Heat Shock Proteins in Glioblastoma Biology: Where Do We Stand?

 , and
, and
Abstract
:1. Introduction
2. HSPs Function in GBM
2.1. HSP90
2.2. HSP70
2.3. HSP60
2.4. Small HSPs (sHSPs)
2.5. Co-Chaperones
3. HSPs and Glioblastoma Stem-like Cells (GSCs)
Other Chaperones
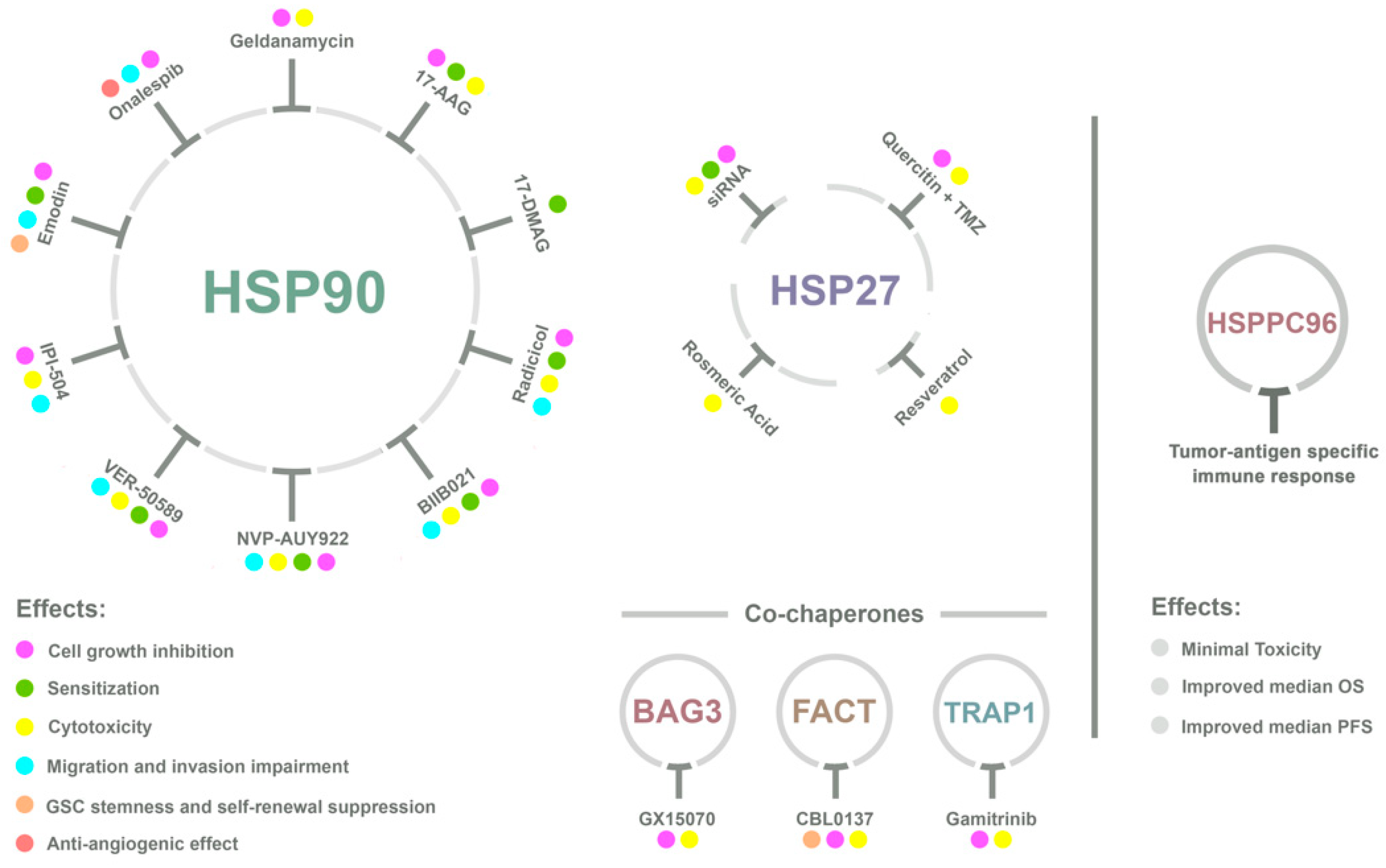
4. HSPs and GBM Therapy
4.1. HSP90
4.2. HSP70 and HSP27
4.3. HSP Vaccines Against GBM
5. Conclusions and Perspectives

{kind=link}
{kind=link}
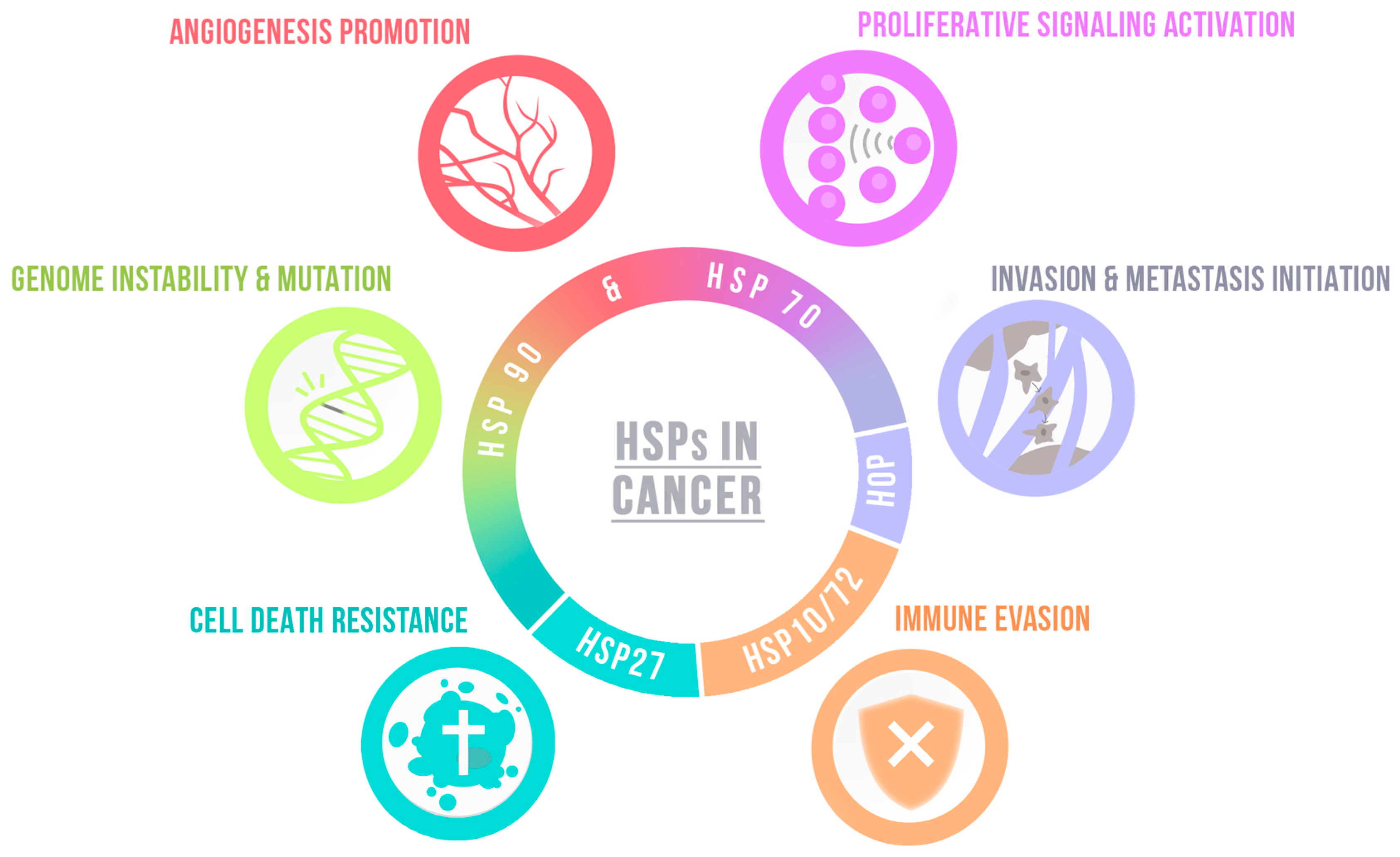
| Hallmark | Chaperone | Function | |
|---|---|---|---|
 | Angiogenesis promotion | HSP90 | Modulates HIF, consequently VEGF [100] |
| HSP47 | Promotes angiogenesis activating TGFβ signaling pathway [105] | ||
 | Proliferative signaling activation | HSC71 | Modulates cyclin D1 expression in GSCs [101] |
| HSP60 | Increases cell growth, regulates ROS production and EMT [83] | ||
| DnaJ | Increases cell growth in vivo [95] | ||
| HOP | Promotes proliferation in vitro and GBM growth in vivo [99] | ||
| Modulates self-renewal and proliferation of GSCs [104] | |||
| FACT | FACT inhibition supresses self-renewal and sternness of GSCs [106,107] | ||
| DAXX | DAXX inhibition decreases tumor growth and improves survival [108] | ||
| TRAP1 | TRAP1 depletion decreases sphere formation of GSCs and tumor growth [114,115] | ||
| Bag3 | Bag3 silencing impairs cell proliferation in vitro and in vivo [93] | ||
 | Invasion and metastasis initiation | HSP90 | Promotes cells migration and invasiveness [61,62,63,64] |
| HSC71 | HSC71 knockdown impairs nestin-dependent cell invasion [101] | ||
| HSP47 | Promotes cell migration and ECM proteins expression in GSCs [105] | ||
| DnaJ | Increase tumor invasiveness [95] | ||
| TRAP1 | TRAP1 knockdown decreases migration by disturbing the Warburg effect [115] | ||
 | Cell death resistance | HSP90 | Modulates apoptosis resistance in vivo and in vitro [65,68] |
| HSP70 | Regulates oxidative stress protection and cell survival [74] | ||
| HSPB5 | Modulates apoptosis resistance [88] | ||
| FACT | FACT inhibiticm increases apoptosis [106,107] | ||
| NPM1 | NPM1 depletion increases apoptosis and susceptibility to chemotherapy [110] | ||
| TRAP1 | TRAP1 loss impairs GSCs survival [114,115] | ||
| Bag3 | Regulates autophagy, promotes apoptosis resistance and survival in vivo [91,92,93,94] | ||
| HSP5A | HSP5A overactivation induces radio-induced apoptosis of GSCs [118] | ||
 | Genome instability & mutation | ATRX | ATRX loss leads to genetic instability and increases tumor aggressiveness [109] |
 | Immune evasion | HSPs contribute to antigen presentation—See Section 4.3. | |
Author Contributions
Funding
Conflicts of Interest
References
- Hartl, F.U.; Bracher, A.; Hayer-Hartl, M. Molecular chaperones in protein folding and proteostasis. Nature 2011, 475, 324. [Google Scholar] [CrossRef] [PubMed]
- Fernandes, C.F.d.L.; Iglesia, R.P.; Melo-Escobar, M.I.; Prado, M.B.; Lopes, M.H. Chaperones and Beyond as Key Players in Pluripotency Maintenance. Front. Cell Dev. Biol. 2019, 7. [Google Scholar] [CrossRef] [PubMed]
- Lianos, G.D.; Alexiou, G.A.; Mangano, A.; Mangano, A.; Rausei, S.; Boni, L.; Dionigi, G.; Roukos, D.H. The role of heat shock proteins in cancer. Cancer Lett. 2015, 360, 114–118. [Google Scholar] [CrossRef] [PubMed]
- Beere, H.M. The stress of dying’: The role of heat shock proteins in the regulation of apoptosis. J. Cell Sci. 2004, 117, 2641–2651. [Google Scholar] [CrossRef] [PubMed]
- Saibil, H. Chaperone machines for protein folding, unfolding and disaggregation. Nat. Rev. Mol. Cell Biol. 2013, 14, 630. [Google Scholar] [CrossRef] [PubMed]
- Ciocca, D.R.; Calderwood, S.K. Heat shock proteins in cancer: Diagnostic, prognostic, predictive, and treatment implications. Cell Stress Chaperones 2005, 10, 86. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef]
- Weinberg, R.; Hanahan, D. The hallmarks of cancer. Cell 2000, 100, 57–70. [Google Scholar]
- Schopf, F.H.; Biebl, M.M.; Buchner, J. The HSP90 chaperone machinery. Nat. Rev. Mol. Cell Biol. 2017, 18, 345. [Google Scholar] [CrossRef]
- Rappa, F.; Farina, F.; Zummo, G.; David, S.; Campanella, C.; Carini, F.; Tomasello, G.; Damiani, P.; Cappello, F.; De Macario, E.C. HSP-molecular chaperones in cancer biogenesis and tumor therapy: An overview. Anticancer Res. 2012, 32, 5139–5150. [Google Scholar]
- Sato, S.; Fujita, N.; Tsuruo, T. Modulation of Akt kinase activity by binding to Hsp90. Proc. Natl. Acad. Sci. USA 2000, 97, 10832. [Google Scholar] [CrossRef] [PubMed]
- Khan, F.; Ricks-Santi, L.; Hudson, T.; Beyene, D.; Naab, T. HSP90 Expression Associated with HER2 Overexpressing Breast Ductal Carcinomas in African American Women. Am. J. Clin. Pathol. 2016, 146. [Google Scholar] [CrossRef]
- Ahsan, A.; Ramanand, S.G.; Whitehead, C.; Hiniker, S.M.; Rehemtulla, A.; Pratt, W.B.; Jolly, S.; Gouveia, C.; Truong, K.; Waes, C.V.; et al. Wild-type EGFR Is Stabilized by Direct Interaction with HSP90 in Cancer Cells and Tumors. Neoplasia 2012, 14, 670-IN671. [Google Scholar] [CrossRef] [PubMed]
- Schulte, T.W.; Blagosklonny, M.V.; Ingui, C.; Neckers, L. Disruption of the Raf-1-Hsp90 molecular complex results in destabilization of Raf-1 and loss of Raf-1-Ras association. J. Biol. Chem. 1995, 270, 24585–24588. [Google Scholar] [CrossRef] [PubMed]
- Sherman, M.Y.; Gabai, V.L. Hsp70 in cancer: Back to the future. Oncogene 2015, 34, 4153. [Google Scholar] [CrossRef]
- Takahashi, S.; Mikami, T.; Watanabe, Y.; Okazaki, M.; Okazaki, Y.; Okazaki, A.; Sato, T.; Asaishi, K.; Hirata, K.; Narimatsu, E.; et al. Correlation of Heat Shock Protein 70 Expression with Estrogen Receptor Levels in Invasive Human Breast Cancer. Am. J. Clin. Pathol. 1994, 101, 519–525. [Google Scholar] [CrossRef]
- Blagosklonny, M.V.; Toretsky, J.; Bohen, S.; Neckers, L. Mutant conformation of p53 translated in vitro or in vivo requires functional HSP90. Proc. Natl. Acad. Sci. USA 1996, 93, 8379–8383. [Google Scholar] [CrossRef]
- Peng, Y.; Chen, L.; Li, C.; Lu, W.; Chen, J. Inhibition of MDM2 by hsp90 contributes to mutant p53 stabilization. J. Biol. Chem. 2001, 276, 40583–40590. [Google Scholar] [CrossRef]
- Wiech, M.; Olszewski, M.B.; Tracz-Gaszewska, Z.; Wawrzynow, B.; Zylicz, M.; Zylicz, A. Molecular mechanism of mutant p53 stabilization: The role of HSP70 and MDM2. PLoS ONE 2012, 7, e51426. [Google Scholar] [CrossRef]
- Khajapeer, K.V.; Baskaran, R. Hsp90 inhibitors for the treatment of chronic myeloid leukemia. Leuk. Res. Treat. 2015, 2015, 757694. [Google Scholar] [CrossRef]
- Kasibhatla, S.; Tseng, B. Why target apoptosis in cancer treatment? Mol. Cancer Ther. 2003, 2, 573–580. [Google Scholar] [PubMed]
- Redlak, M.J.; Miller, T.A. Targeting PI3K/Akt/HSP90 signaling sensitizes gastric cancer cells to deoxycholate-induced apoptosis. Dig. Dis. Sci. 2011, 56, 323–329. [Google Scholar] [CrossRef] [PubMed]
- Pandey, P.; Saleh, A.; Nakazawa, A.; Kumar, S.; Srinivasula, S.M.; Kumar, V.; Weichselbaum, R.; Nalin, C.; Alnemri, E.S.; Kufe, D.; et al. Negative regulation of cytochrome c-mediated oligomerization of Apaf-1 and activation of procaspase-9 by heat shock protein 90. EMBO J. 2000, 19, 4310–4322. [Google Scholar] [CrossRef] [PubMed]
- Stankiewicz, A.R.; Lachapelle, G.; Foo, C.P.; Radicioni, S.M.; Mosser, D.D. Hsp70 inhibits heat-induced apoptosis upstream of mitochondria by preventing Bax translocation. J. Biol. Chem. 2005, 280, 38729–38739. [Google Scholar] [CrossRef]
- Xanthoudakis, S.; Roy, S.; Rasper, D.; Hennessey, T.; Aubin, Y.; Cassady, R.; Tawa, P.; Ruel, R.; Rosen, A.; Nicholson, D.W. Hsp60 accelerates the maturation of pro-caspase-3 by upstream activator proteases during apoptosis. EMBO J. 1999, 18, 2049–2056. [Google Scholar] [CrossRef]
- Mosser, D.D.; Caron, A.W.; Bourget, L.; Meriin, A.B.; Sherman, M.Y.; Morimoto, R.I.; Massie, B. The Chaperone Function of hsp70 Is Required for Protection against Stress-Induced Apoptosis. Mol. Cell Biol. 2000, 20, 7146. [Google Scholar] [CrossRef]
- Tezel, G.; Wax, M.B. The Mechanisms of hsp27 Antibody-Mediated Apoptosis in Retinal Neuronal Cells. J. Neurosci. 2000, 20, 3552. [Google Scholar] [CrossRef]
- GARRIDO, C.; BRUEY, J.-M.; FROMENTIN, A.; HAMMANN, A.; ARRIGO, A.P.; SOLARY, E. HSP27 inhibits cytochrome c-dependent activation of procaspase-9. FASEB J. 1999, 13, 2061–2070. [Google Scholar] [CrossRef]
- Zhou, J.; Schmid, T.; Frank, R.; Brüne, B. PI3K/Akt is required for heat shock proteins to protect hypoxia-inducible factor 1α from pVHL-independent degradation. J. Biol. Chem. 2004, 279, 13506–13513. [Google Scholar] [CrossRef]
- Guo, W.; Yang, Z.; Xia, Q.; Liu, J.; Yu, Y.; Li, J.; Zuo, Z.; Zhang, D.; Li, X.; Shi, X.; et al. Arsenite stabilizes HIF-1α protein through p85α-mediated up-regulation of inducible Hsp70 protein expression. Cell Mol. Life Sci. 2011, 68, 475–488. [Google Scholar] [CrossRef]
- Minet, E.; Mottet, D.; Michel, G.; Roland, I.; Raes, M.; Remacle, J.; Michiels, C. Hypoxia-induced activation of HIF-1: Role of HIF-1α-Hsp90 interaction. FEBS Lett. 1999, 460, 251–256. [Google Scholar] [CrossRef]
- Eschricht, S.; Jarr, K.-U.; Kuhn, C.; Lehmann, L.; Kreusser, M.; Katus, H.A.; Frey, N.; Chorianopoulos, E. Heat-shock-protein 90 protects from downregulation of HIF-1α in calcineurin-induced myocardial hypertrophy. J. Mol. Cell Cardiol. 2015, 85, 117–126. [Google Scholar] [CrossRef] [PubMed]
- Muenst, S.; Laubli, H.; Soysal, S.D.; Zippelius, A.; Tzankov, A.; Hoeller, S. The immune system and cancer evasion strategies: Therapeutic concepts. J. Intern. Med. 2016, 279, 541–562. [Google Scholar] [CrossRef] [PubMed]
- Akyol, S.; Gercel-Taylor, C.; Reynolds, L.C.; Taylor, D.D. HSP-10 in ovarian cancer: Expression and suppression of T-cell signaling. Gynecol. Oncol. 2006, 101, 481–486. [Google Scholar] [CrossRef]
- Stocki, P.; Wang, X.N.; Dickinson, A.M. Inducible heat shock protein 70 reduces T cell responses and stimulatory capacity of monocyte-derived dendritic cells. J. Biol. Chem. 2012, 287, 12387–12394. [Google Scholar] [CrossRef] [Green Version]
- Shevtsov, M.; Multhoff, G. Heat shock protein–peptide and HSP-based immunotherapies for the treatment of cancer. Front. Immunol. 2016, 7, 171. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.; Huang, W. Fusion proteins of Hsp70 with tumor-associated antigen acting as a potent tumor vaccine and the C-terminal peptide-binding domain of Hsp70 being essential in inducing antigen-independent anti-tumor response in vivo. Cell Stress Chaperones 2006, 11, 216–226. [Google Scholar] [CrossRef]
- Hantschel, M.; Pfister, K.; Jordan, A.; Scholz, R.; Andreesen, R.; Schmitz, G.; Schmetzer, H.; Hiddemann, W.; Multhoff, G. Hsp70 plasma membrane expression on primary tumor biopsy material and bone marrow of leukemic patients. Cell Stress Chaperones 2000, 5, 438–442. [Google Scholar] [CrossRef] [Green Version]
- Becker, B.; Multhoff, G.; Farkas, B.; Wild, P.J.; Landthaler, M.; Stolz, W.; Vogt, T. Induction of Hsp90 protein expression in malignant melanomas and melanoma metastases. Exp. Dermatol. 2004, 13, 27–32. [Google Scholar] [CrossRef]
- Ethun, C.G.; Postlewait, L.M.; Lopez-Aguiar, A.G.; Zhelnin, K.; Krasinskas, A.; El-Rayes, B.F.; Russell, M.C.; Kooby, D.A.; Staley, C.A.; Cardona, K. HSP90 expression and early recurrence in gastroenteropancreatic neuroendocrine tumors: Potential for novel therapeutic targets. Am. Soc. Clin. Oncol. 2017. [Google Scholar] [CrossRef]
- Kalluri, R.; Weinberg, R.A. The basics of epithelial-mesenchymal transition. J. Clin. Investig. 2009, 119, 1420–1428. [Google Scholar] [CrossRef] [Green Version]
- Eustace, B.K.; Sakurai, T.; Stewart, J.K.; Yimlamai, D.; Unger, C.; Zehetmeier, C.; Lain, B.; Torella, C.; Henning, S.W.; Beste, G. Functional proteomic screens reveal an essential extracellular role for hsp90α in cancer cell invasiveness. Nat. Cell Biol. 2004, 6, 507. [Google Scholar] [CrossRef]
- Armstrong, H.K.; Gillis, J.L.; Johnson, I.R.; Nassar, Z.D.; Moldovan, M.; Levrier, C.; Sadowski, M.C.; Chin, M.Y.; Guns, E.S.T.; Tarulli, G. Dysregulated fibronectin trafficking by Hsp90 inhibition restricts prostate cancer cell invasion. Sci. Rep. 2018, 8, 2090. [Google Scholar] [CrossRef]
- Teng, Y.; Ngoka, L.; Mei, Y.; Lesoon, L.; Cowell, J.K. HSP90 and HSP70 proteins are essential for stabilization and activation of WASF3 metastasis-promoting protein. J. Biol. Chem. 2012, 287, 10051–10059. [Google Scholar] [CrossRef] [Green Version]
- Walsh, N.; Larkin, A.; Swan, N.; Conlon, K.; Dowling, P.; McDermott, R.; Clynes, M. RNAi knockdown of Hop (Hsp70/Hsp90 organising protein) decreases invasion via MMP-2 down regulation. Cancer Lett. 2011, 306, 180–189. [Google Scholar] [CrossRef] [Green Version]
- Alexander, B.M.; Cloughesy, T.F. Adult glioblastoma. J. Clin. Oncol. 2017, 35, 2402–2409. [Google Scholar] [CrossRef]
- Holland, E.C. Glioblastoma multiforme: The terminator. Proc. Natl. Acad. Sci. USA 2000, 97, 6242–6244. [Google Scholar] [CrossRef] [Green Version]
- Piccirillo, S.; Reynolds, B.A.; Zanetti, N.; Lamorte, G.; Binda, E.; Broggi, G.; Brem, H.; Olivi, A.; Dimeco, F.; Vescovi, A.L. Bone morphogenetic proteins inhibit the tumorigenic potential of human brain tumour-initiating cells. Nature 2006, 444, 761. [Google Scholar] [CrossRef]
- McLendon, R.E.; Rich, J.N. Glioblastoma stem cells: A neuropathologist’s view. J. Oncol. 2011, 2011. [Google Scholar] [CrossRef]
- Lathia, J.D.; Mack, S.C.; Mulkearns-Hubert, E.E.; Valentim, C.L.; Rich, J.N. Cancer stem cells in glioblastoma. Genes Dev. 2015, 29, 1203–1217. [Google Scholar] [CrossRef] [Green Version]
- Fidoamore, A.; Cristiano, L.; Antonosante, A.; d’Angelo, M.; Di Giacomo, E.; Astarita, C.; Giordano, A.; Ippoliti, R.; Benedetti, E.; Cimini, A. Glioblastoma stem cells microenvironment: The paracrine roles of the niche in drug and radioresistance. Stem Cells Int. 2016, 2016. [Google Scholar] [CrossRef] [Green Version]
- Persano, L.; Rampazzo, E.; Della Puppa, A.; Pistollato, F.; Basso, G. The three-layer concentric model of glioblastoma: Cancer stem cells, microenvironmental regulation, and therapeutic implications. Sci. World J. 2011, 11, 1829–1841. [Google Scholar] [CrossRef]
- Xu, H.S.; Qin, X.L.; Zong, H.L.; He, X.G.; Cao, L. Cancer stem cell markers in glioblastoma - an update. Eur. Rev. Med. Pharmacol. Sci. 2017, 21, 3207–3211. [Google Scholar]
- Guerra-Rebollo, M.; Garrido, C.; Sánchez-Cid, L.; Soler-Botija, C.; Meca-Cortés, O.; Rubio, N.; Blanco, J. Targeting of replicating CD133 and OCT4/SOX2 expressing glioma stem cells selects a cell population that reinitiates tumors upon release of therapeutic pressure. Sci. Rep. 2019, 9, 9549. [Google Scholar] [CrossRef]
- Stupp, R.; Mason, W.P.; van den Bent, M.J.; Weller, M.; Fisher, B.; Taphoorn, M.J.; Belanger, K.; Brandes, A.A.; Marosi, C.; Bogdahn, U.; et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N. Engl. J. Med. 2005, 352, 987–996. [Google Scholar] [CrossRef]
- Stupp, R.; Brada, M.; van den Bent, M.J.; Tonn, J.-C.; Pentheroudakis, G.E.S.M.O. High-grade glioma: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann. Oncol. 2014, 25, iii93–iii101. [Google Scholar] [CrossRef]
- Lee, C.Y. Strategies of temozolomide in future glioblastoma treatment. OncoTargets Ther. 2017, 10, 265. [Google Scholar] [CrossRef] [Green Version]
- Ban, H.S.; Han, T.-S.; Hur, K.; Cho, H.-S. Epigenetic Alterations of Heat Shock Proteins (HSPs) in Cancer. Int. J. Mol. Sci. 2019, 20, 4758. [Google Scholar] [CrossRef] [Green Version]
- Hoter, A.; Naim, H.Y. Heat Shock Proteins and Ovarian Cancer: Important Roles and Therapeutic Opportunities. Cancers 2019, 11, 1389. [Google Scholar] [CrossRef] [Green Version]
- Yang, I.; Fang, S.; Parsa, A.T. Heat shock proteins in glioblastomas. Neurosurg. Clin. 2010, 21, 111–123. [Google Scholar] [CrossRef]
- Memmel, S.; Sisario, D.; Zöller, C.; Fiedler, V.; Katzer, A.; Heiden, R.; Becker, N.; Eing, L.; Ferreira, F.L.; Zimmermann, H. Migration pattern, actin cytoskeleton organization and response to PI3K-, mTOR-, and Hsp90-inhibition of glioblastoma cells with different invasive capacities. Oncotarget 2017, 8, 45298. [Google Scholar] [CrossRef] [PubMed]
- Gopal, U.; Bohonowych, J.E.; Lema-Tome, C.; Liu, A.; Garrett-Mayer, E.; Wang, B.; Isaacs, J.S. A novel extracellular Hsp90 mediated co-receptor function for LRP1 regulates EphA2 dependent glioblastoma cell invasion. PLoS ONE 2011, 6, e17649. [Google Scholar] [CrossRef] [PubMed]
- Thuringer, D.; Hammann, A.; Benikhlef, N.; Fourmaux, E.; Bouchot, A.; Wettstein, G.; Solary, E.; Garrido, C. Transactivation of the epidermal growth factor receptor by heat shock protein 90 via Toll-like receptor 4 contributes to the migration of glioblastoma cells. J. Biol. Chem. 2011, 286, 3418–3428. [Google Scholar] [CrossRef] [Green Version]
- Snigireva, A.V.; Vrublevskaya, V.V.; Skarga, Y.Y.; Morenkov, O.S. Cell surface heparan sulfate proteoglycans are involved in the extracellular Hsp90-stimulated migration and invasion of cancer cells. Cell Stress Chaperones 2019, 24, 309–322. [Google Scholar] [CrossRef]
- Wachsberger, P.R.; Lawrence, Y.R.; Liu, Y.; Rice, B.; Feo, N.; Leiby, B.; Dicker, A.P. Hsp90 inhibition enhances PI-3 kinase inhibition and radiosensitivity in glioblastoma. J. Cancer Res. Clin. Oncol. 2014, 140, 573–582. [Google Scholar] [CrossRef]
- Deng, X.; Song, L.; Zhao, W.; Wei, Y.; Guo, X.-b. HAX-1 protects glioblastoma cells from apoptosis through the Akt1 pathway. Front. Cell. Neurosci. 2017, 11, 420. [Google Scholar] [CrossRef] [Green Version]
- Fadeel, B.; Grzybowska, E. HAX-1: A multifunctional protein with emerging roles in human disease. Biochim. Biophys. Acta (BBA) Gen. Subj. 2009, 1790, 1139–1148. [Google Scholar] [CrossRef]
- Lam, C.K.; Zhao, W.; Cai, W.; Vafiadaki, E.; Florea, S.M.; Ren, X.; Liu, Y.; Robbins, N.; Zhang, Z.; Zhou, X. Novel role of HAX-1 in ischemic injury protection involvement of heat shock protein 90. Circ. Res. 2013, 112, 79–89. [Google Scholar] [CrossRef] [Green Version]
- Furnari, F.B.; Cloughesy, T.F.; Cavenee, W.K.; Mischel, P.S. Heterogeneity of epidermal growth factor receptor signalling networks in glioblastoma. Nat. Rev. Cancer 2015, 15, 302–310. [Google Scholar] [CrossRef] [Green Version]
- Sartori, E.; Langer, R.; Vassella, E.; Hewer, E.; Schucht, P.; Zlobec, I.; Berezowska, S. Low co-expression of epidermal growth factor receptor and its chaperone heat shock protein 90 is associated with worse prognosis in primary glioblastoma, IDH-wild-type. Oncol. Rep. 2017, 38, 2394–2400. [Google Scholar] [CrossRef] [Green Version]
- Kumar, S.; Stokes III, J.; Singh, U.P.; Gunn, K.S.; Acharya, A.; Manne, U.; Mishra, M. Targeting Hsp70: A possible therapy for cancer. Cancer Lett. 2016, 374, 156–166. [Google Scholar] [CrossRef] [Green Version]
- Boudesco, C.; Cause, S.; Jego, G.; Garrido, C. Hsp70: A cancer target inside and outside the cell. In Chaperones. Methods in Molecular Biology; Calderwood, S., Prince, T., Eds.; Humana Press: New York, NY, USA, 2018; vol. 1709, pp. 371–396. [Google Scholar]
- Thorsteinsdottir, J.; Stangl, S.; Fu, P.; Guo, K.; Albrecht, V.; Eigenbrod, S.; Erl, J.; Gehrmann, M.; Tonn, J.-C.; Multhoff, G. Overexpression of cytosolic, plasma membrane bound and extracellular heat shock protein 70 (Hsp70) in primary glioblastomas. J. Neuro Oncol. 2017, 135, 443–452. [Google Scholar] [CrossRef]
- Lazarev, V.F.; Nikotina, A.D.; Mikhaylova, E.R.; Nudler, E.; Polonik, S.G.; Guzhova, I.V.; Margulis, B.A. Hsp70 chaperone rescues C6 rat glioblastoma cells from oxidative stress by sequestration of aggregating GAPDH. Biochem. Biophys. Res. Commun. 2016, 470, 766–771. [Google Scholar] [CrossRef]
- Reindl, J.; Shevtsov, M.; Dollinger, G.; Stangl, S.; Multhoff, G. Membrane Hsp70-supported cell-to-cell connections via tunneling nanotubes revealed by live-cell STED nanoscopy. Cell Stress Chaperones 2019, 24, 213–221. [Google Scholar] [CrossRef] [Green Version]
- Ibrahim, I.M.; Abdelmalek, D.H.; Elfiky, A.A. GRP78: A cell’s response to stress. Life Sci. 2019. [Google Scholar] [CrossRef]
- Lee, A.S. Glucose-regulated proteins in cancer: Molecular mechanisms and therapeutic potential. Nat. Rev. Cancer 2014, 14, 263. [Google Scholar] [CrossRef] [Green Version]
- Viswanath, A.N.I.; Lim, J.W.; Seo, S.H.; Lee, J.Y.; Lim, S.M.; Pae, A.N. GRP78-targeted in-silico virtual screening of novel anticancer agents. Chem. Biol. Drug Des. 2018, 92, 1555–1566. [Google Scholar] [CrossRef]
- Horwich, A.L.; Willison, K.R. Protein folding in the cell: Functions of two families of molecular chaperone, hsp 60 and TF55-TCP1. Philos. Trans. R. Soc. Lond. Ser. B Biol. Sci. 1993, 339, 313–325, discussion 325–316. [Google Scholar] [CrossRef]
- Marino Gammazza, A.; Macaluso, F.; di Felice, V.; Cappello, F.; Barone, R. Hsp60 in skeletal muscle fiber biogenesis and homeostasis: From physical exercise to skeletal muscle pathology. Cells 2018, 7, 224. [Google Scholar] [CrossRef] [Green Version]
- Ekspresyonu, B.T.I.Ş.P. Expression of heat shock proteins in brain tumors. Turk. Neurosurg 2014, 24, 745–749. [Google Scholar]
- Ghosh, J.C.; Siegelin, M.D.; Dohi, T.; Altieri, D.C. Heat shock protein 60 regulation of the mitochondrial permeability transition pore in tumor cells. Cancer Res. 2010, 70, 8988–8993. [Google Scholar] [CrossRef] [Green Version]
- Tang, H.; Li, J.; Liu, X.; Wang, G.; Luo, M.; Deng, H. Down-regulation of HSP60 Suppresses the Proliferation of Glioblastoma Cells via the ROS/AMPK/mTOR Pathway. Sci. Rep. 2016, 6, 28388. [Google Scholar] [CrossRef] [Green Version]
- Carra, S.; Alberti, S.; Arrigo, P.A.; Benesch, J.L.; Benjamin, I.J.; Boelens, W.; Bartelt-Kirbach, B.; Brundel, B.; Buchner, J.; Bukau, B.; et al. The growing world of small heat shock proteins: From structure to functions. Cell Stress Chaperones 2017, 22, 601–611. [Google Scholar] [CrossRef]
- Zhenying, L.; Shengnan, Z.; Dan, L.; Cong, L. A Structural View of αB-crystallin Assembly and Amyloid Aggregation. Protein Pept. Lett. 2017, 24, 315–321. [Google Scholar] [CrossRef]
- Choi, S.K.; Kam, H.; Kim, K.Y.; Park, S.I.; Lee, Y.S. Targeting Heat Shock Protein 27 in Cancer: A Druggable Target for Cancer Treatment? Cancers (Basel) 2019, 11, 1195. [Google Scholar] [CrossRef] [Green Version]
- Kato, K.; Goto, S.; Hasegawa, K.; Shinohara, H.; Inaguma, Y. Responses to heat shock of αB crystallin and HSP28 in U373 MG human glioma cells. Biochim. Biophys. Acta (BBA) Mol. Cell Res. 1993, 1175, 257–262. [Google Scholar] [CrossRef]
- Hermisson, M.; Strik, H.; Rieger, J.; Dichgans, J.; Meyermann, R.; Weller, M. Expression and functional activity of heat shock proteins in human glioblastoma multiforme. Neurology 2000, 54, 1357. [Google Scholar] [CrossRef]
- Bohush, A.; Bieganowski, P. Hsp90 and Its Co-Chaperones in Neurodegenerative Diseases. Int. J. Mol. Sci. 2019, 20, 4976. [Google Scholar] [CrossRef] [Green Version]
- Joshi, S.; Wang, T.; Araujo, T.L.S.; Sharma, S.; Brodsky, J.L.; Chiosis, G. Adapting to stress — chaperome networks in cancer. Nat. Rev. Cancer 2018, 18, 562–575. [Google Scholar] [CrossRef]
- Festa, M.; Del Valle, L.; Khalili, K.; Franco, R.; Scognamiglio, G.; Graziano, V.; De Laurenzi, V.; Turco, M.C.; Rosati, A. BAG3 protein is overexpressed in human glioblastoma and is a potential target for therapy. Am. J. Pathol. 2011, 178, 2504–2512. [Google Scholar] [CrossRef] [Green Version]
- Merabova, N.; Sariyer, I.K.; Saribas, A.S.; Knezevic, T.; Gordon, J.; Turco, M.C.; Rosati, A.; Weaver, M.; Landry, J.; Khalili, K. WW Domain of BAG3 Is Required for the Induction of Autophagy in Glioma Cells. J. Cell Physiol. 2015, 230, 831–841. [Google Scholar] [CrossRef] [Green Version]
- Karpel-Massler, G.; Shu, C.; Chau, L.; Banu, M.; Halatsch, M.-E.; Westhoff, M.-A.; Ramirez, Y.; Ross, A.H.; Bruce, J.N.; Canoll, P.; et al. Combined inhibition of Bcl-2/Bcl-xL and Usp9X/Bag3 overcomes apoptotic resistance in glioblastoma in vitro and in vivo. Oncotarget 2015, 6, 14507. [Google Scholar] [CrossRef] [Green Version]
- Antonietti, P.; Linder, B.; Hehlgans, S.; Mildenberger, I.C.; Burger, M.C.; Fulda, S.; Steinbach, J.P.; Gessler, F.; Rodel, F.; Mittelbronn, M.; et al. Interference with the HSF1/HSP70/BAG3 pathway primes glioma cells to matrix detachment and BH3 mimetic-induced apoptosis. Mol. Cancer Ther. 2016, 16, 156–168. [Google Scholar] [CrossRef] [Green Version]
- Meshalkina, D.A.; Shevtsov, M.A.; Dobrodumov, A.V.; Komarova, E.Y.; Voronkina, I.V.; Lazarev, V.F.; Margulis, B.A.; Guzhova, I.V. Knock-down of Hdj2/DNAJA1 co-chaperone results in an unexpected burst of tumorigenicity of C6 glioblastoma cells. Oncotarget 2016, 7, 22050. [Google Scholar] [CrossRef] [Green Version]
- Erlich, R.B.; Kahn, S.A.; Lima, F.R.S.; Muras, A.G.; Martins, R.A.P.; Linden, R.; Chiarini, L.B.; Martins, V.R.; Moura Neto, V. STI1 promotes glioma proliferation through MAPK and PI3K pathways. Glia 2007, 55, 1690–1698. [Google Scholar] [CrossRef]
- Soares, I.N.; Caetano, F.A.; Pinder, J.; Roz Rodrigues, B.; Beraldo, F.H.; Ostapchenko, V.G.; Durette, C.; Schenatto Pereira, G.; Lopes, M.H.; Queiroz-Hazarbassanov, N.; et al. Regulation of stress-inducible phosphoprotein 1 nuclear retention by PIAS1. Mol. Cell. Proteom. 2013. [Google Scholar] [CrossRef] [Green Version]
- Horibe, T.; Torisawa, A.; Kohno, M.; Kawakami, K. Molecular mechanism of cytotoxicity induced by Hsp90-targeted Antp-TPR hybrid peptide in glioblastoma cells. Mol. Cancer 2012, 11, 59. [Google Scholar] [CrossRef] [Green Version]
- Lopes, M.H.; Santos, T.G.; Rodrigues, B.R.; Queiroz-Hazarbassanov, N.; Cunha, I.W.; Wasilewska-Sampaio, A.P.; Costa-Silva, B.; Marchi, F.A.; Bleggi-Torres, L.F.; Sanematsu, P.I.; et al. Disruption of prion protein-HOP engagement impairs glioblastoma growth and cognitive decline and improves overall survival. Oncogene 2015, 34, 3305–3314. [Google Scholar] [CrossRef]
- Filatova, A.; Seidel, S.; Böğürcü, N.; Gräf, S.; Garvalov, B.K.; Acker, T. Acidosis Acts through HSP90 in a PHD/VHL-Independent Manner to Promote HIF Function and Stem Cell Maintenance in Glioma. Cancer Res. 2016, 76, 5845. [Google Scholar] [CrossRef] [Green Version]
- Matsuda, Y.; Ishiwata, T.; Yoshimura, H.; Hagio, M.; Arai, T. Inhibition of nestin suppresses stem cell phenotype of glioblastomas through the alteration of post-translational modification of heat shock protein HSPA8/HSC71. Cancer Lett. 2015, 357, 602–611. [Google Scholar] [CrossRef]
- Ronci, M.; Catanzaro, G.; Pieroni, L.; Po, A.; Besharat, Z.M.; Greco, V.; Levi Mortera, S.; Screpanti, I.; Ferretti, E.; Urbani, A. Proteomic analysis of human sonic hedgehog (SHH) medulloblastoma stem-like cells. Mol. Biosyst. 2015, 11, 1603–1611. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salimi, L.; Rahbarghazi, R.; Jafarian, V.; Biray Avci, Ç.; Goker Bagca, B.; Pinar Ozates, N.; Khaksar, M.; Nourazarian, A. Heat shock protein 70 modulates neural progenitor cells dynamics in human neuroblastoma SH-SY5Y cells exposed to high glucose content. J. Cell. Biochem. 2018, 119, 6482–6491. [Google Scholar] [CrossRef] [PubMed]
- Iglesia, R.P.; Prado, M.B.; Cruz, L.; Martins, V.R.; Santos, T.G.; Lopes, M.H. Engagement of cellular prion protein with the co-chaperone Hsp70/90 organizing protein regulates the proliferation of glioblastoma stem-like cells. Stem Cell Res. Ther. 2017, 8, 76. [Google Scholar] [CrossRef] [PubMed]
- Jiang, X.; Zhou, T.; Wang, Z.; Qi, B.; Xia, H. HSP47 Promotes Glioblastoma Stemlike Cell Survival by Modulating Tumor Microenvironment Extracellular Matrix through TGF-β Pathway. ACS Chem. Neurosci. 2017, 8, 128–134. [Google Scholar] [CrossRef]
- Dermawan, J.K.T.; Hitomi, M.; Silver, D.J.; Wu, Q.; Sandlesh, P.; Sloan, A.E.; Purmal, A.A.; Gurova, K.V.; Rich, J.N.; Lathia, J.D.; et al. Pharmacological targeting of the histone chaperone complex FACT preferentially eliminates glioblastoma stem cells and prolongs survival in preclinical models. Cancer Res. 2016. [Google Scholar] [CrossRef] [Green Version]
- Barone, T.A.; Burkhart, C.A.; Safina, A.; Haderski, G.; Gurova, K.V.; Purmal, A.A.; Gudkov, A.V.; Plunkett, R.J. Anticancer drug candidate CBL0137, which inhibits histone chaperone FACT, is efficacious in preclinical orthotopic models of temozolomide-responsive and -resistant glioblastoma. Neuro-Oncology 2016, 19, 186–196. [Google Scholar] [CrossRef] [Green Version]
- Benitez, J.A.; Ma, J.; D’Antonio, M.; Boyer, A.; Camargo, M.F.; Zanca, C.; Kelly, S.; Khodadadi-Jamayran, A.; Jameson, N.M.; Andersen, M.; et al. PTEN regulates glioblastoma oncogenesis through chromatin-associated complexes of DAXX and histone H3.3. Nat. Commun. 2017, 8, 15223. [Google Scholar] [CrossRef]
- Koschmann, C.; Calinescu, A.-A.; Nunez, F.J.; Mackay, A.; Fazal-Salom, J.; Thomas, D.; Mendez, F.; Kamran, N.; Dzaman, M.; Mulpuri, L.; et al. ATRX loss promotes tumor growth and impairs nonhomologous end joining DNA repair in glioma. Sci. Transl. Med. 2016, 8, 328ra328. [Google Scholar] [CrossRef] [Green Version]
- Holmberg Olausson, K.; Elsir, T.; Moazemi Goudarzi, K.; Nister, M.; Lindstrom, M.S. NPM1 histone chaperone is upregulated in glioblastoma to promote cell survival and maintain nucleolar shape. Sci Rep. 2015, 5, 16495. [Google Scholar] [CrossRef]
- Vlashi, E.; Lagadec, C.; Vergnes, L.; Matsutani, T.; Masui, K.; Poulou, M.; Popescu, R.; Della Donna, L.; Evers, P.; Dekmezian, C.; et al. Metabolic state of glioma stem cells and nontumorigenic cells. Proc. Natl. Acad. Sci. USA 2011, 108, 16062. [Google Scholar] [CrossRef] [Green Version]
- Janiszewska, M.; Suva, M.L.; Riggi, N.; Houtkooper, R.H.; Auwerx, J.; Clement-Schatlo, V.; Radovanovic, I.; Rheinbay, E.; Provero, P.; Stamenkovic, I. Imp2 controls oxidative phosphorylation and is crucial for preserving glioblastoma cancer stem cells. Genes Dev. 2012, 26, 1926–1944. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kang, B.H.; Altieri, D.C. Compartmentalized cancer drug discovery targeting mitochondrial Hsp90 chaperones. Oncogene 2009, 28, 3681–3688. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, J.; Liu, Y.; Cho, K.; Dong, X.; Teng, L.; Han, D.; Liu, H.; Chen, X.; Chen, X.; Hou, X.; et al. Downregulation of TRAP1 sensitizes glioblastoma cells to temozolomide chemotherapy through regulating metabolic reprogramming. NeuroReport 2016, 27, 136–144. [Google Scholar] [CrossRef] [PubMed]
- Park, H.-K.; Hong, J.-H.; Oh, Y.T.; Kim, S.S.; Yin, J.; Lee, A.-J.; Chae, Y.C.; Kim, J.H.; Park, S.-H.; Park, C.-K.; et al. Interplay between TRAP1 and sirtuin-3 modulates mitochondrial respiration and oxidative stress to maintain stemness of glioma stem cells. Cancer Res. 2019, 79, 1369–1382. [Google Scholar] [CrossRef] [Green Version]
- Voos, W.; Röttgers, K. Molecular chaperones as essential mediators of mitochondrial biogenesis. Biochim. Biophys. Acta (BBA) Mol. Cell Res. 2002, 1592, 51–62. [Google Scholar] [CrossRef] [Green Version]
- Tsuge Ishida, C.; Shu, C.; Halatsch, M.-E.; Westhoff, M.-A.; Altieri, D.C.; Karpel-Massler, G.; David Siegelin, M. Mitochondrial matrix chaperone and c-myc inhibition causes enhanced lethality in glioblastoma. Oncotarget 2017, 8, 37140. [Google Scholar]
- Shah, S.S.; Rodriguez, G.A.; Musick, A.; Walters, W.M.; de Cordoba, N.; Barbarite, E.; Marlow, M.M.; Marples, B.; Prince, J.S.; Komotar, R.J.; et al. Targeting Glioblastoma Stem Cells with 2-Deoxy-D-Glucose (2-DG) Potentiates Radiation-Induced Unfolded Protein Response (UPR). Cancers (Basel) 2019, 11, 159. [Google Scholar] [CrossRef] [Green Version]
- Morihiro, Y.; Yasumoto, Y.; Vaidyan, L.K.; Sadahiro, H.; Uchida, T.; Inamura, A.; Sharifi, K.; Ideguchi, M.; Nomura, S.; Tokuda, N.; et al. Fatty acid binding protein 7 as a marker of glioma stem cells. Pathol. Int. 2013, 63, 546–553. [Google Scholar] [CrossRef]
- Touat, M.; Idbaih, A.; Sanson, M.; Ligon, K.L. Glioblastoma targeted therapy: Updated approaches from recent biological insights. Ann. Oncol. 2017, 28, 1457–1472. [Google Scholar] [CrossRef]
- Eroglu, Z.; Chen, Y.A.; Gibney, G.T.; Weber, J.S.; Kudchadkar, R.R.; Khushalani, N.I.; Markowitz, J.; Brohl, A.S.; Tetteh, L.F.; Ramadan, H.; et al. Combined BRAF and HSP90 inhibition in patients with unresectable BRAFV600E-mutant melanoma. Clin. Cancer Res. 2018. [Google Scholar] [CrossRef] [Green Version]
- Felip, E.; Barlesi, F.; Besse, B.; Chu, Q.; Gandhi, L.; Kim, S.W.; Carcereny, E.; Sequist, L.V.; Brunsvig, P.; Chouaid, C.; et al. Phase 2 Study of the HSP-90 Inhibitor AUY922 in Previously Treated and Molecularly Defined Patients with Advanced Non-Small Cell Lung Cancer. J. Thorac. Oncol. Off. Publ. Int. Assoc. Study Lung Cancer 2018, 13, 576–584. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hendriks, L.E.L.; Dingemans, A.C. Heat shock protein antagonists in early stage clinical trials for NSCLC. Expert Opin. Investig. Drugs 2017, 26, 541–550. [Google Scholar] [CrossRef] [PubMed]
- Powers, M.V.; Workman, P. Inhibitors of the heat shock response: Biology and pharmacology. FEBS Lett. 2007, 581, 3758–3769. [Google Scholar] [CrossRef] [Green Version]
- Miekus, K.; Kijowski, J.; Sekula, M.; Majka, M. 17AEP-GA, an HSP90 antagonist, is a potent inhibitor of glioblastoma cell proliferation, survival, migration and invasion. Oncol Rep. 2012, 28, 1903–1909. [Google Scholar] [CrossRef] [Green Version]
- García-Morales, P.; Carrasco-García, E.; Ruiz-Rico, P.; Martínez-Mira, R.; Menéndez-Gutiérrez, M.P.; Ferragut, J.A.; Saceda, M.; Martínez-Lacaci, I. Inhibition of Hsp90 function by ansamycins causes downregulation of cdc2 and cdc25c and G2/M arrest in glioblastoma cell lines. Oncogene 2007, 26, 7185–7193. [Google Scholar] [CrossRef] [Green Version]
- Premkumar, D.R.; Arnold, B.; Pollack, I.F. Cooperative inhibitory effect of ZD1839 (Iressa) in combination with 17-AAG on glioma cell growth. Mol. Carcinog. 2006, 45, 288–301. [Google Scholar] [CrossRef]
- Jane, E.P.; Pollack, I.F. The heat shock protein antagonist 17-AAG potentiates the activity of enzastaurin against malignant human glioma cells. Cancer Lett. 2008, 268, 46–55. [Google Scholar] [CrossRef] [Green Version]
- Premkumar, D.R.; Arnold, B.; Jane, E.P.; Pollack, I.F. Synergistic interaction between 17-AAG and phosphatidylinositol 3-kinase inhibition in human malignant glioma cells. Mol. Carcinog. 2006, 45, 47–59. [Google Scholar] [CrossRef]
- Wang, X.; Chen, M.; Zhou, J.; Zhang, X. HSP27, 70 and 90, anti-apoptotic proteins, in clinical cancer therapy (Review). Int. J. Oncol. 2014, 45, 18–30. [Google Scholar] [CrossRef]
- Sauvageot, C.M.-E.; Weatherbee, J.L.; Kesari, S.; Winters, S.E.; Barnes, J.; Dellagatta, J.; Ramakrishna, N.R.; Stiles, C.D.; Kung, A.L.-J.; Kieran, M.W.; et al. Efficacy of the HSP90 inhibitor 17-AAG in human glioma cell lines and tumorigenic glioma stem cells. Neuro-Oncology 2009, 11, 109–121. [Google Scholar] [CrossRef] [Green Version]
- Ikegaki, N.; Shimada, H.; Fox, A.M.; Regan, P.L.; Jacobs, J.R.; Hicks, S.L.; Rappaport, E.F.; Tang, X.X. Transient treatment with epigenetic modifiers yields stable neuroblastoma stem cells resembling aggressive large-cell neuroblastomas. Proc. Natl. Acad. Sci. USA 2013, 110, 6097–6102. [Google Scholar] [CrossRef] [Green Version]
- Ohba, S.; Hirose, Y.; Yoshida, K.; Yazaki, T.; Kawase, T. Inhibition of 90-kD heat shock protein potentiates the cytotoxicity of chemotherapeutic agents in human glioma cells. J. Neurosurg. 2010, 112, 33–42. [Google Scholar] [CrossRef]
- Garcia-Carbonero, R.; Carnero, A.; Paz-Ares, L. Inhibition of HSP90 molecular chaperones: Moving into the clinic. Lancet. Oncol. 2013, 14, e358–369. [Google Scholar] [CrossRef]
- Pedersen, K.S.; Kim, G.P.; Foster, N.R.; Wang-Gillam, A.; Erlichman, C.; McWilliams, R.R. Phase II trial of gemcitabine and tanespimycin (17AAG) in metastatic pancreatic cancer: A Mayo Clinic Phase II Consortium study. Investig. New Drugs 2015, 33, 963–968. [Google Scholar] [CrossRef] [Green Version]
- Choi, E.J.; Cho, B.J.; Lee, D.J.; Hwang, Y.H.; Chun, S.H.; Kim, H.H.; Kim, I.A. Enhanced cytotoxic effect of radiation and temozolomide in malignant glioma cells: Targeting PI3K-AKT-mTOR signaling, HSP90 and histone deacetylases. BMC Cancer 2014, 14, 17. [Google Scholar] [CrossRef] [Green Version]
- Gaspar, N.; Sharp, S.Y.; Pacey, S.; Jones, C.; Walton, M.; Vassal, G.; Eccles, S.; Pearson, A.; Workman, P. Acquired Resistance to 17-Allylamino-17-Demethoxygeldanamycin (17-AAG, Tanespimycin) in Glioblastoma Cells. Cancer Res. 2009. [Google Scholar] [CrossRef]
- Lundgren, K.; Zhang, H.; Brekken, J.; Huser, N.; Powell, R.E.; Timple, N.; Busch, D.J.; Neely, L.; Sensintaffar, J.L.; Yang, Y.C.; et al. BIIB021, an orally available, fully synthetic small-molecule inhibitor of the heat shock protein Hsp90. Mol. Cancer 2009, 8, 921–929. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beckner, M.E.; Fellows-Mayle, W.; Zhang, Z.; Agostino, N.R.; Kant, J.A.; Day, B.W.; Pollack, I.F. Identification of ATP citrate lyase as a positive regulator of glycolytic function in glioblastomas. Int. J. Cancer 2010, 126, 2282–2295. [Google Scholar] [CrossRef] [Green Version]
- Eccles, S.A.; Massey, A.; Raynaud, F.I.; Sharp, S.Y.; Box, G.; Valenti, M.; Patterson, L.; de Haven Brandon, A.; Gowan, S.; Boxall, F.; et al. NVP-AUY922: A Novel Heat Shock Protein 90 Inhibitor Active against Xenograft Tumor Growth, Angiogenesis, and Metastasis. Cancer Res. 2008, 68, 2850. [Google Scholar] [CrossRef]
- Hartmann, S.; Gunther, N.; Biehl, M.; Katzer, A.; Kuger, S.; Worschech, E.; Sukhorukov, V.L.; Krohne, G.; Zimmermann, H.; Flentje, M.; et al. Hsp90 inhibition by NVP-AUY922 and NVP-BEP800 decreases migration and invasion of irradiated normoxic and hypoxic tumor cell lines. Cancer Lett. 2013, 331, 200–210. [Google Scholar] [CrossRef]
- Stingl, L.; Stühmer, T.; Chatterjee, M.; Jensen, M.R.; Flentje, M.; Djuzenova, C.S. Novel HSP90 inhibitors, NVP-AUY922 and NVP-BEP800, radiosensitise tumour cells through cell-cycle impairment, increased DNA damage and repair protraction. Br. J. Cancer 2010, 102, 1578. [Google Scholar] [CrossRef]
- Gaspar, N.; Sharp, S.Y.; Eccles, S.A.; Gowan, S.; Popov, S.; Jones, C.; Pearson, A.; Vassal, G.; Workman, P. Mechanistic evaluation of the novel HSP90 inhibitor NVP-AUY922 in adult and pediatric glioblastoma. Mol. Cancer 2010, 9, 1219–1233. [Google Scholar] [CrossRef]
- Di, K.; Keir, S.T.; Alexandru-Abrams, D.; Gong, X.; Nguyen, H.; Friedman, H.S.; Bota, D.A. Profiling Hsp90 differential expression and the molecular effects of the Hsp90 inhibitor IPI-504 in high-grade glioma models. J. Neurooncol. 2014, 120, 473–481. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.; Lee, J.S.; Jung, J.; Lim, I.; Lee, J.Y.; Park, M.J. Emodin suppresses maintenance of stemness by augmenting proteosomal degradation of epidermal growth factor receptor/epidermal growth factor receptor variant III in glioma stem cells. Stem Cells Dev. 2015, 24, 284–295. [Google Scholar] [CrossRef] [Green Version]
- Graham, B.; Curry, J.; Smyth, T.; Fazal, L.; Feltell, R.; Harada, I.; Coyle, J.; Williams, B.; Reule, M.; Angove, H.; et al. The heat shock protein 90 inhibitor, AT13387, displays a long duration of action in vitro and in vivo in non-small cell lung cancer. Cancer Sci. 2012, 103, 522–527. [Google Scholar] [CrossRef]
- Do, K.; Speranza, G.; Chang, L.-C.; Polley, E.C.; Bishop, R.; Zhu, W.; Trepel, J.B.; Lee, S.; Lee, M.-J.; Kinders, R.J.; et al. Phase I study of the heat shock protein 90 (Hsp90) inhibitor onalespib (AT13387) administered on a daily for 2 consecutive days per week dosing schedule in patients with advanced solid tumors. Investig. New Drugs 2015, 33, 921–930. [Google Scholar] [CrossRef]
- Canella, A.; Welker, A.M.; Yoo, J.Y.; Xu, J.; Abas, F.S.; Kesanakurti, D.; Nagarajan, P.; Beattie, C.E.; Sulman, E.P.; Liu, J.; et al. Efficacy of Onalespib, a Long-Acting Second-Generation HSP90 Inhibitor, as a Single Agent and in Combination with Temozolomide against Malignant Gliomas. Clin. Cancer Res. 2017, 23, 6215. [Google Scholar] [CrossRef] [Green Version]
- Aloy, M.-T.; Hadchity, E.; Bionda, C.; Diaz-Latoud, C.; Claude, L.; Rousson, R.; Arrigo, A.-P.; Rodriguez-Lafrasse, C. Protective Role of Hsp27 Protein Against Gamma Radiation–Induced Apoptosis and Radiosensitization Effects of Hsp27 Gene Silencing in Different Human Tumor Cells. Int. J. Radiat. Oncol. Biol. Phys. 2008, 70, 543–553. [Google Scholar] [CrossRef]
- Jakubowicz-Gil, J.; Langner, E.; Badziul, D.; Wertel, I.; Rzeski, W. Silencing of Hsp27 and Hsp72 in glioma cells as a tool for programmed cell death induction upon temozolomide and quercetin treatment. Toxicol. Appl. Pharmacol. 2013, 273, 580–589. [Google Scholar] [CrossRef]
- Belkacemi, L.; Hebb, M.O. HSP27 knockdown produces synergistic induction of apoptosis by HSP90 and kinase inhibitors in glioblastoma multiforme. Anticancer Res. 2014, 34, 4915–4927. [Google Scholar]
- Kim, E.J.; Choi, C.H.; Park, J.Y.; Kang, S.K.; Kim, Y.K. Underlying Mechanism of Quercetin-induced Cell Death in Human Glioma Cells. Neurochem. Res. 2008, 33, 971–979. [Google Scholar] [CrossRef] [PubMed]
- Barbarisi, M.; Iaffaioli, R.V.; Armenia, E.; Schiavo, L.; De Sena, G.; Tafuto, S.; Barbarisi, A.; Quagliariello, V. Novel nanohydrogel of hyaluronic acid loaded with quercetin alone and in combination with temozolomide as new therapeutic tool, CD44 targeted based, of glioblastoma multiforme. J. Cell. Physiol. 2018, 233, 6550–6564. [Google Scholar] [CrossRef] [PubMed]
- Sengelen, A.; Onay-Ucar, E. Rosmarinic acid and siRNA combined therapy represses Hsp27 (HSPB1) expression and induces apoptosis in human glioma cells. Cell Stress Chaperones 2018, 23, 885–896. [Google Scholar] [CrossRef]
- Onay Ucar, E.; Sengelen, A. Resveratrol and siRNA in combination reduces Hsp27 expression and induces caspase-3 activity in human glioblastoma cells. Cell Stress Chaperones 2019, 24, 763–775. [Google Scholar] [CrossRef]
- Zamin, L.L.; Filippi-Chiela, E.C.; Vargas, J.; Demartini, D.R.; Meurer, L.; Souza, A.P.; Bonorino, C.; Salbego, C.; Lenz, G. Quercetin promotes glioma growth in a rat model. Food Chem. Toxicol. Int. J. Publ. Br. Ind. Biol. Res. Assoc. 2014, 63, 205–211. [Google Scholar] [CrossRef]
- Anil, S.; Mitchel, S.B. Basic concepts of immunology and neuroimmunology. Neurosurg. Focus FOC 2000, 9, 1–6. [Google Scholar] [CrossRef]
- Ampie, L.; Choy, W.; Lamano, J.B.; Fakurnejad, S.; Bloch, O.; Parsa, A.T. Heat shock protein vaccines against glioblastoma: From bench to bedside. J. Neurooncol. 2015, 123, 441–448. [Google Scholar] [CrossRef] [Green Version]
- Basu, S.; Binder, R.J.; Ramalingam, T.; Srivastava, P.K. CD91 Is a Common Receptor for Heat Shock Proteins gp96, hsp90, hsp70, and Calreticulin. Immunity 2001, 14, 303–313. [Google Scholar] [CrossRef] [Green Version]
- Binder, R.J.; Srivastava, P.K. Essential role of CD91 in re-presentation of gp96-chaperoned peptides. Proc. Natl. Acad. Sci. USA 2004, 101, 6128. [Google Scholar] [CrossRef] [Green Version]
- Panjwani, N.N.; Popova, L.; Srivastava, P.K. Heat Shock Proteins gp96 and hsp70 Activate the Release of Nitric Oxide by APCs. J. Immunol. 2002, 168, 2997. [Google Scholar] [CrossRef] [Green Version]
- Srivastava, P. Roles of heat-shock proteins in innate and adaptive immunity. Nat. Rev. Immunol. 2002, 2, 185–194. [Google Scholar] [CrossRef] [PubMed]
- Crane, C.A.; Han, S.J.; Ahn, B.J.; Oehlke, J.; Kivett, V.; Fedoroff, A.; Butowski, N.; Chang, S.M.; Clarke, J.; Berger, M.S.; et al. Individual patient-specific immunity against high-grade glioma after vaccination with autologous tumor derived peptides bound to the 96 KD chaperone protein. Clin. Cancer Res. 2013, 19, 205–241. [Google Scholar] [CrossRef] [PubMed]
- Bloch, O.; Crane, C.A.; Fuks, Y.; Kaur, R.; Aghi, M.K.; Berger, M.S.; Butowski, N.A.; Chang, S.M.; Clarke, J.L.; McDermott, M.W.; et al. Heat-shock protein peptide complex-96 vaccination for recurrent glioblastoma: A phase II, single-arm trial. Neuro Oncol. 2014, 16, 274–279. [Google Scholar] [CrossRef] [PubMed]
- Ji, N.; Zhang, Y.; Liu, Y.; Xie, J.; Wang, Y.; Hao, S.; Gao, Z. Heat shock protein peptide complex-96 vaccination for newly diagnosed glioblastoma: A phase I, single-arm trial. JCI Insight 2018, 3. [Google Scholar] [CrossRef] [Green Version]
- Bloch, O.; Lim, M.; Sughrue, M.E.; Komotar, R.J.; Abrahams, J.M.; O’Rourke, D.M.; D’Ambrosio, A.; Bruce, J.N.; Parsa, A.T. Autologous Heat Shock Protein Peptide Vaccination for Newly Diagnosed Glioblastoma: Impact of Peripheral PD-L1 Expression on Response to Therapy. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2017, 23, 3575–3584. [Google Scholar] [CrossRef] [Green Version]


© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Iglesia, R.P.; Fernandes, C.F.d.L.; Coelho, B.P.; Prado, M.B.; Melo Escobar, M.I.; Almeida, G.H.D.R.; Lopes, M.H. Heat Shock Proteins in Glioblastoma Biology: Where Do We Stand? Int. J. Mol. Sci. 2019, 20, 5794. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20225794
Iglesia RP, Fernandes CFdL, Coelho BP, Prado MB, Melo Escobar MI, Almeida GHDR, Lopes MH. Heat Shock Proteins in Glioblastoma Biology: Where Do We Stand? International Journal of Molecular Sciences. 2019; 20(22):5794. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20225794
Chicago/Turabian StyleIglesia, Rebeca Piatniczka, Camila Felix de Lima Fernandes, Bárbara Paranhos Coelho, Mariana Brandão Prado, Maria Isabel Melo Escobar, Gustavo Henrique Doná Rodrigues Almeida, and Marilene Hohmuth Lopes. 2019. "Heat Shock Proteins in Glioblastoma Biology: Where Do We Stand?" International Journal of Molecular Sciences 20, no. 22: 5794. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20225794