Evaluation of the Major Steps in the Conventional Protocol for the Alkaline Comet Assay

 , , and
, , and
Abstract
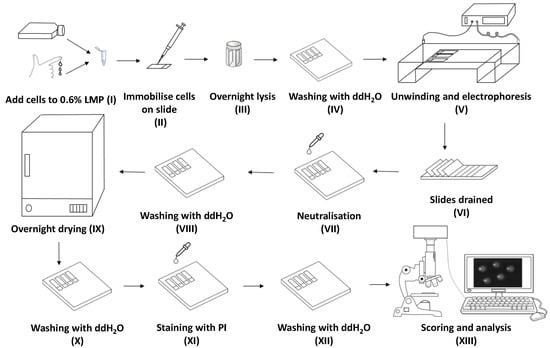
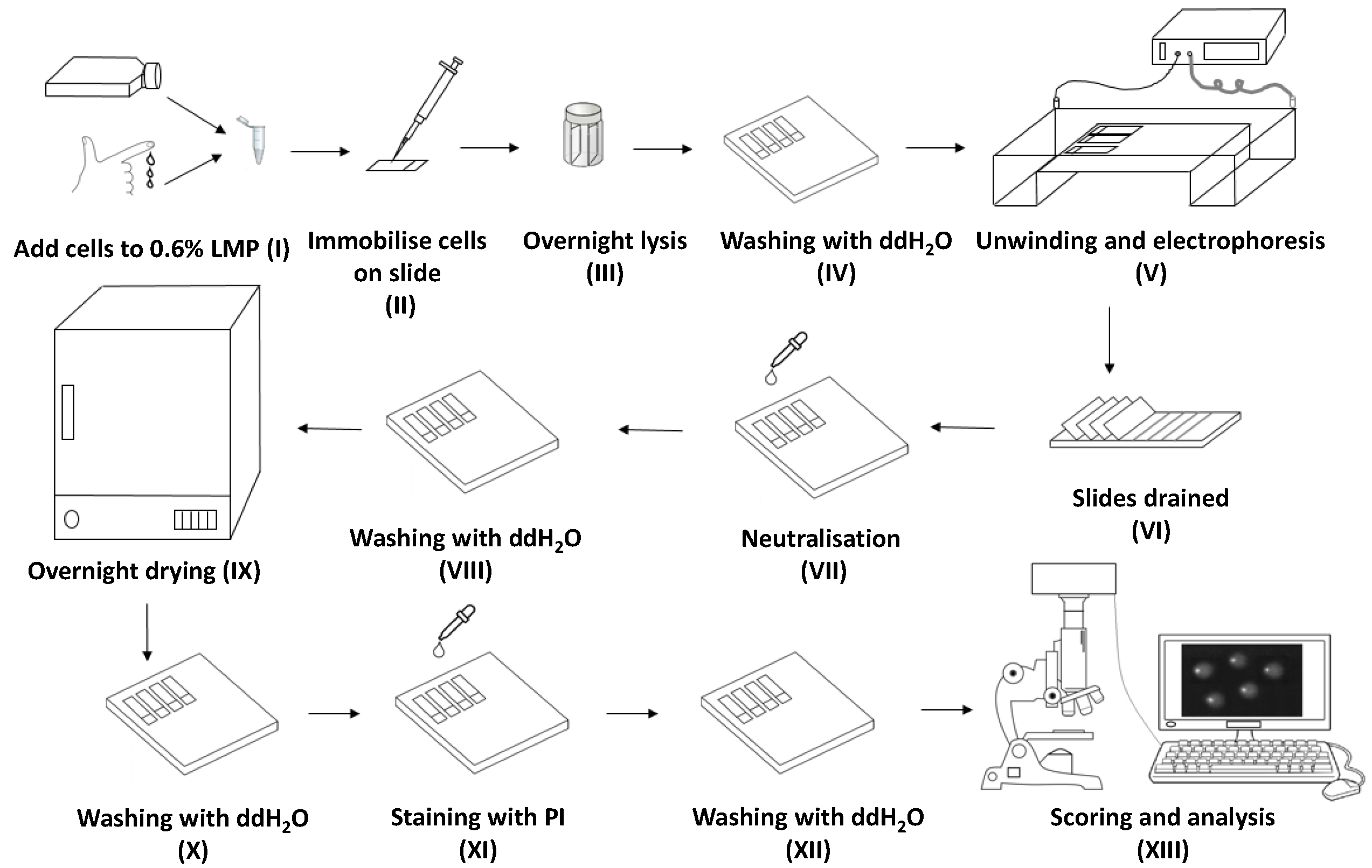
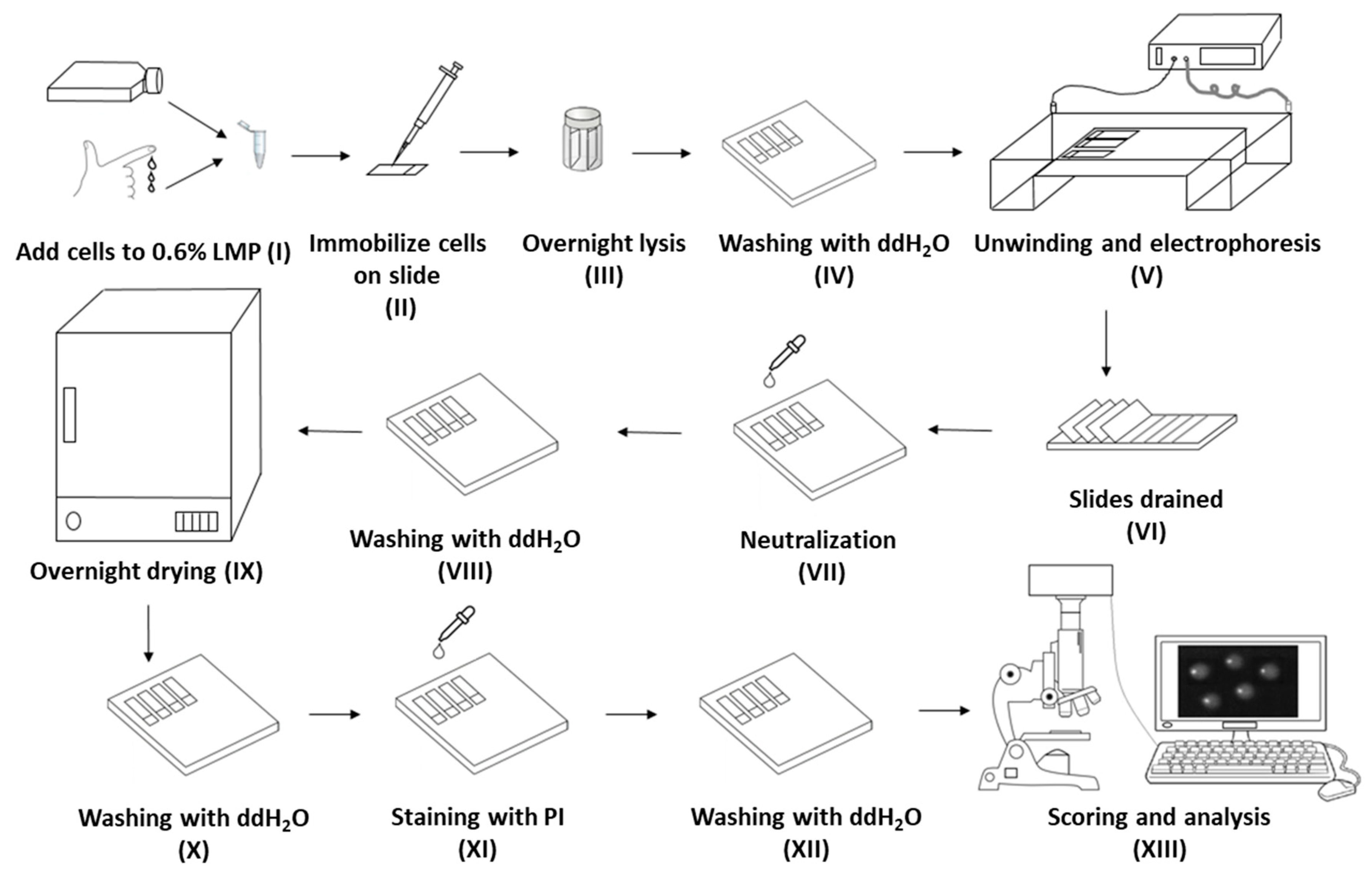
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
Clinical Significance
2. Results
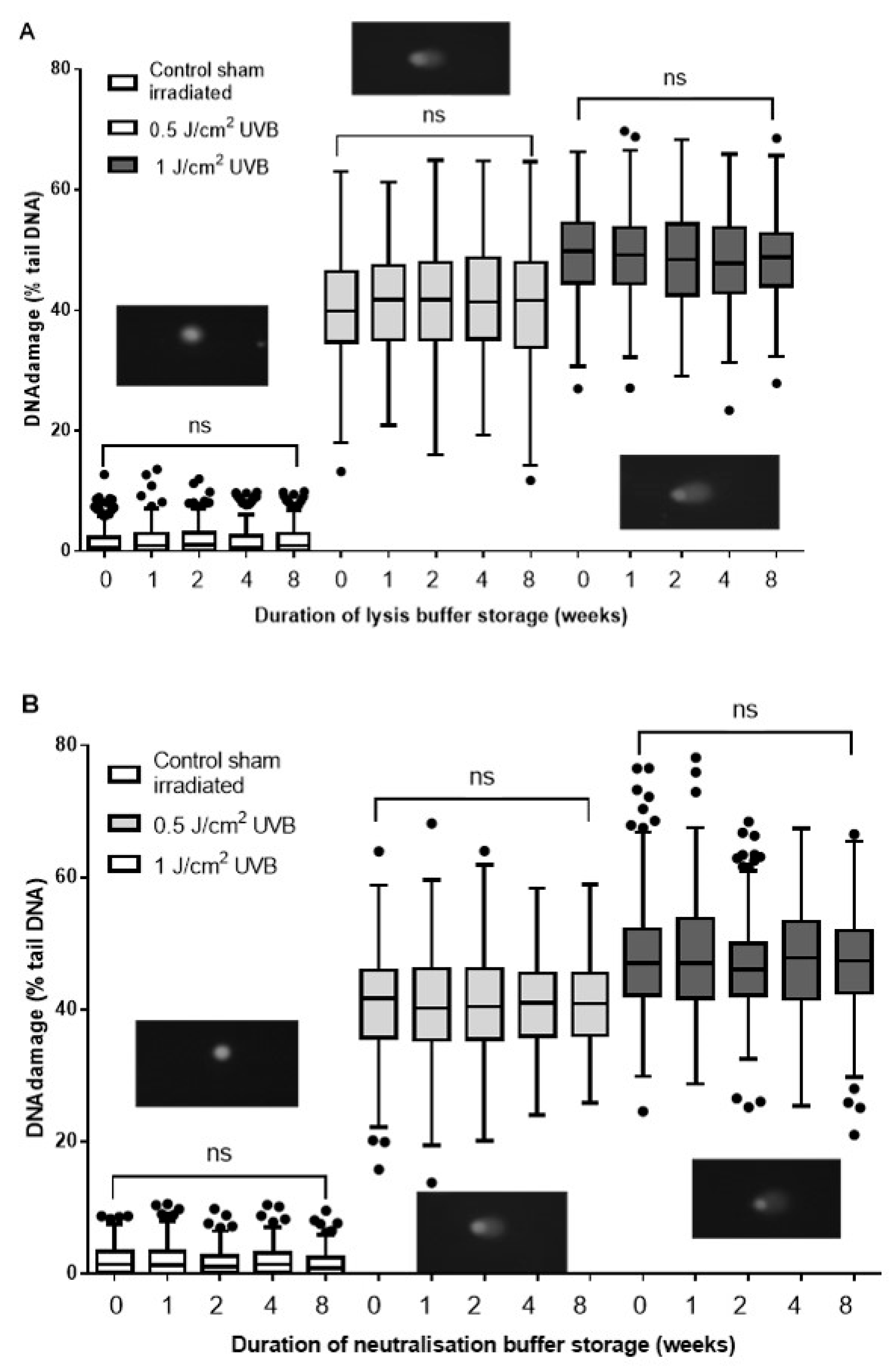
2.1. Evaluation of Lysis Buffer Shelf Life
2.2. Evaluation of Neutralization Buffer Shelf Life
2.2.1. Evaluation of the Effect of Lysis Buffer Temperature, and Incubation Duration
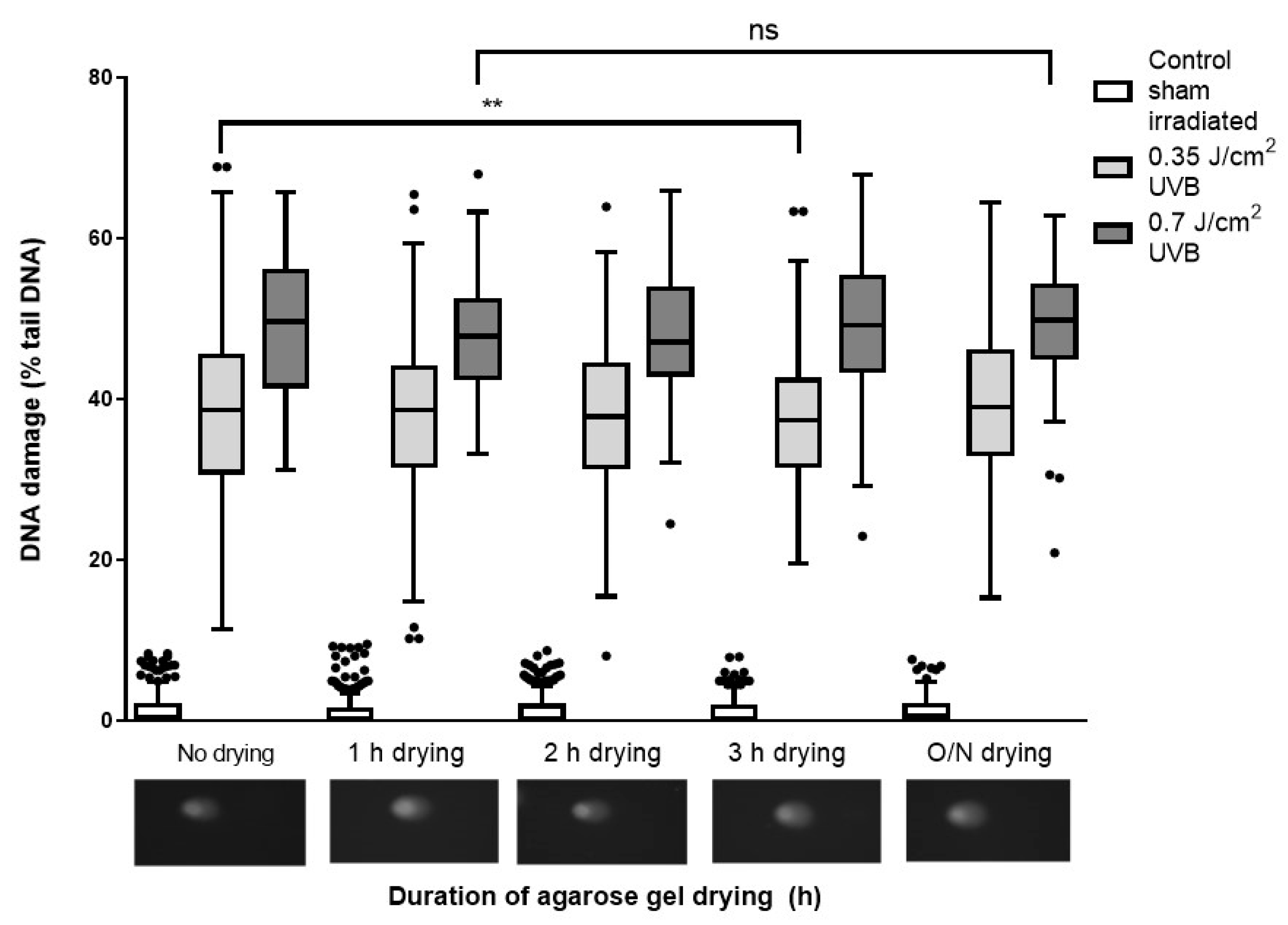
2.2.2. Evaluation of Airdrying Slides for Different Times between Neutralization and Staining
2.2.3. Evaluation of Ambient vs. Subdued Illumination on the ACA
3. Discussion
4. Material and Methods
4.1. Lines and Culture Conditions
4.2. Cell Treatments
4.3. Alkaline Comet Assay (ACA)
4.4. Modified ACA Protocols
4.4.1. Evaluation of Lysis Buffer Shelf Life
4.4.2. Evaluation of Neutralization Buffer Shelf Life
4.4.3. Evaluation of the Effect of Lysis Buffer Temperature and Incubation Duration
4.4.4. Evaluation of Airdrying Slides for Different Times between Neutralization and Staining
4.4.5. Evaluation of Ambient vs. Subdued Illumination on the ACA
4.5. Statistics
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Collins, A.R. The comet assay for DNA damage and repair: Principles, applications, and limitations. Mol. Biotechnol. 2004, 26, 249–261. [Google Scholar] [CrossRef]
- Sabine, A.S.L.; Amaya, A.; Andrew, R.C. The comet assay: Past, present, and future. Front. Genet. 2015, 6, 266. [Google Scholar]
- Cadet, J.; Wagner, J.R. DNA Base Damage by Reactive Oxygen Species, Oxidizing Agents, and UV Radiation. Cold Spring Harb. Perspect. Biol. 2013, 5, a012559. [Google Scholar] [CrossRef] [PubMed]
- Azqueta, A.; Dusinska, M. The use of the comet assay for the evaluation of the genotoxicity of nanomaterials. Front. Genet. 2015, 6, 239. [Google Scholar] [CrossRef]
- Collins, A.; Koppen, G.; Valdiglesias, V.; Dusinska, M.; Kruszewski, M.; Møller, P.; Rojas, E.; Dhawan, A.; Benzie, I.; Coskun, E.; et al. The comet assay as a tool for human biomonitoring studies: The ComNet project. Mutat. Res. Rev. Mutat. Res. 2014, 759, 27–39. [Google Scholar] [CrossRef]
- Valverde, M.; Rojas, E. Environmental and occupational biomonitoring using the Comet assay. Mutat. Res. 2009, 681, 93–109. [Google Scholar] [CrossRef]
- Frenzilli, G.; Nigro, M.; Lyons, B.P. The Comet assay for the evaluation of genotoxic impact in aquatic environments. Mutat. Res. 2009, 681, 80–92. [Google Scholar] [CrossRef]
- Koppen, G.; Azqueta, A.; Pourrut, B.; Brunborg, G.; Collins, A.R.; Langie, S.A.S. The next three decades of the comet assay: A report of the 11th International Comet Assay Workshop. Mutagenesis 2017, 32, 397–408. [Google Scholar] [CrossRef]
- Moneef, M.A.L.; Sherwood, B.T.; Bowman, K.J.; Kockelbergh, R.C.; Symonds, R.P.; Steward, W.P.; Mellon, J.K.; Jones, G.D.D. Measurements using the alkaline comet assay predict bladder cancer cell radiosensitivity. Br. J. Cancer 2003, 89, 2271–2276. [Google Scholar] [CrossRef]
- Karbaschi, M.; Cooke, M.S. Novel method for the high-throughput processing of slides for the comet assay. Sci. Rep. 2014, 4, 7200. [Google Scholar] [CrossRef]
- Wood, D.K.; Weingeist, D.M.; Bhatia, S.N.; Engelward, B.P. Single cell trapping and DNA damage analysis using microwell arrays. Proc. Natl. Acad. Sci. USA 2010, 107, 10008–10013. [Google Scholar] [CrossRef]
- Watson, C.; Ge, J.; Cohen, J.; Pyrgiotakis, G.; Engelward, B.P.; Demokritou, P. High-throughput screening platform for engineered nanoparticle-mediated genotoxicity using CometChip technology. ACS Nano 2014, 8, 2118–2133. [Google Scholar] [CrossRef]
- Gutzkow, K.B.; Langleite, T.M.; Meier, S.; Graupner, A.; Collins, A.R.; Brunborg, G. High-throughput comet assay using 96 minigels. Mutagenesis 2013, 28, 333–340. [Google Scholar] [CrossRef]
- Cooke, M.S.; Bhansali, S.; Karbaschi, M.; Shah, P. Devices and Methods for High-Throughput Assay. U.S. Patent US 9,897,572 B2, 20 February 2018. [Google Scholar]
- Braafladt, S.; Reipa, V.; Atha, D.H. The Comet Assay: Automated Imaging Methods for Improved Analysis and Reproducibility. Sci. Rep. 2016, 6, 32162. [Google Scholar] [CrossRef]
- OECD. Test No. 489: In Vivo Mammalian Alkaline Comet Assay; OECD: Paris, France, 2016. [Google Scholar]
- Gunasekarana, V.; Raj, G.V.; Chand, P. A comprehensive review on clinical applications of comet assay. J. Clin. Diagn. Res. 2015, 9, GE01–GE05. [Google Scholar] [CrossRef]
- Fikrova, P.; Stetina, R.; Hronek, M.; Hyspler, R.; Ticha, A.; Zadak, Z. Application of the comet assay method in clinical studies. Wien. Klin. Wochenschr. 2011, 123, 693–699. [Google Scholar] [CrossRef]
- Jiang, Y.; Liu, Y.; Hu, H. Studies on DNA Damage Repair and Precision Radiotherapy for Breast Cancer. Adv. Exp. Med. Biol. 2017, 1026, 105–123. [Google Scholar]
- Schabath, M.B.; Spitz, M.R.; Grossman, H.B.; Zhang, K.; Dinney, C.P.; Zheng, P.J.; Wu, X. Genetic instability in bladder cancer assessed by the comet assay. J. Natl. Cancer Inst. 2003, 95, 540–547. [Google Scholar] [CrossRef]
- Kopjar, N.; Milas, I.; Garaj-Vrhovac, V.; Gamulin, M. Alkaline comet assay study with breast cancer patients: Evaluation of baseline and chemotherapy-induced DNA damage in non-target cells. Clin. Exp. Med. 2006, 6, 177–190. [Google Scholar] [CrossRef]
- Santos, R.A.; Teixeira, A.C.; Mayorano, M.B.; Carrara, H.H.; Andrade, J.M.; Takahashi, C.S. Basal levels of DNA damage detected by micronuclei and comet assays in untreated breast cancer patients and healthy women. Clin. Exp. Med. 2010, 10, 87–92. [Google Scholar] [CrossRef]
- McKenna, D.J.; McKeown, S.R.; McKelvey-Martin, V.J. Potential use of the comet assay in the clinical management of cancer. Mutagenesis 2008, 23, 183–190. [Google Scholar] [CrossRef]
- Marples, B.; Longhurst, D.; Eastham, A.M.; West, C.M. The ratio of initial/residual DNA damage predicts intrinsic radiosensitivity in seven cervix carcinoma cell lines. Br. J. Cancer 1998, 77, 1108–1114. [Google Scholar] [CrossRef]
- Bowman, K.J.; Al-Moneef, M.M.; Sherwood, B.T.; Colquhoun, A.J.; Goddard, J.C.; Griffiths, T.R.; Payne, D.; Singh, S.; Butterworth, P.C.; Khan, M.A.; et al. Comet assay measures of DNA damage are predictive of bladder cancer cell treatment sensitivity in vitro and outcome in vivo. Int. J. Cancer 2014, 134, 1102–1111. [Google Scholar] [CrossRef]
- Colquhoun, A.J.; Jones, G.D.; Moneef, M.A.; Bowman, K.J.; Kockelbergh, R.C.; Symonds, R.P.; Steward, W.P.; Mellon, J.K. Improving and predicting radiosensitivity in muscle invasive bladder cancer. J. Urol. 2003, 169, 1983–1992. [Google Scholar] [CrossRef]
- Anderson, D.; Mojgan, N.; Andrew, S.; Badie, J.; John, G.; Rohit, C.; Richard, L.; Michel, S.; Francoise, S. Using a Modified Lymphocyte Genome Sensitivity (LGS) test or TumorScan test to detect cancer at an early stage in each individual. FASEB J. 2019, 1, 32–39. [Google Scholar] [CrossRef]
- Collins, A.R.; Oscoz, A.A.; Brunborg, G.; Gaivão, I.; Giovannelli, L.; Kruszewski, M.; Smith, C.C.; Stetina, R. The comet assay: Topical issues. Mutagenesis 2008, 23, 143–151. [Google Scholar] [CrossRef]
- Enciso, J.M.; Sanchez, O.; Lopez de Cerain, A.; Azqueta, A. Does the duration of lysis affect the sensitivity of the in vitro alkaline comet assay? Mutagenesis 2015, 30, 21–28. [Google Scholar] [CrossRef]
- Moller, P. The comet assay: Ready for 30 more years. Mutagenesis 2018, 33, 1–7. [Google Scholar] [CrossRef]
- Olive, P.L.; Banath, J.P. The comet assay: A method to measure DNA damage in individual cells. Nat. Protoc. 2006, 1, 23–29. [Google Scholar] [CrossRef]
- Banath, J.P.; Fushiki, M.; Olive, P.L. Rejoining of DNA single- and double-strand breaks in human white blood cells exposed to ionizing radiation. Int. J. Radiat. Biol. 1998, 73, 649–660. [Google Scholar] [CrossRef]
- Ward, J.F. Biochemistry of DNA lesions. Radiat. Res. 1985, 104 (Suppl. 8), S103–S111. [Google Scholar] [CrossRef]
- Olive, P.L. The comet assay. An overview of techniques. Methods Mol. Biol. 2002, 203, 179–194. [Google Scholar]
- Enciso, J.M.; Lopez de Cerain, A.; Pastor, L.; Azqueta, A.; Vettorazzi, A. Is oxidative stress involved in the sex-dependent response to ochratoxin A renal toxicity? Food Chem. Toxicol. 2018, 116, 379–387. [Google Scholar] [CrossRef]
- Tice, R.R.; Agurell, E.; Anderson, D.; Burlinson, B.; Hartmann, A.; Kobayashi, H.; Miyamae, Y.; Rojas, E.; Ryu, J.C.; Sasaki, Y.F. Single cell gel/comet assay: Guidelines for in vitro and in vivo genetic toxicology testing. Environ. Mol. Mutagen. 2000, 35, 206–221. [Google Scholar] [CrossRef]
- Collins, A.R. The comet assay: A heavenly method! Mutagenesis 2015, 30, 1–4. [Google Scholar] [CrossRef] [Green Version]
- Decome, L.; De Méo, M.; Geffard, A.; Doucet, O.; Duménil, G.; Botta, A. Evaluation of photolyase (Photosome) repair activity in human keratinocytes after a single dose of ultraviolet B irradiation using the comet assay. J. Photochem. Photobiol. B 2005, 79, 101–108. [Google Scholar] [CrossRef]
- Baumgartner, A.; Cemeli, E.; Anderson, D. The comet assay in male reproductive toxicology. Cell Biol. Toxicol. 2009, 25, 81–98. [Google Scholar] [CrossRef]
- Rojas, E.; Valverde, M.; Sordo, M.; Ostrosky-Wegman, P. DNA damage in exfoliated buccal cells of smokers assessed by the single cell gel electrophoresis assay. Mutat. Res. 1996, 370, 115–120. [Google Scholar] [CrossRef]
- Enciso, M.; Sarasa, J.; Agarwal, A.; Fernandez, J.L.; Gosalvez, J. A two-tailed Comet assay for assessing DNA damage in spermatozoa. Reprod. Biomed. Online 2009, 18, 609–616. [Google Scholar] [CrossRef]
- Azqueta, A.; Gutzkow, K.B.; Brunborg, G.; Collins, A.R. Towards a more reliable comet assay: Optimising agarose concentration, unwinding time and electrophoresis conditions. Mutat. Res. 2011, 724, 41–45. [Google Scholar] [CrossRef]
- Gyori, B.M.; Venkatachalam, G.; Thiagarajan, P.S.; Hsu, D.; Clement, M.V. OpenComet: An automated tool for comet assay image analysis. Redox Biol. 2014, 2, 457–465. [Google Scholar] [CrossRef] [Green Version]
- Ganesan, M.; Agilan, B.; Kanimozhi, G.; Karthikeyan, R.; Veeramani, K.P.; Illiyas, M.M.; Rajendra, P.N. Modified comet assays for the detection of cyclobutane pyrimidine dimers and oxidative base damages. J. Radiat. Cancer Res. 2017, 8, 82–86. [Google Scholar]
- Gombocz, E.; Chrambach, A. Rehydratable agarose gels: Use of low EEO agarose without charged additives. Appl. Theor. Electrophor. 1989, 1, 109–113. [Google Scholar]
- Wischermann, K.; Boukamp, P.; Schmezer, P. Improved alkaline comet assay protocol for adherent HaCaT keratinocytes to study UVA-induced DNA damage. Mutat. Res. 2007, 630, 122–128. [Google Scholar] [CrossRef]
- Karbaschi, M.; Macip, S.; Mistry, V.; Abbas, H.H.K.; Delinassios, G.J.; Evans, M.D.; Young, A.R.; Cooke, M.S. Rescue of cells from apoptosis increases DNA repair in UVB exposed cells: Implications for the DNA damage response. Toxicol. Res. 2015, 4, 725–738. [Google Scholar] [CrossRef] [Green Version]
- Knopper, L.D.; Mineau, P.; McNamee, J.P.; Lean, D.R. Use of comet and micronucleus assays to measure genotoxicity in meadow voles (Microtus pennsylvanicus) living in golf course ecosystems exposed to pesticides. Ecotoxicology 2005, 14, 323–335. [Google Scholar] [CrossRef]
- Azqueta, A.; Muruzabal, D.; Boutet-Robinet, E.; Milic, M.; Dusinska, M.; Brunborg, G.; Møller, P.; Collins, A.R. Technical recommendations to perform the alkaline standard and enzyme-modified comet assay in human biomonitoring studies. Mutat. Res. 2019, 843, 24–32. [Google Scholar] [CrossRef]





© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Karbaschi, M.; Ji, Y.; Abdulwahed, A.M.S.; Alohaly, A.; Bedoya, J.F.; Burke, S.L.; Boulos, T.M.; Tempest, H.G.; Cooke, M.S. Evaluation of the Major Steps in the Conventional Protocol for the Alkaline Comet Assay. Int. J. Mol. Sci. 2019, 20, 6072. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20236072
Karbaschi M, Ji Y, Abdulwahed AMS, Alohaly A, Bedoya JF, Burke SL, Boulos TM, Tempest HG, Cooke MS. Evaluation of the Major Steps in the Conventional Protocol for the Alkaline Comet Assay. International Journal of Molecular Sciences. 2019; 20(23):6072. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20236072
Chicago/Turabian StyleKarbaschi, Mahsa, Yunhee Ji, Abdulhadi Mohammed S. Abdulwahed, Alhanoof Alohaly, Juan F. Bedoya, Shanna L. Burke, Thomas M. Boulos, Helen G. Tempest, and Marcus S. Cooke. 2019. "Evaluation of the Major Steps in the Conventional Protocol for the Alkaline Comet Assay" International Journal of Molecular Sciences 20, no. 23: 6072. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20236072