Unravelling the Regions of Mutant F508del-CFTR More Susceptible to the Action of Four Cystic Fibrosis Correctors


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. Effect of Correctors on the Expression of M1N1, M2N2, ∆NBD2 and CFTR mRNAs.
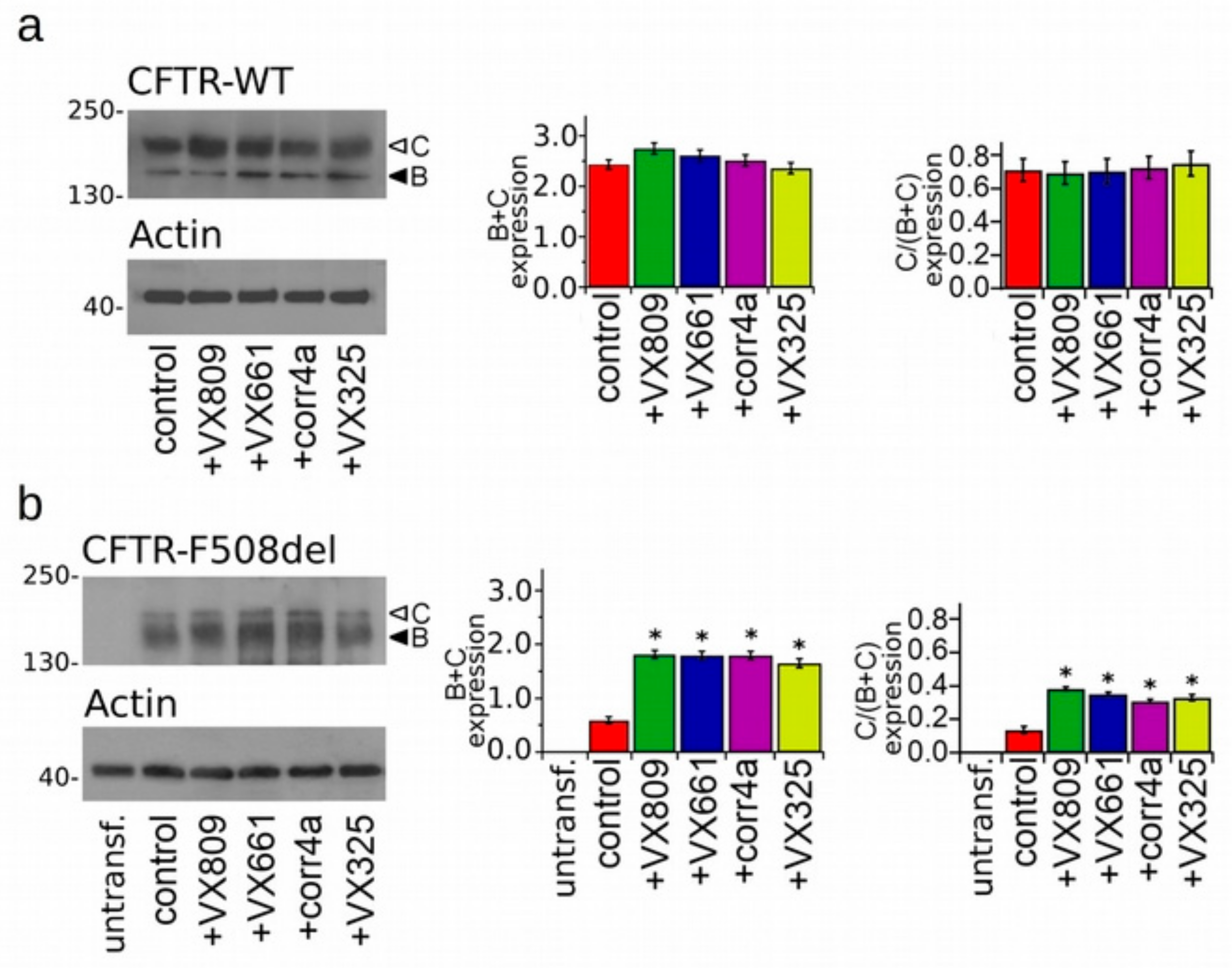
2.2. Effects of Correctors on the Functional Expression of Full Length WT- and F508del-Proteins.
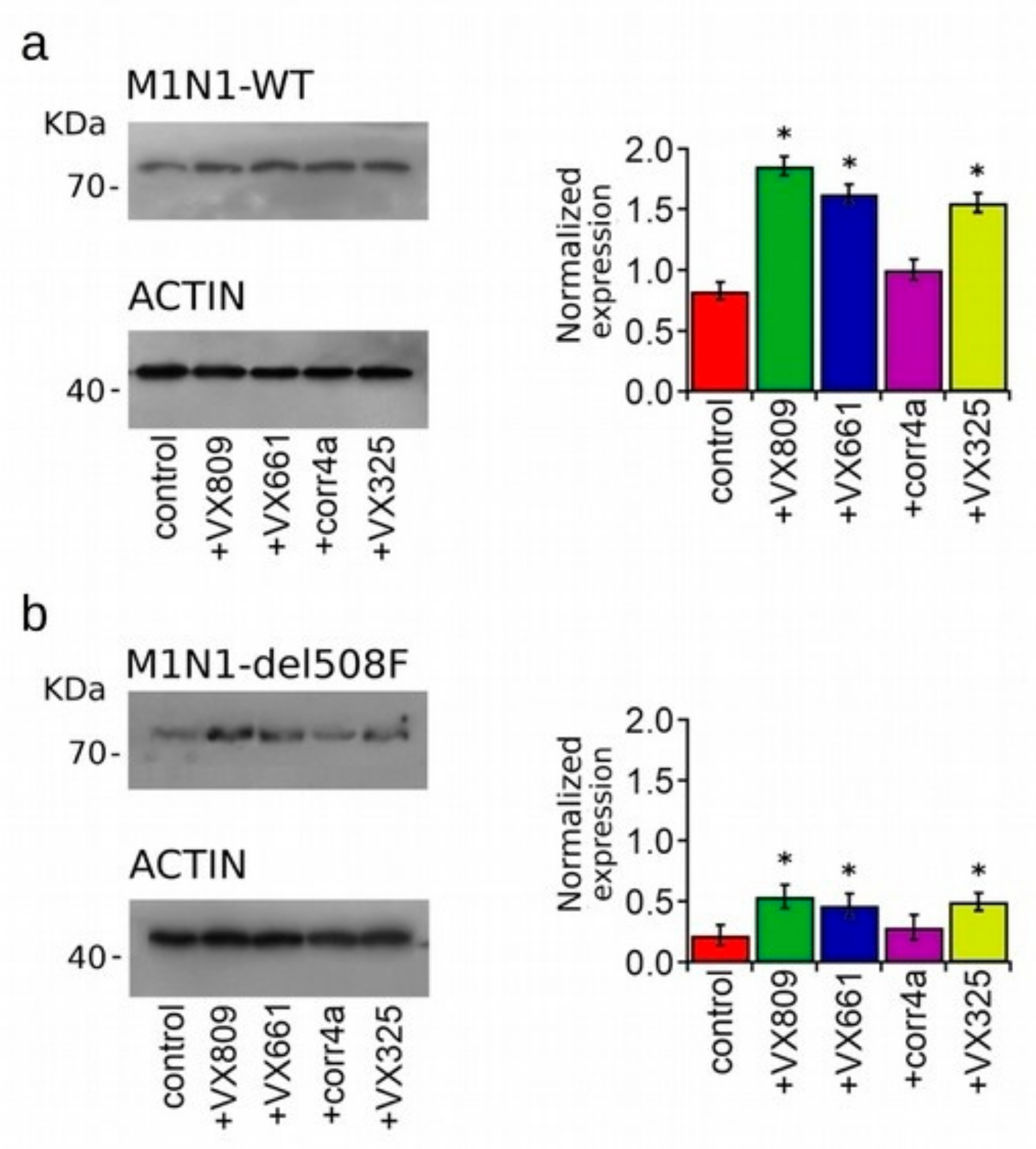
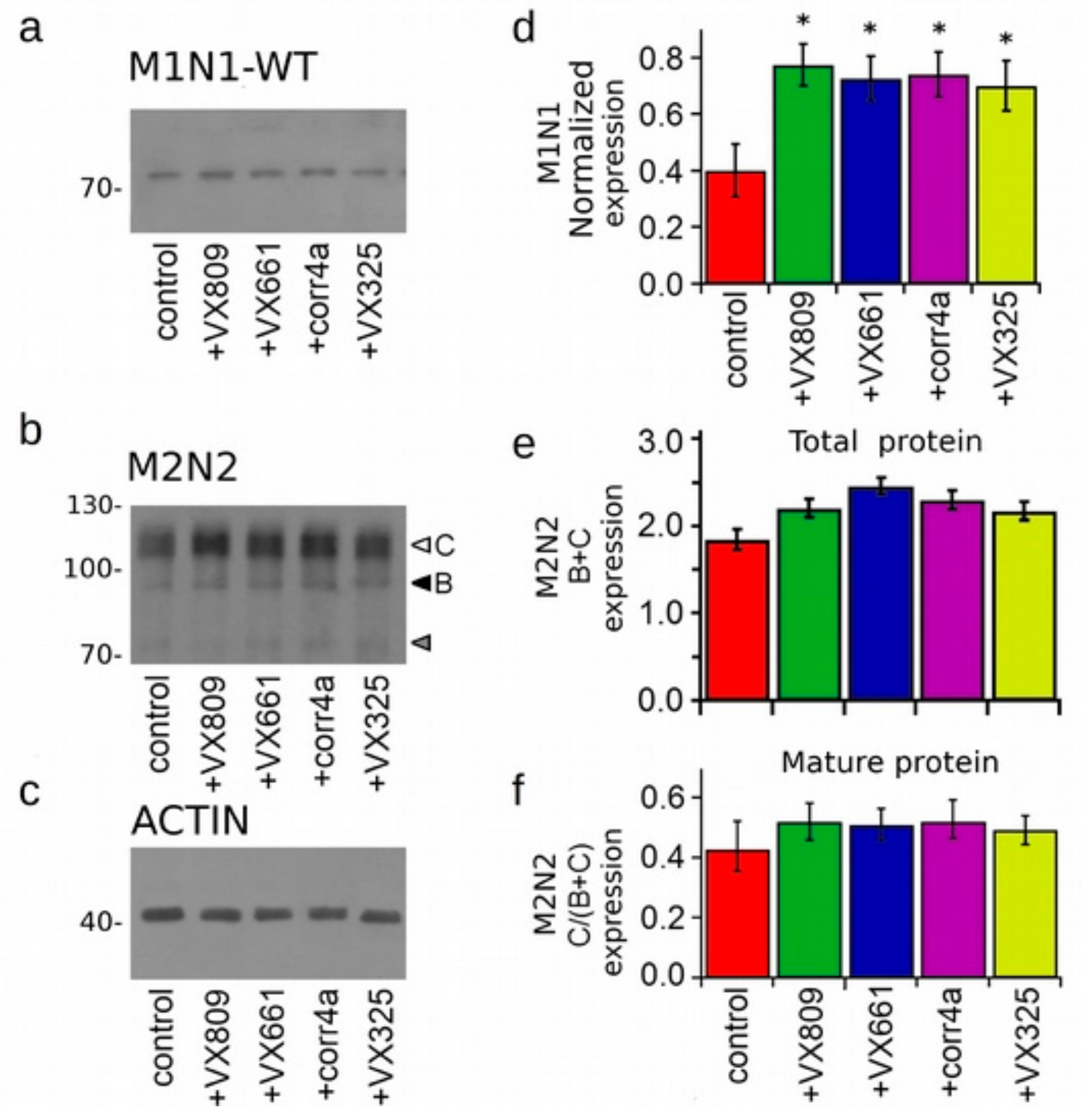
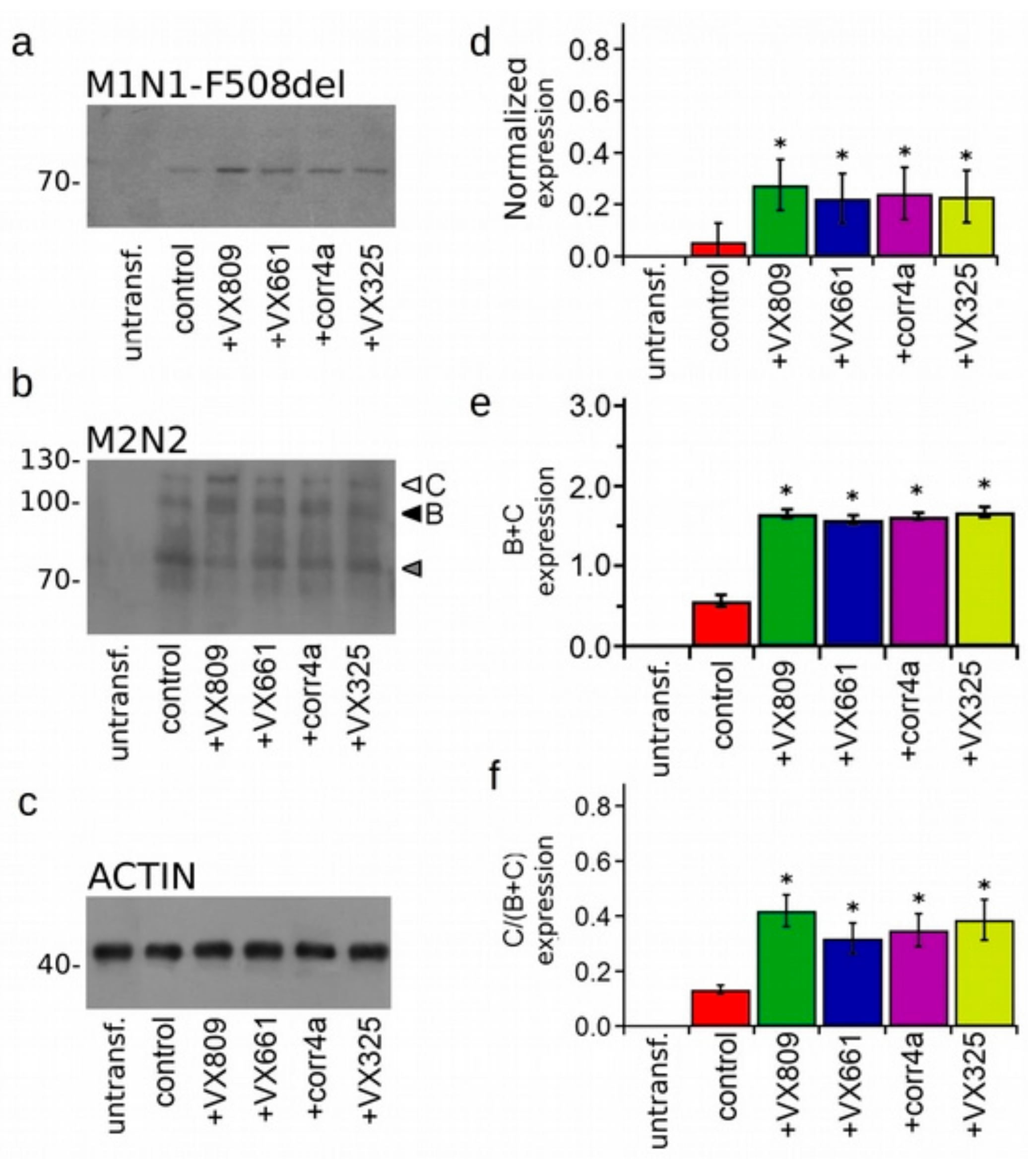
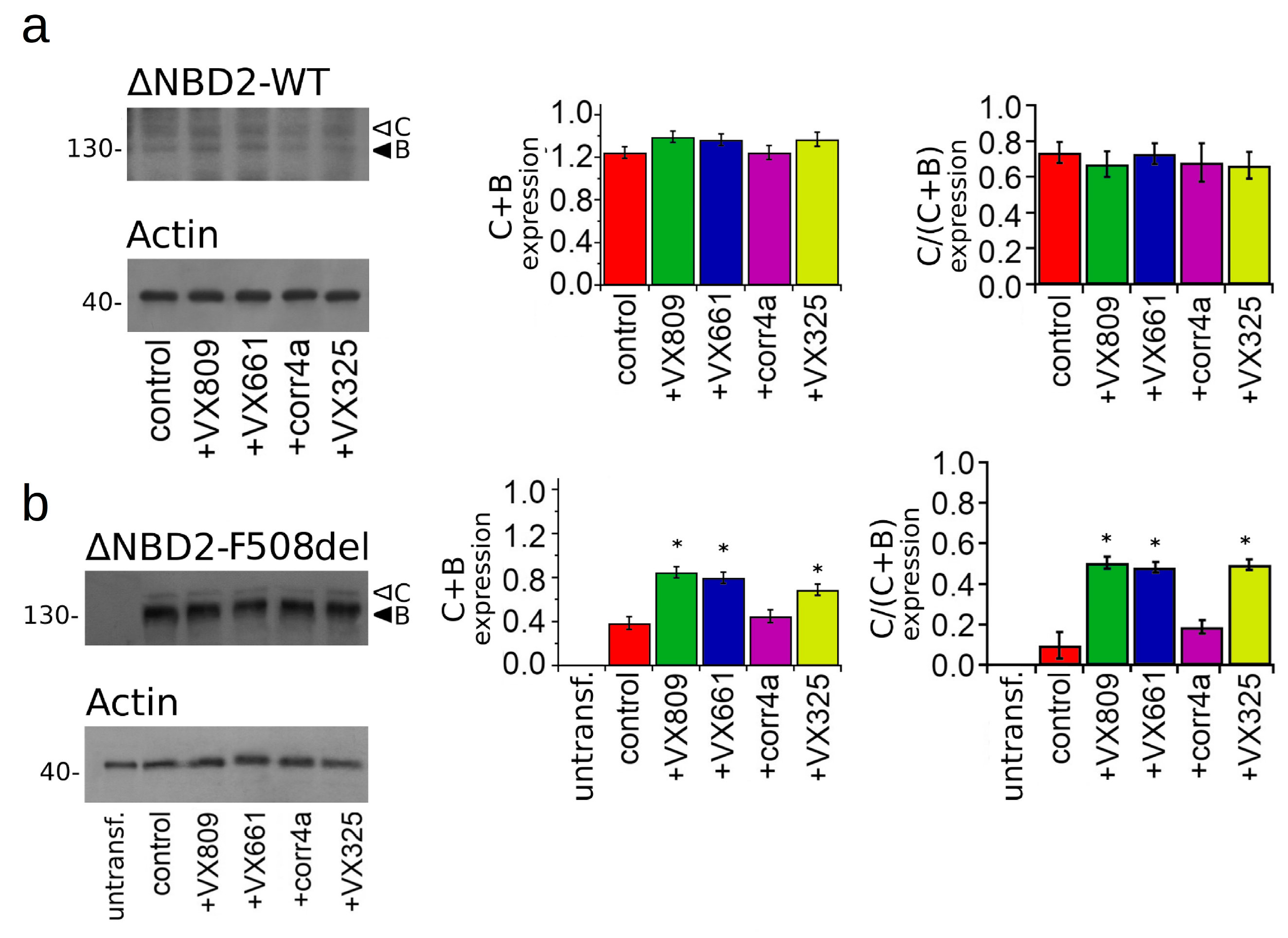
2.3. Effect of Correctors on the Expression of M1N1, M1N1 + M2N2, and ∆NBD2 Polypeptides.
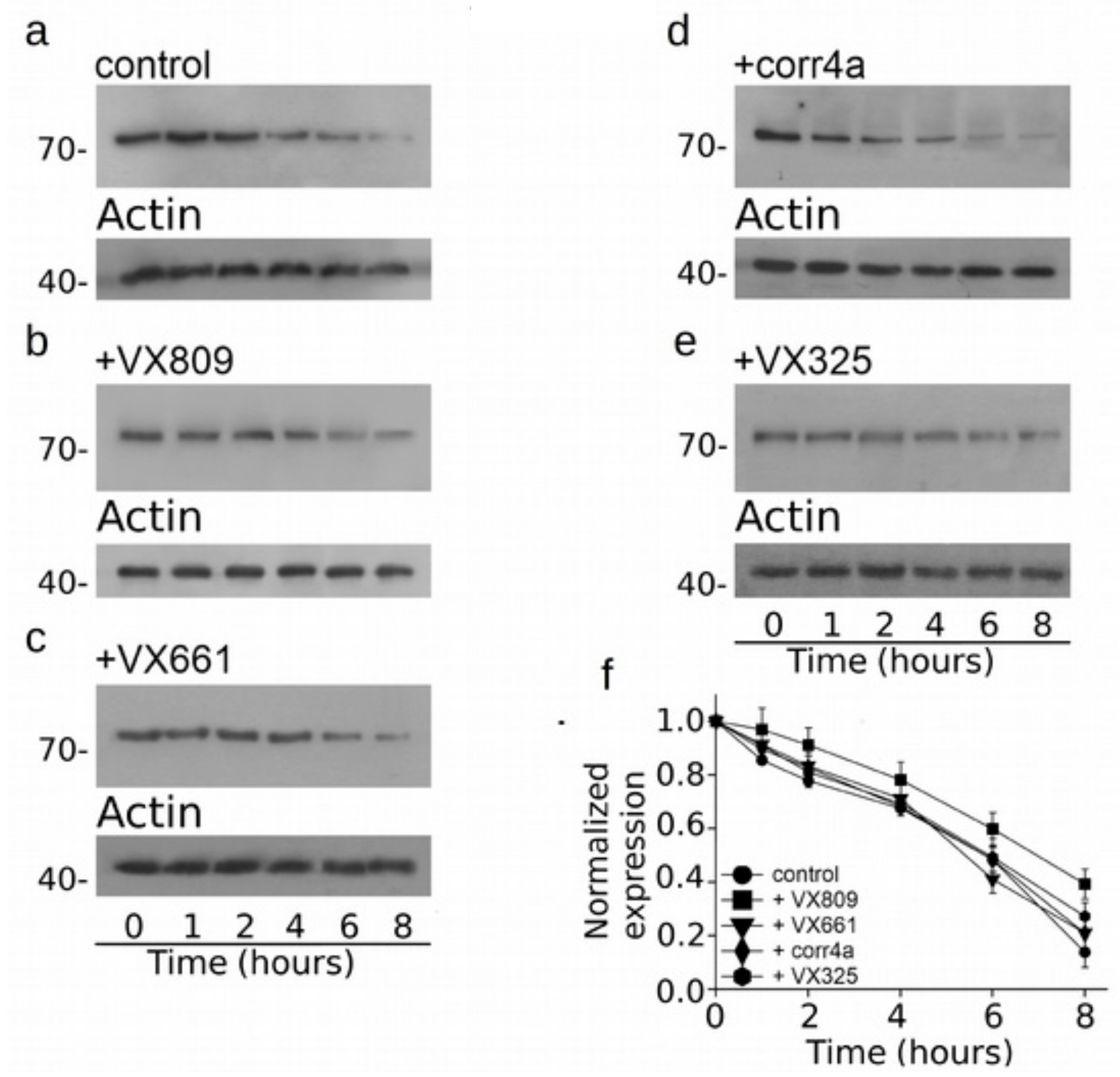
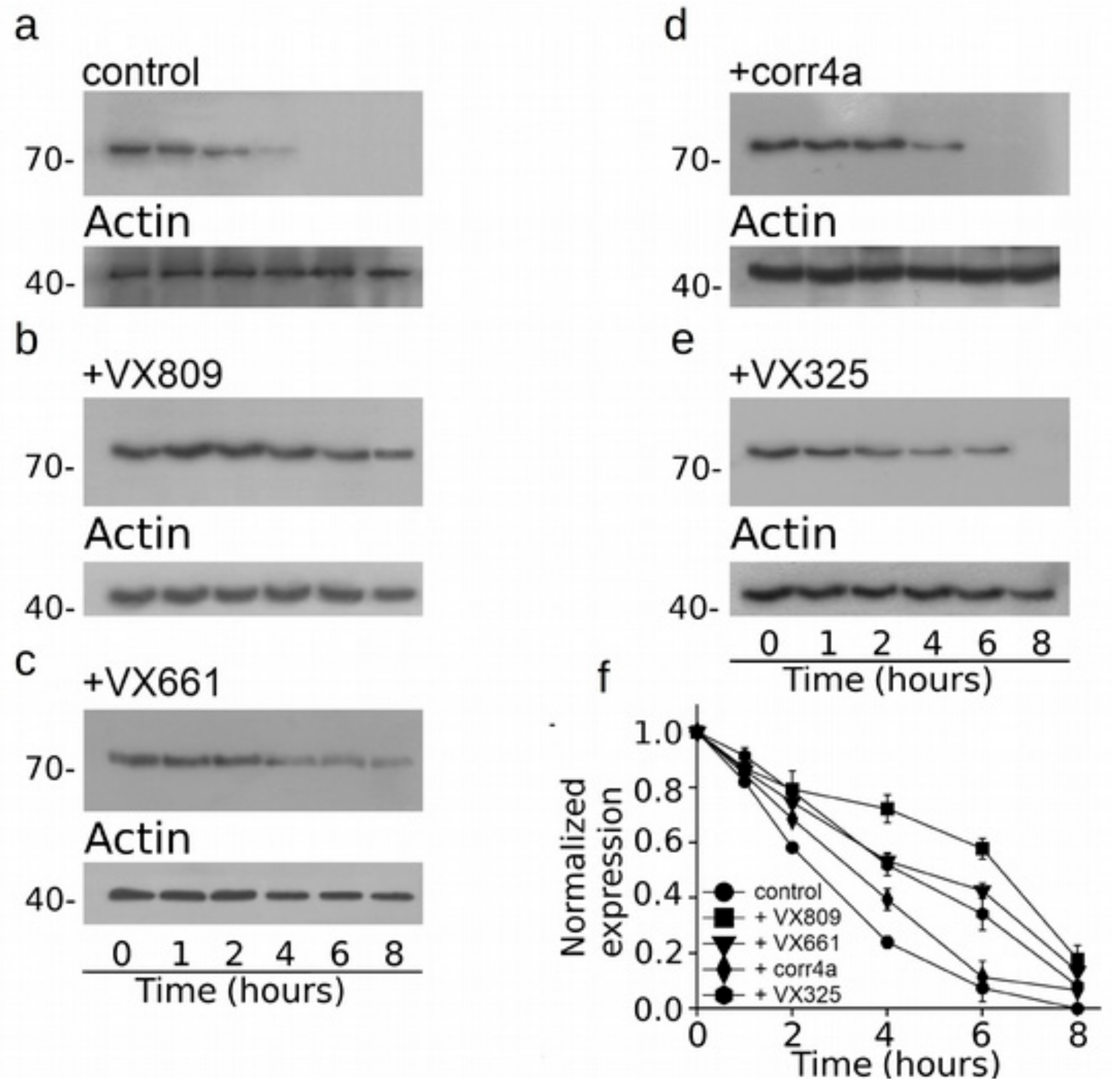
2.4. Effect of Correctors on the Stabilization of CFTR N-Half.
3. Discussion
4. Materials and Methods
4.1. Chemicals
4.2. Cell Culture
4.3. Generation and Expression of CFTR Constructs
4.4. RNA Isolation, Reverse Transcription, and Quantitative Real-Time Polymerase Chain Reaction
4.5. Western Blot
4.6. Iodide Influx Assay
4.7. Cycloheximide Chase Assay
4.8. Statistics
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Riordan, J.R.; Rommens, J.M.; Kerem, B.; Alon, N.; Rozmahel, R.; Grzelczak, Z.; Zielenski, J.; Lok, S.; Plavsic, N.; Chou, J.L.; et al. Identification of the cystic fibrosis gene: Cloning and characterization of complementary DNA. Science 1989, 245, 1066–1073. [Google Scholar] [CrossRef] [PubMed]
- Bobadilla, J.L.; Macek, M.J.; Fine, J.P.; Farrell, P.M. Cystic fibrosis: A worldwide analysis of CFTR mutations-correlation with incidence data and application to screening. Hum. Mutat. 2002, 19, 575–606. [Google Scholar] [CrossRef] [PubMed]
- Castellani, C.; Assael, B.M. Cystic fibrosis: A clinical view. Cell. Mol. Life Sci. 2017, 74, 129–140. [Google Scholar] [CrossRef]
- Moran, O. On the structural organization of the intracellular domains of CFTR. Int. J. Biochem. Cell Biol. 2014, 52, 7–14. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.J.; Skach, W.R. Mechanisms of CFTR folding at the endoplasmic reticulum. Front. Pharm. 2012, 3, 201. [Google Scholar] [CrossRef] [PubMed]
- Farinha, C.M.; Canato, S. From the endoplasmic reticulum to the plasma membrane: Mechanisms of CFTR folding and trafficking. Cell. Mol. Life Sci. 2017, 74, 39–55. [Google Scholar] [CrossRef] [PubMed]
- Farinha, C.M.; Amaral, M.D. Most F508del-CFTR is targeted to degradation at an early folding checkpoint and independently of calnexin. Mol. Cell. Biol. 2005, 25, 5242–5252. [Google Scholar] [CrossRef]
- Ward, C.L.; Kopito, R.R. Intracellular turnover of cystic fibrosis transmembrane conductance regulator. Inefficient processing and rapid degradation of wild-type and mutant proteins. J. Biol. Chem. 1994, 269, 25710–25718. [Google Scholar]
- Skach, W.R. Defects in processing and trafficking of the cystic fibrosis transmembrane conductance regulator. Kidney Int. 2000, 57, 825–831. [Google Scholar] [CrossRef] [Green Version]
- Jensen, T.J.; Loo, M.A.; Pind, S.; Williams, D.B.; Goldberg, A.L.; Riordan, J.R. Multiple proteolytic systems, including the proteasome, contribute to CFTR processing. Cell 1995, 83, 129–135. [Google Scholar] [CrossRef] [Green Version]
- Denning, G.M.; Ostedgaard, L.S.; Welsh, M.J. Abnormal localization of cystic fibrosis transmembrane conductance regulator in primary cultures of cystic fibrosis airway epithelia. J. Cell Biol. 1992, 118, 551–559. [Google Scholar] [CrossRef] [PubMed]
- Dalemans, W.; Barbry, P.; Champigny, G.; Jallat, S.; Dott, K.; Dreyer, D.; Crystal, R.G.; Pavirani, A.; Lecocq, J.P.; Lazdunski, M. Altered chloride ion channel kinetics associated with the delta F508 cystic fibrosis mutation. Nature 1991, 354, 526–528. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Zeltwanger, S.; Hu, S.; Hwang, T.C. Deletion of phenylalanine 508 causes attenuated phosphorylation-dependent activation of CFTR chloride channels. J. Physiol. (Lond.) 2000, 524, 637–648. [Google Scholar] [CrossRef] [PubMed]
- Howard, M.; Jilling, T.; DuVall, M.; Frizzell, R.A. cAMP-regulated trafficking of epitope-tagged CFTR. Kidney Int. 1996, 49, 1642–1648. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brown, C.R.; Hong-Brown, L.Q.; Biwersi, J.; Verkman, A.S.; Welch, W.J. Chemical chaperones correct the mutant phenotype of the delta F508 cystic fibrosis transmembrane conductance regulator protein. Cell Stress Chaperones 1996, 1, 117–125. [Google Scholar] [CrossRef]
- Sato, S.; Ward, C.L.; Krouse, M.E.; Wine, J.J.; Kopito, R.R. Glycerol reverses the misfolding phenotype of the most common cystic fibrosis mutation. J. Biol. Chem. 1996, 271, 635–638. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.-M.; Wang, X.-T.; Yue, H.; Leung, S.W.; Thibodeau, P.H.; Thomas, P.J.; Guggino, S.E. Organic solutes rescue the functional defect in delta F508 cystic fibrosis transmembrane conductance regulator. J. Biol. Chem. 2003, 278, 51232–51242. [Google Scholar] [CrossRef]
- Pedemonte, N.; Lukacs, G.L.; Du, K.; Caci, E.; Zegarra-Moran, O.; Galietta, L.J.; Verkman, A.S. Small-molecule correctors of defective DeltaF508-CFTR cellular processing identified by high-throughput screening. J. Clin. Invest. 2005, 115, 2564–2571. [Google Scholar] [CrossRef]
- Van Goor, F.; Straley, K.S.; Cao, D.; González, J.; Hadida, S.; Hazlewood, A.; Joubran, J.; Knapp, T.; Makings, L.R.; Miller, M.; et al. Rescue of DeltaF508-CFTR trafficking and gating in human cystic fibrosis airway primary cultures by small molecules. Am. J. Physiol. Lung Cell Mol. Physiol. 2006, 290, L1117–L1130. [Google Scholar] [CrossRef]
- Carlile, G.W.; Robert, R.; Zhang, D.; Teske, K.A.; Luo, Y.; Hanrahan, J.W.; Thomas, D.Y. Correctors of protein trafficking defects identified by a novel high-throughput screening assay. Chembiochem 2007, 8, 1012–1020. [Google Scholar] [CrossRef]
- Kalid, O.; Mense, M.; Fischman, S.; Shitrit, A.; Bihler, H.; Ben-Zeev, E.; Schutz, N.; Pedemonte, N.; Thomas, P.J.; Bridges, R.J.; et al. Small molecule correctors of F508del-CFTR discovered by structure-based virtual screening. J. Comput. Aided Mol. Des. 2010, 24, 971–991. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, S.; Sui, J.; Cotard, S.; Fung, B.; Andersen, J.; Zhu, P.; El Messadi, N.; Lehar, J.; Lee, M.; Staunton, J. Identification of synergistic combinations of F508del cystic fibrosis transmembrane conductance regulator (CFTR) modulators. Assay Drug Dev. Technol. 2010, 8, 669–684. [Google Scholar] [CrossRef] [PubMed]
- Van Goor, F.; Hadida, S.; Grootenhuis, P.D.J.; Burton, B.; Stack, J.H.; Straley, K.S.; Decker, C.J.; Miller, M.; McCartney, J.; Olson, E.R.; et al. Correction of the F508del-CFTR protein processing defect in vitro by the investigational drug VX-809. Proc. Natl. Acad. Sci. USA 2011, 108, 18843–18848. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brodlie, M.; Haq, I.J.; Roberts, K.; Elborn, J.S. Targeted therapies to improve CFTR function in cystic fibrosis. Genome Med. 2015, 7, 101. [Google Scholar] [CrossRef] [Green Version]
- Clancy, J.P.; Rowe, S.M.; Accurso, F.J.; Aitken, M.L.; Amin, R.S.; Ashlock, M.A.; Ballmann, M.; Boyle, M.P.; Bronsveld, I.; Campbell, P.W.; et al. Results of a phase IIa study of VX-809, an investigational CFTR corrector compound, in subjects with cystic fibrosis homozygous for the F508del-CFTR mutation. Thorax 2012, 67, 12–18. [Google Scholar] [CrossRef]
- Zegarra-Moran, O.; Galietta, L.J.V. CFTR pharmacology. Cell. Mol. Life Sci. 2017, 74, 117–128. [Google Scholar] [CrossRef]
- Okiyoneda, T.; Veit, G.; Dekkers, J.F.; Bagdany, M.; Soya, N.; Xu, H.; Roldan, A.; Verkman, A.S.; Kurth, M.; Simon, A.; et al. Mechanism-based corrector combination restores ΔF508-CFTR folding and function. Nat. Chem. Biol. 2013, 9, 444–454. [Google Scholar] [CrossRef]
- Veit, G.; Avramescu, R.G.; Chiang, A.N.; Houck, S.A.; Cai, Z.; Peters, K.W.; Hong, J.S.; Pollard, H.B.; Guggino, W.B.; Balch, W.E.; et al. From CFTR biology toward combinatorial pharmacotherapy: Expanded classification of cystic fibrosis mutations. Mol. Biol. Cell 2016, 27, 424–433. [Google Scholar] [CrossRef]
- Veit, G.; Xu, H.; Dreano, E.; Avramescu, R.G.; Bagdany, M.; Beitel, L.K.; Roldan, A.; Hancock, M.A.; Lay, C.; Li, W.; et al. Structure-guided combination therapy to potently improve the function of mutant CFTRs. Nat. Med. 2018, 24, 1732–1742. [Google Scholar] [CrossRef]
- Wang, Y.; Loo, T.W.; Bartlett, M.C.; Clarke, D.M. Correctors promote maturation of cystic fibrosis transmembrane conductance regulator (CFTR)-processing mutants by binding to the protein. J. Biol. Chem. 2007, 282, 33247–33251. [Google Scholar] [CrossRef]
- Wang, Y.; Loo, T.W.; Bartlett, M.C.; Clarke, D.M. Modulating the folding of P-glycoprotein and cystic fibrosis transmembrane conductance regulator truncation mutants with pharmacological chaperones. Mol. Pharm. 2007, 71, 751–758. [Google Scholar] [CrossRef] [PubMed]
- He, L.; Kota, P.; Aleksandrov, A.A.; Cui, L.; Jensen, T.; Dokholyan, N.V.; Riordan, J.R. Correctors of ΔF508 CFTR restore global conformational maturation without thermally stabilizing the mutant protein. FASEB J. 2013, 27, 536–545. [Google Scholar] [CrossRef] [PubMed]
- Loo, T.W.; Bartlett, M.C.; Clarke, D.M. Correctors enhance maturation of DeltaF508 CFTR by promoting interactions between the two halves of the molecule. Biochemistry 2009, 48, 9882–9890. [Google Scholar] [CrossRef] [PubMed]
- Loo, T.W.; Bartlett, M.C.; Clarke, D.M. Corrector VX-809 stabilizes the first transmembrane domain of CFTR. Biochem. Pharm. 2013, 86, 612–619. [Google Scholar] [CrossRef] [PubMed]
- Loo, T.W.; Bartlett, M.C.; Clarke, D.M. Bithiazole correctors rescue CFTR mutants by two different mechanisms. Biochemistry 2013, 52, 5161–5163. [Google Scholar] [CrossRef]
- Loo, T.W.; Clarke, D.M. Corrector VX-809 promotes interactions between cytoplasmic loop one and the first nucleotide-binding domain of CFTR. Biochem. Pharm. 2017, 136, 24–31. [Google Scholar] [CrossRef]
- Phuan, P.-W.; Veit, G.; Tan, J.; Roldan, A.; Finkbeiner, W.E.; Lukacs, G.L.; Verkman, A.S. Synergy-based small-molecule screen using a human lung epithelial cell line yields ΔF508-CFTR correctors that augment VX-809 maximal efficacy. Mol. Pharm. 2014, 86, 42–51. [Google Scholar] [CrossRef]
- Ren, H.Y.; Grove, D.E.; De La Rosa, O.; Houck, S.A.; Sopha, P.; Van Goor, F.; Hoffman, B.J.; Cyr, D.M. VX-809 corrects folding defects in cystic fibrosis transmembrane conductance regulator protein through action on membrane-spanning domain 1. Mol. Biol. Cell 2013, 24, 3016–3024. [Google Scholar] [CrossRef]
- Serohijos, A.W.; Hegedus, T.; Riordan, J.R.; Dokholyan, N.V. Diminished self-chaperoning activity of the DeltaF508 mutant of CFTR results in protein misfolding. PLoS Comput. Biol. 2008, 4, e1000008. [Google Scholar]
- Farinha, C.M.; King-Underwood, J.; Sousa, M.; Correia, A.R.; Henriques, B.J.; Roxo-Rosa, M.; Da Paula, A.C.; Williams, J.; Hirst, S.; Gomes, C.M.; et al. Revertants, low temperature, and correctors reveal the mechanism of F508del-CFTR rescue by VX-809 and suggest multiple agents for full correction. Chem. Biol. 2013, 20, 943–955. [Google Scholar] [CrossRef]
- Farinha, C.M.; Sousa, M.; Canato, S.; Schmidt, A.; Uliyakina, I.; Amaral, M.D. Increased efficacy of VX-809 in different cellular systems results from an early stabilization effect of F508del-CFTR. Pharm. Res. Perspect. 2015, 3, e00152. [Google Scholar] [CrossRef] [PubMed]
- Grove, D.E.; Rosser, M.F.N.; Ren, H.Y.; Naren, A.P.; Cyr, D.M. Mechanisms for rescue of correctable folding defects in CFTRDelta F508. Mol. Biol. Cell 2009, 20, 4059–4069. [Google Scholar] [CrossRef] [PubMed]
- Sampson, H.M.; Robert, R.; Liao, J.; Matthes, E.; Carlile, G.W.; Hanrahan, J.W.; Thomas, D.Y. Identification of a NBD1-binding pharmacological chaperone that corrects the trafficking defect of F508del-CFTR. Chem. Biol. 2011, 18, 231–242. [Google Scholar] [CrossRef] [PubMed]
- Yu, W.; Chiaw, P.K.; Bear, C.E. Probing conformational rescue induced by a chemical corrector of F508del-cystic fibrosis transmembrane conductance regulator (CFTR) mutant. J. Biol. Chem. 2011, 286, 24714–24725. [Google Scholar] [CrossRef] [PubMed]
- Powers, E.T.; Morimoto, R.I.; Dillin, A.; Kelly, J.W.; Balch, W.E. Biological and chemical approaches to diseases of proteostasis deficiency. Annu. Rev. Biochem. 2009, 78, 959–991. [Google Scholar] [CrossRef] [PubMed]
- Mu, T.-W.; Ong, D.S.T.; Wang, Y.-J.; Balch, W.E.; Yates, J.R.; Segatori, L.; Kelly, J.W. Chemical and biological approaches synergize to ameliorate protein-folding diseases. Cell 2008, 134, 769–781. [Google Scholar] [CrossRef] [PubMed]
- Thomas, P.; Smart, T.G. HEK293 cell line: A vehicle for the expression of recombinant proteins. J. Pharm. Toxicol Methods 2005, 51, 187–200. [Google Scholar] [CrossRef] [PubMed]
- Lemtiri-Chlieh, F.; Ali, R. Characterization of heterologously expressed transporter genes by patch- and voltage-clamp methods: Application to cyclic nucleotide-dependent responses. Methods Mol. Biol. 2013, 1016, 67–93. [Google Scholar]
- Ooi, A.; Wong, A.; Esau, L.; Lemtiri-Chlieh, F.; Gehring, C. A Guide to Transient Expression of Membrane Proteins in HEK-293 Cells for Functional Characterization. Front. Physiol. 2016, 7, 300. [Google Scholar] [CrossRef] [Green Version]
- Riordan, J.R. CFTR function and prospects for therapy. Annu. Rev. Biochem. 2008, 77, 701–726. [Google Scholar] [CrossRef]
- Proesmans, M.; Vermeulen, F.; De Boeck, K. What’s new in cystic fibrosis? From treating symptoms to correction of the basic defect. Eur. J. Pediatr. 2008, 167, 839–849. [Google Scholar] [CrossRef] [PubMed]
- Kerem, E. Pharmacologic therapy for stop mutations: How much CFTR activity is enough? Curr. Opin. Pulm. Med. 2004, 10, 547–552. [Google Scholar] [CrossRef] [PubMed]
- Ramalho, A.S.; Beck, S.; Meyer, M.; Penque, D.; Cutting, G.R.; Amaral, M.D. Five percent of normal cystic fibrosis transmembrane conductance regulator mRNA ameliorates the severity of pulmonary disease in cystic fibrosis. Am. J. Respir. Cell Mol. Biol. 2002, 27, 619–627. [Google Scholar] [CrossRef] [PubMed]
- Loo, T.W.; Bartlett, M.C.; Clarke, D.M. Rescue of DeltaF508 and other misprocessed CFTR mutants by a novel quinazoline compound. Mol. Pharm. 2005, 2, 407–413. [Google Scholar] [CrossRef] [PubMed]
- Donaldson, S.H.; Pilewski, J.M.; Griese, M.; Cooke, J.; Viswanathan, L.; Tullis, E.; Davies, J.C.; Lekstrom-Himes, J.A.; Wang, L.T. VX11-661-101 Study Group Tezacaftor/Ivacaftor in Subjects with Cystic Fibrosis and F508del/F508del-CFTR or F508del/G551D-CFTR. Am. J. Respir. Crit. Care Med. 2018, 197, 214–224. [Google Scholar] [CrossRef] [PubMed]
- Grasemann, H.; Ratjen, F. Emerging therapies for cystic fibrosis lung disease. Expert Opin. Emerg. Drugs 2010, 15, 653–659. [Google Scholar] [CrossRef] [PubMed]
- Hudson, R.P.; Dawson, J.E.; Chong, P.A.; Yang, Z.; Millen, L.; Thomas, P.J.; Brouillette, C.G.; Forman-Kay, J.D. Direct binding of the corrector VX-809 to human CFTR NBD1: Evidence of an allosteric coupling between the binding site and the NBD1:CL4 interface. Mol. Pharm. 2017, 92, 124–135. [Google Scholar] [CrossRef]
- Mense, M.; Vergani, P.; White, D.M.; Altberg, G.; Nairn, A.C.; Gadsby, D.C. In vivo phosphorylation of CFTR promotes formation of a nucleotide-binding domain heterodimer. EMBO J. 2006, 25, 4728–4739. [Google Scholar] [CrossRef]
- Chan, K.W.; Csanády, L.; Seto-Young, D.; Nairn, A.C.; Gadsby, D.C. Severed molecules functionally define the boundaries of the cystic fibrosis transmembrane conductance regulator’s NH(2)-terminal nucleotide binding domain. J. Gen. Physiol. 2000, 116, 163–180. [Google Scholar] [CrossRef]
- Csanády, L.; Chan, K.W.; Nairn, A.C.; Gadsby, D.C. Functional roles of nonconserved structural segments in CFTR’s NH2-terminal nucleotide binding domain. J. Gen. Physiol. 2005, 125, 43–55. [Google Scholar] [CrossRef]
- Rich, D.P.; Gregory, R.J.; Anderson, M.P.; Manavalan, P.; Smith, A.E.; Welsh, M.J. Effect of deleting the R domain on CFTR-generated chloride channels. Science 1991, 253, 205–207. [Google Scholar] [CrossRef] [PubMed]
- Ostedgaard, L.S.; Zabner, J.; Vermeer, D.W.; Rokhlina, T.; Karp, P.H.; Stecenko, A.A.; Randak, C.; Welsh, M.J. CFTR with a partially deleted R domain corrects the cystic fibrosis chloride transport defect in human airway epithelia in vitro and in mouse nasal mucosa in vivo. Proc. Natl. Acad. Sci. USA 2002, 99, 3093–3098. [Google Scholar] [CrossRef] [PubMed]
- Laselva, O.; Molinski, S.; Casavola, V.; Bear, C.E. The investigational Cystic Fibrosis drug Trimethylangelicin directly modulates CFTR by stabilizing the first membrane-spanning domain. Biochem. Pharm. 2016, 119, 85–92. [Google Scholar] [CrossRef] [PubMed]
- Tamanini, A.; Borgatti, M.; Finotti, A.; Piccagli, L.; Bezzerri, V.; Favia, M.; Guerra, L.; Lampronti, I.; Bianchi, N.; Dall’Acqua, F.; et al. Trimethylangelicin reduces IL-8 transcription and potentiates CFTR function. Am. J. Physiol. Lung Cell Mol. Physiol. 2011, 300, L380–L390. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Favia, M.; Mancini, M.T.; Bezzerri, V.; Guerra, L.; Laselva, O.; Abbattiscianni, A.C.; Debellis, L.; Reshkin, S.J.; Gambari, R.; Cabrini, G.; et al. Trimethylangelicin promotes the functional rescue of mutant F508del CFTR protein in cystic fibrosis airway cells. Am. J. Physiol. Lung Cell Mol. Physiol. 2014, 307, L48–L61. [Google Scholar] [CrossRef] [Green Version]
- Galietta, L.; Haggie, P.; Verkman, A. Green fluorescent protein-based halide indicators with improved chloride and iodide affinities. FEBS Lett. 2001, 499, 220–224. [Google Scholar] [CrossRef] [Green Version]
- Galietta, L.V.; Jayaraman, S.; Verkman, A.S. Cell-based assay for high-throughput quantitative screening of CFTR chloride transport agonists. Am. J. Physiol. Cell Physiol. 2001, 281, C1734–C1742. [Google Scholar] [CrossRef]
- Caci, E.; Caputo, A.; Hinzpeter, A.; Arous, N.; Fanen, P.; Sonawane, N.; Verkman, A.S.; Ravazzolo, R.; Zegarra-Moran, O.; Galietta, L.J.V. Evidence for direct CFTR inhibition by CFTR(inh)-172 based on Arg347 mutagenesis. Biochem. J. 2008, 413, 135–142. [Google Scholar] [CrossRef]








© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Amico, G.; Brandas, C.; Moran, O.; Baroni, D. Unravelling the Regions of Mutant F508del-CFTR More Susceptible to the Action of Four Cystic Fibrosis Correctors. Int. J. Mol. Sci. 2019, 20, 5463. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20215463
Amico G, Brandas C, Moran O, Baroni D. Unravelling the Regions of Mutant F508del-CFTR More Susceptible to the Action of Four Cystic Fibrosis Correctors. International Journal of Molecular Sciences. 2019; 20(21):5463. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20215463
Chicago/Turabian StyleAmico, Giulia, Chiara Brandas, Oscar Moran, and Debora Baroni. 2019. "Unravelling the Regions of Mutant F508del-CFTR More Susceptible to the Action of Four Cystic Fibrosis Correctors" International Journal of Molecular Sciences 20, no. 21: 5463. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20215463