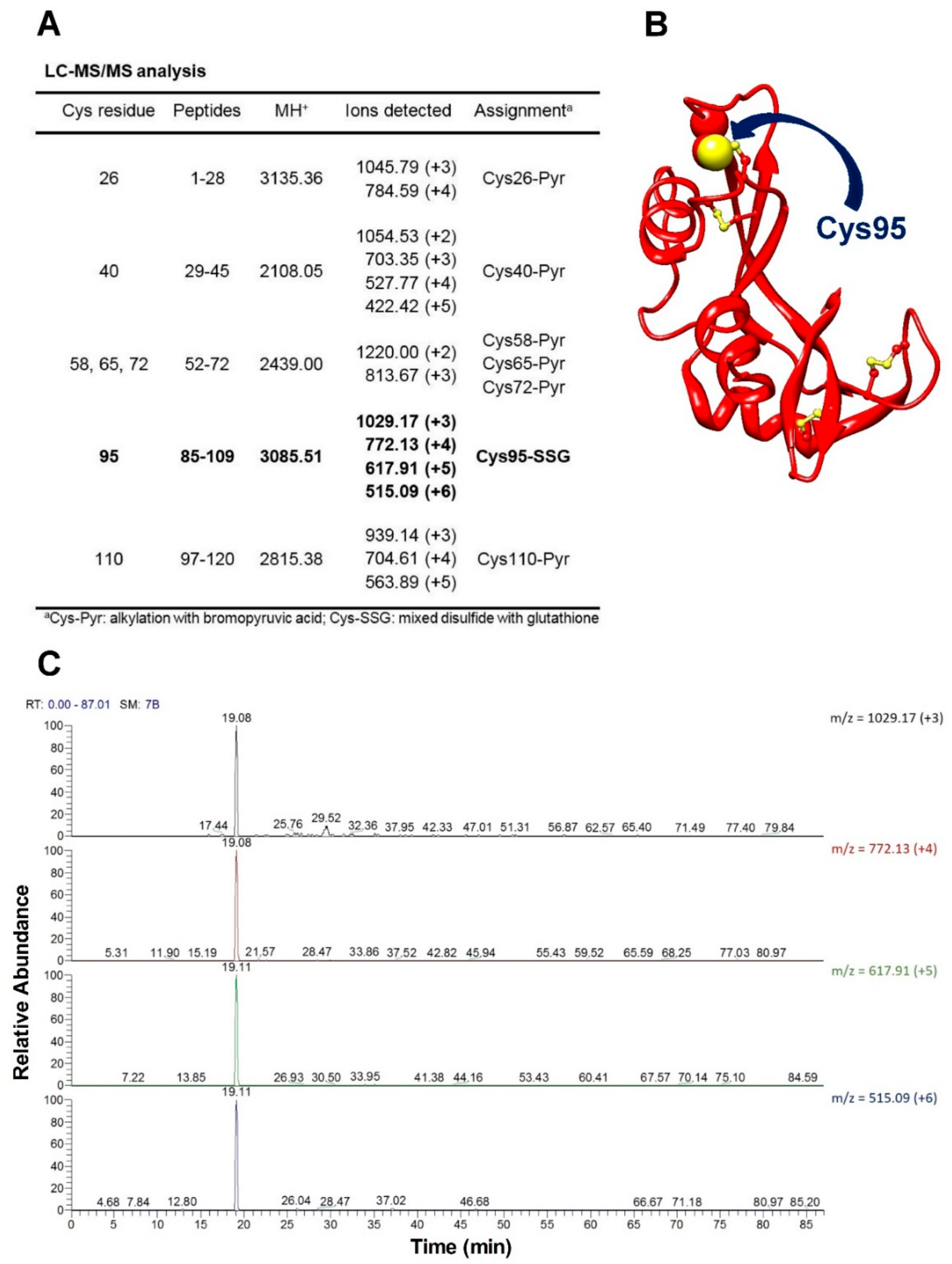
The novelty of the present study is that all the previous investigations about the oxidative folding of rRNase did not evaluate the intrinsic reactivity of the involved cysteines and no comparison has been made with the reactivity of a free cysteine or an unperturbed protein cysteine. Thus, our findings disclose a fascinating aspect, still now completely unknown, about a protein whose oxidative folding probably has been the most studied in the last century. The most hyper-reactive cysteine in the rRNase was identified as Cys95 by mass spectrometry, which revealed the presence of the sole Cys95-SG mixed disulfide after only 10 s incubation with 0.4 mM GSSG.
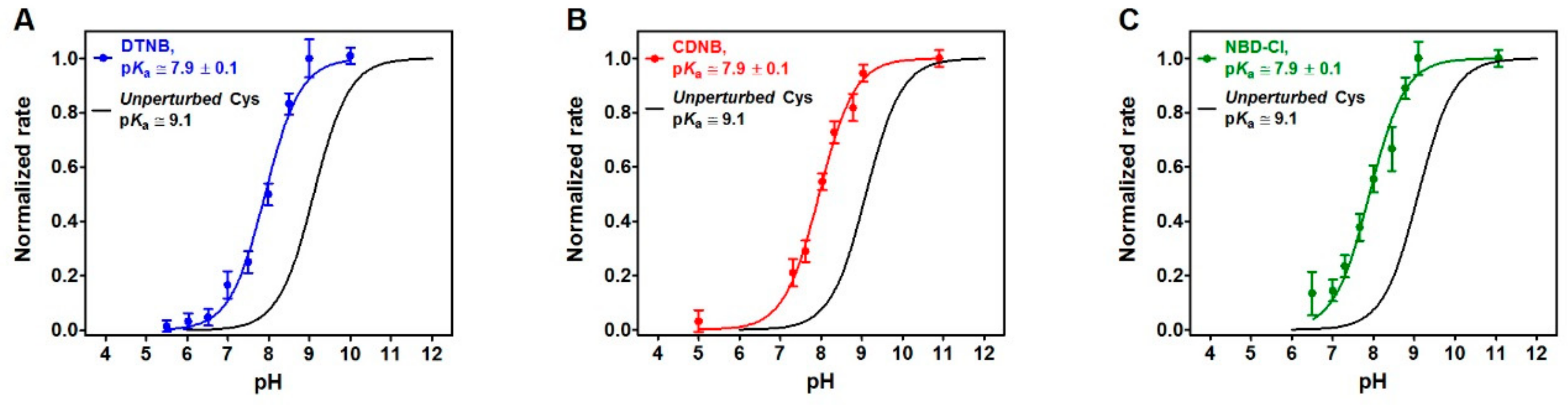
It is generally accepted that the hyper-reactivity of protein cysteines is modulated by two distinct properties: solvent exposure and p
Ka values [
23]. A lowered p
Ka from 9.1 to 7.9, as measured for rRNase cysteines using DTNB, CDNB and NBD-Cl as thiol reagents (
Figure 4), could account for only about 15 times higher reactivity. The hundred times increased reactivity toward DTNB of all the protein cysteines and the 45 times higher reactivity toward GSSG for the five protein cysteines could only be explained by assuming the existence of a weak electrostatic positive environment near most of the protein cysteines given that DTNB and GSSG are negatively charged disulfides. This possibility may also explain the average lower p
Ka values. However, the extraordinary hyper-reactivity observed for Cys95 toward GSSG requires further assumptions. In fact, in a thiol-disulfide reaction, the enhanced reactivity of a cysteine due to a very low p
Ka of its sulfhydryl group cannot exceed 40 or 50 times at pH 7.4 [
17]. A completely novel factor could be the occurrence of a transient protein-GSSG complex which may promote a productive encounter between the –SH group of Cys95 and GSSG, reminiscent of the encounter of two substrates in the active site of an enzyme. The existence of such a transient complex is strongly suggested by the saturation curve shown in
Figure 5C. The plot shows an S
0.5 of 0.12 mM, a value which warrants most of the rRNase to be “saturated” by GSSG at the physiological concentrations of this disulfide in the endoplasmic compartment (0.4 mM), and then to be glutathionylated at the maximal velocity. The cooperative-like behavior shown in
Figure 5C cannot be easily explained. In fact, the RNase is not a multimeric enzyme so the autocatalytic behavior can be tentatively justified assuming that the molten globule displays two binding sites for GSSG (one of them modulating the affinity of the other), or a transient assembly of two enzyme monomers to give a dimeric structure. Interestingly, it was demonstrated that cooperativity requires neither multiple ligand binding events nor multimeric assemblies [
24].
The specificity of the above proposed interaction between GSSG and rRNase is indirectly supported by the absence of hyper-reactivity toward other natural disulfides like cystamine, homocystine, and cystine (
Table 1) and by the absence of a saturation behavior of the other cysteines in their reaction with GSSG (
Figure 5D). All these findings may help to enlighten novel details about the oxidative folding of this enzyme occurring in the cell. In fact, it may be tentatively proposed that the early event of this process may be the glutathionylation of Cys95 possibly finalized to give a hierarchic formation of the natural disulfides restricting the occurrence of incorrect disulfides. This glutathionylated residue could remain as such until all the other native disulfides are formed, as suggested by previous studies indicating the Cys95-Cys40 as the last disulfide in the temporal sequence of the folding [
10,
11]. This covalent modification of Cys95 could serve to “freeze” this residue to avoid its reaction with other cysteines or, more probably, to induce a useful conformational change. In fact, CD spectra show that the glutathionylation of Cys95 triggers a slight but evident structuration of the enzyme (
Figure 6B). The importance of Cys95 and its glutathionylation in the correct folding is indirectly indicated by previous in vivo experiments using a mutated enzyme with Cys95 and its natural counterpart Cys40 replaced by Ala [
25]. The consequence of this mutation on the folding mechanism in a living cell was surprising: although these two cysteines are the last residues to form the proper disulfide, their substitution causes a deep change in the oxidative pathway [
25]. We can now speculate that glutathionylation of Cys95 and the consequent structural change of the enzyme possibly avoids this non-natural route. Thus, the role may be different from the one suggested for Cys110 which may function as an internal catalyst and is able to promote the reshuffling of disulfides to speed the attainment of the native conformation [
26]. Obviously, the proposed role of Cys95 related to its hyper-reactivity toward GSSG must be confirmed by future studies. Unfortunately, the obvious experiment in vivo consisting of the depletion of GSSG by inhibiting its biosynthetic pathway has been done in the past [
27], but the presence of relevant levels of oxidized proteins has led to the hasty and perhaps incorrect conclusion that GSSG does not have a role in oxidative folding. In fact, as discussed by Bass and coworkers [
28] in the absence of glutathione, all the cell environment becomes strongly oxidant and a secondary oxidative pathway may substitute GSSG in its role.
The presence of one single hyper-reactive cysteine toward GSSG in the rRNase is a property very similar to that reported for the reduced form of lysozyme [
17] and serum albumin [
16]. This may suggest a more general property of the molten globule conformation of proteins whose cysteines are devoted to structural and not to catalytic roles. This transient and still now unknown structure is far from a disordered or amorphous conformation, but it appears as a more “organized” status able to perform specific functions like to bind GSSG near Cys95 with high affinity. Accordingly, a relevant portion of the ordered structure of the rRNase is present in 0.2 M urea (
Figure 6A). The structure of the RNase molten-globule is unknown and the contribution of a docking simulation (based on the native structure) to identify residues involved in the RNase-GSSG complex is clearly limited. However, assuming that the structure of the molten-globule is similar to the one of the native crystal structure [
21], a preliminary docking analysis indicates four residues possibly involved in hydrogen bond interactions with GSSG bound near Cys95 (i.e., Lys31, Thr36, Lys37, and Pro93).
While the hyper-reactivity of Cys94 in lysozyme and its fast conversion into a glutathionylated residue seem to be finalized to prevent the deleterious aggregation which occurs when the protein is in a fully reduced status, a similar role cannot be ascribed for rRNase which do not aggregate in such conditions. As proposed above, this particular property may limit the formation of incorrect disulfides in the nascent phase. However, other functions may be taken into account. For example, it was demonstrated that in the human small copper chaperone Cox17, the conformational switch between disordered and folded states is controlled by the formation of a single disulfide bond [
29]; a single fast glutathionylation may have a similar role. Finally, we cannot forget the moderate hyper-reactivity found in the remaining cysteines which are greatly more reactive when compared to the cysteines of BPTI, always cited in the literature as a model of hyper-reactivity of structural cysteines [
15,
16]. Whatever the finality, this phenomenon confirms the presence in the protein science of still new and fascinating mechanisms that may help to clarify more in detail the still obscure event of oxidative folding.
4. Materials and Methods
4.1. Chemicals and Reagents
Ribonuclease A (RNase) from bovine pancreas (Type XII-A, 75–125 Kunitz units/mg protein) was used. The commercial stock of the RNase used in all the experiments was ≥99% pure according to our HPLC/MS analysis. l-cysteine (Cys), l-cystine, l-glutathione (GSH), oxidized glutathione (GSSG), homocystine, cystamine, 1-chloro-2,4-dinitrobenzene (CDNB), 5,5′-dithiobis(2-nitrobenzoic acid) (DTNB), 4-chloro-7-nitrobenzofurazane (NBD-Cl), dithiothreitol (DTT), ethylendiaminetetraacetic acid (EDTA), bromopyruvic acid, and all other reagents were from Sigma-Aldrich (St. Louis, MO, USA).
4.2. RNase Reduction
The RNase concentration was evaluated by an extinction coefficient of 9440 M
−1 cm
−1 at 280 nm [
30]. The reductions of the RNase at 37 °C and 60 °C were performed as follows: 0.05 mM final concentration in 0.01 M sodium borate buffer, pH 8.5, with 8 M urea, EDTA 1 mM final concentration, and DTT (RNase:DTT, 1:10). The pH was adjusted to 8.5 with 0.1 M NaOH. At fixed times, the content of reduced cysteines was evaluated by reacting the rRNase (1.25 μM) with 20 μM DTNB at pH 5.0 (ε
M TNBS
− = 11800 M
−1 cm
−1 at pH 5.0) and 0.2 M urea taking advantage of the reactivity of all the eight protein cysteines with this reagent (see
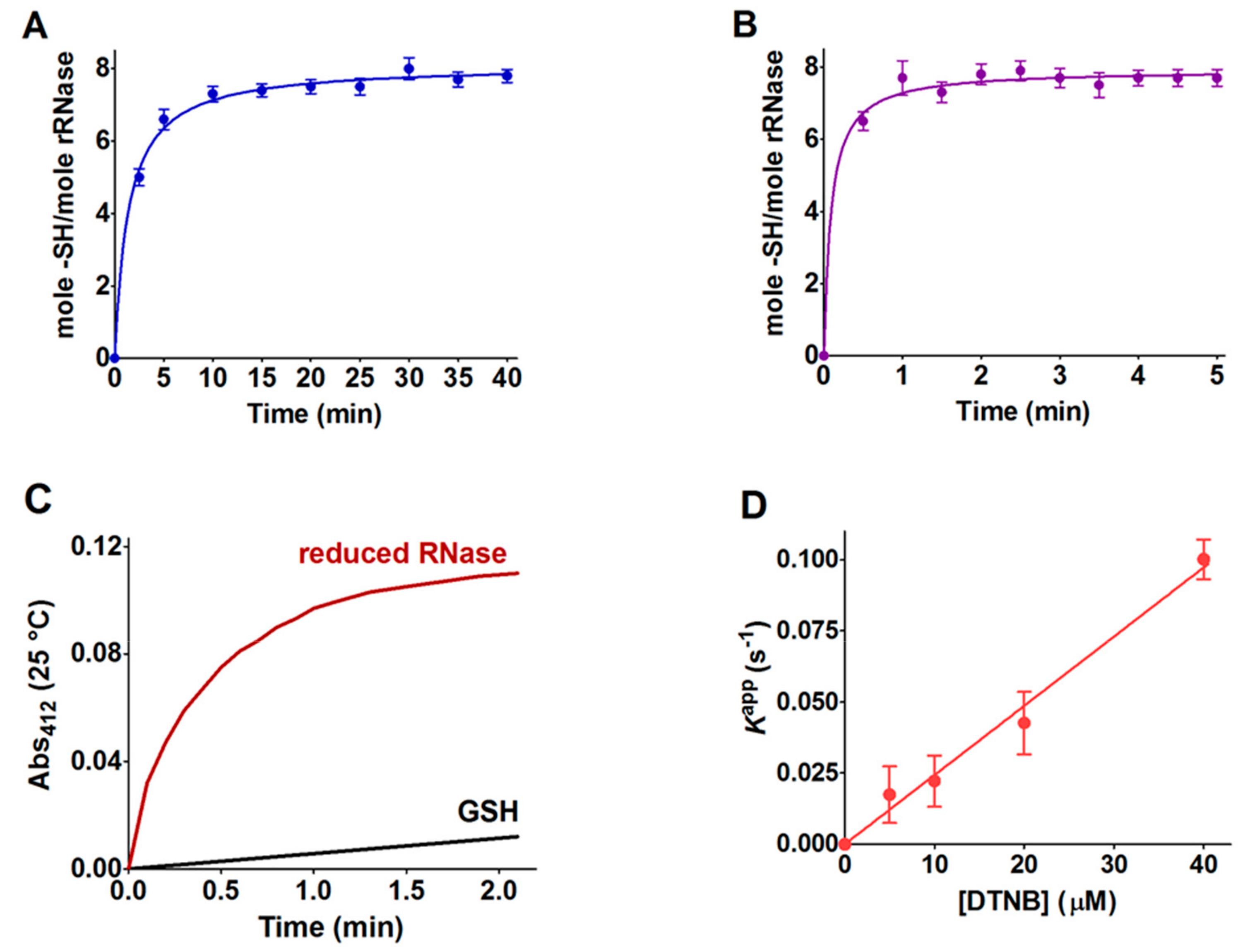
Table 1). Within the manuscript, the term rRNase refers to that obtained by reduction with DTT after 40 and 5 min at 37 and 60 °C, respectively. DTT stock solutions were titrated with DTNB prior to use.
An alternative procedure was applied for the preparation of the rRNase for experimental measures, with the conditions for the reduction being slightly different. Briefly, 0.1 mM of the RNase was dissolved in 0.01 M sodium borate buffer, pH 8.5, 8 M urea, EDTA 1 mM, and DTT (RNase:DTT, 1:100) at 37 °C for 30 min. After reduction, the rRNase (0.1 mM) was passed through a Sephadex G-25 gravity flow column (1 × 20 cm) equilibrated with 20 mM sodium phosphate buffer, pH 7.4, containing 2 M urea and 1 mM EDTA. The eluted protein in a single fraction (≈40 μM) without DTT was used for the determination of the second-order rate constants, circular dichroism spectra, and pKa values.
4.3. Reactivity of rRNase Cysteines toward GSSG
The interaction of the rRNase with GSSG (0.4 mM, same concentration of endoplasmic reticulum) [
31] was measured by incubating 1.25 μM of protein with GSSG in 0.01 M potassium phosphate buffer pH 7.4, 0.2 M urea at 25 °C. The loss of hyper-reactive cysteines due to reaction with GSSG was estimated by the titration of the –SH group with DTNB. The reactivity of free cysteines with GSSG was evaluated as reported in our previous study [
16,
17].
4.4. Reactivity of rRNase toward Several Disulfides and Thiol Reagents
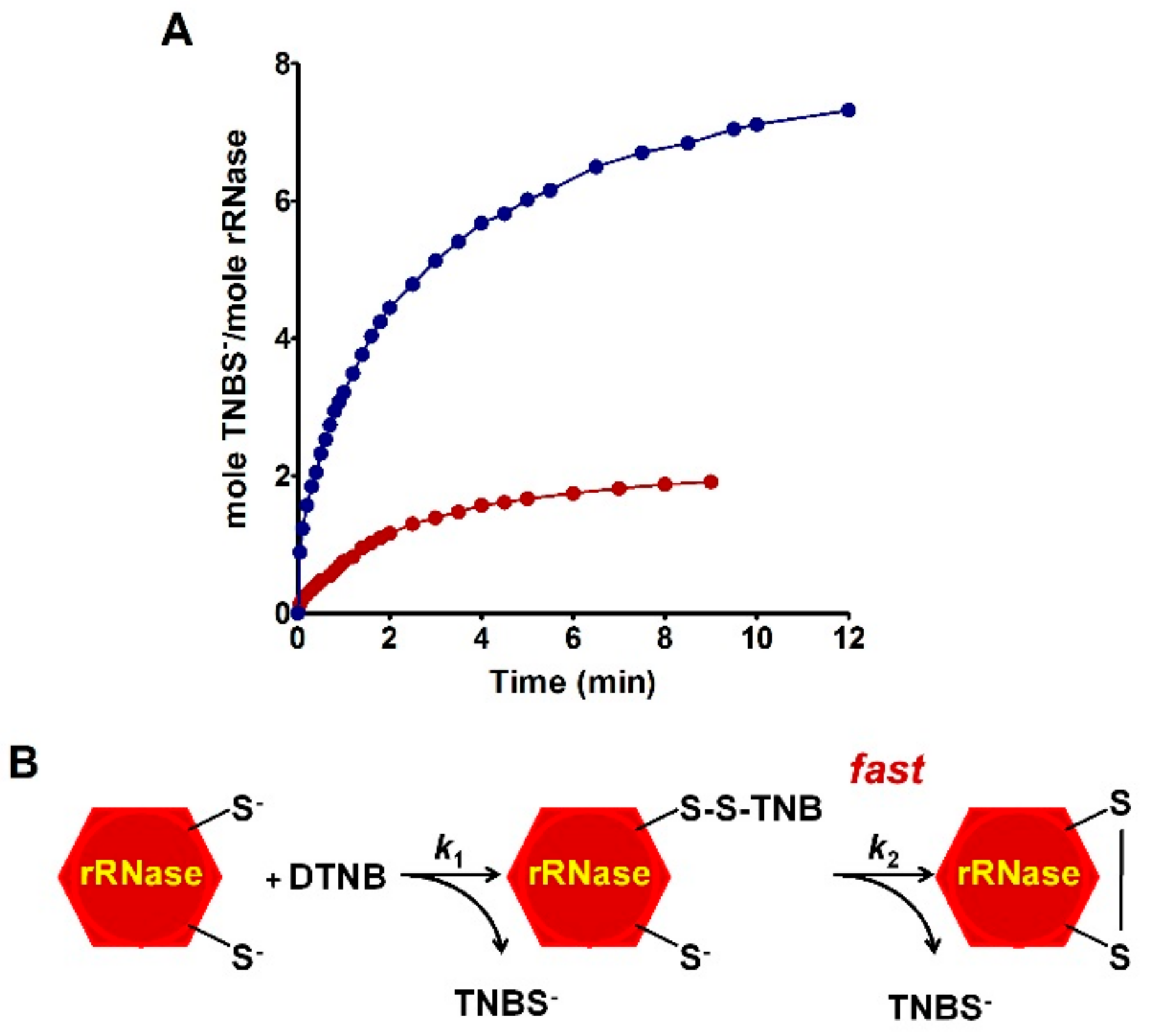
The reactivity of the sulfhydryl groups of the rRNase (1.25 µM final concentration) toward DTNB (20 µM) was evaluated in a continuous spectrophotometric assay at 412 nm where TNBS– absorbs (εM TNBS– = 11800 M−1 cm−1 at pH 5.0). The first-order kinetic constants were evaluated on the basis of t1/2 at different DTNB concentrations. The reactivity of the rRNase (1.25 µM) toward homocystine (0.4 mM), cystine (0.2 mM), and cystamine (0.2 mM) was determined in 10 mM potassium phosphate buffer, pH 7.4, 0.2 M urea. At fixed times, aliquots were placed in 0.1 M acetate buffer, pH 5.0, 0.2 M urea and the disappearance of the reactive cysteines of the rRNase was determined using DTNB as the titrant (25 °C).
The reactivity toward CDNB was evaluated using continuous spectrophotometry at 340 nm where the Cys-DNB adduct absorbs (ԑ
M = 9600 M
−1 cm
−1) [
16]. The rRNase (1.25 µM) was reacted with 0.4 mM CDNB in 0.1 M potassium phosphate buffer, pH 7.4, 0.2 M urea (25 °C). A slight turbidity due to the CDNB-modified enzyme was subtracted by each determination.
The reaction of the rRNase (1.25 µM) toward NBD-Cl (20 µM) was determined spectrophotometrically at 419 nm, where the Cys-NBD adduct absorbs (ԑ
M = 13000 M
−1 cm
−1) [
32], in 0.1 M potassium phosphate buffer, pH 7.4, 0.2 M urea (25 °C).
Second-order kinetic constants of the reaction between free GSH (0.1 mM) and homocystine(0.4 mM) were determined by using 0.1 M potassium phosphate buffer, pH 7.4, to determine the amount of homocysteine released as a consequence of the reaction at fixed times. Homocysteine was determined by adding NaOH (20 mM final concentration) to the mixture and after the reaction with 0.3 mM bromopyruvate. The corresponding product is a cyclic ketimine sulfur compound (cystathionine ketimine) absorbing at 296 nm (ԑ
M = 3200 M
−1 cm
−1) [
33]. Second-order kinetic constants for the reaction of free cysteine toward GSSG and free GSH toward all other reagents were derived from our previous study [
17]. GSH solutions were freshly prepared and the amount of GSSG was less than 1% as assayed by standard analytical procedures.
4.5. pKa Determination
The average pKa of cysteines of the rRNase (1.25 μM) was calculated in a solution of 0.02 M Britton–Robinson buffer (pH varying from 4.0 to 11.0) and 0.2 M urea. The reactivity of these cysteines (8 –SH/mole rRNase) was measured with DTNB (20 μM), NBD-Cl (20 μM), and CDNB (1 mM). Only in the case of DTNB, the reaction was followed using an SFA-12 Rapid Kinetics Accessory (Hi-Tech Scientific, Bradford-on-Avon, UK). Below pH 7.0, appropriate TNBS– extinction coefficients at 412 nm were considered. Normalized rates were calculated from observed initial velocities normalized to maximum velocities calculated at full deprotonation. The initial velocities of the reaction of the rRNase with the compounds were recorded spectrophotometrically in a continuous assay. Finally, pKa were calculated by a curve fitting analysis.
4.6. Circular Dichroism Spectroscopy
CD spectra of the native RNase and rRNase in the presence of 0.2 M or 8 M urea were measured at 1.25 μM protein concentration in 10 mM potassium phosphate buffer, pH 7.4, 25 °C. A control sample of rRNase (6.5 μM) was alkylated with 1 mM bromopyruvate at pH 7.4 in 0.2 M urea (25 °C). A test sample of rRNase (6.5 μM) was incubated with 0.4 mM GSSG for 10 s and then alkylated with 1 mM bromopyruvate at pH 7.4 in 0.2 M urea (25 °C). Both these samples were eluted onto Sephadex G-25 to clean samples from GSSG and bromopyruvate. CD spectra of these two samples were measured at 1.25 μM protein concentration in 10 mM potassium phosphate buffer, pH 7.4, 0.2 M urea at 25 °C. The setting panel of the spectropolarimeter Jasco J-715 (Easton, MD) was: slit 2 nm, sensibility 50 mdeg, range 250–205 nm, resolution 0.2 nm, using a quartz cuvette of 0.5 cm light path.
4.7. Effect of Urea Concentration on the Hyper-Reactivity
The effect of urea on hyper-reactivity of the rRNase was assayed using DTNB as a thiol reagent. In a typical experiment, the rRNase (1.25 μM) was incubated with 0.01 M potassium phosphate buffer, pH 7.4, in the presence of variable concentrations of urea (from 0.2 M to 8 M). After five minutes of incubation, the rate of reaction with DTNB (20 μM) was measured spectrophotometrically at 412 nm in 0.1 M acetate buffer, pH 5.0 (25 °C).
4.8. Mass Spectrometry Identification of Hyper-Reactive Cysteine
The rRNase (1.25 μM) was incubated with GSSG (0.4 mM) in 0.01 M potassium phosphate buffer, pH 7.4. The reaction was stopped after 10 s by adding 1 mM bromopyruvate which alkylated residual protein cysteines within 1–2 s. Then the sample was lyophilized. A reduced RNase solution (1.25 μM) was immediately alkylated with bromopyruvate and used as a control. Samples were resuspended in 0.1% trifluoroacetic acid (TFA) and desalted by reverse-phase HPLC on a Phenomenex Jupiter C4 column (250 mm × 2.0 mm, 300 Å pore size) with a linear gradient from 10% to 95% of solvent B (0.07% TFA in 95% acetonitrile) in 30 min, at a flow rate of 200 μL/min using Agilent Technologies 1100 HPLC (Agilent Technologies, Santa Clara, CA, USA). Protein fractions were collected and lyophilized.
Controlled pepsin hydrolysis was carried out by dissolving the samples in 5% formic acid, pH 2.5 and adding pepsin at an enzyme to substrate ratio of 1:50 w/w at 37 °C for 2 h. Samples were then lyophilized and resuspended in 0.2% formic acid. Peptic hydrolysis was controlled by MALDI-MS analyses of the resulting peptide mixture and the samples were then directly analyzed by nanoLC/MS-MS on an LTQ-XL Orbitrap mass spectrometer equipped with a nanoHPLC (ThermoFisher, Waltham, MA, USA). Peptides containing modified cysteine residues were selected using the ion-extraction chromatograms of the corresponding multiply charged ions and the assignments were confirmed by manual inspection of their fragmentation spectra.
4.9. Statistical and Graphical Analysis
Data are represented as means ± standard deviation (S.D.). Data were obtained from independent experiments (from three to ten) performed in different days by the same operators using the same instruments. Statistical significance of the differences between second-order kinetic constants of the rRNase and free Cys/free GSH toward reagents was analyzed by
t-tests. The same statistical analysis was applied for p
Ka values.
p-value < 0.05 was considered statistically significant. Statistical analyses were performed using the computer software package, MedCalc (Mariakerke, Belgium). The propagation of uncertainties for the quotients ‘enhanced reactivity’ was analyzed according to the classical statistical methods [
20]. The equations used to fit kinetic data are:
| Hyperbolic curve: | | (1) |
| pKa curve: | | (2) |
| Two-phase decay: | | (3) |
| Sigmoidal curve: | | (4) |
| One-phase decay: | . | (5) |
The graphic and results visualization were obtained using GraphPad Prism software v5.0 (La Jolla, CA, USA).

 and
and 







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}