RankerGUI: A Computational Framework to Compare Differential Gene Expression Profiles Using Rank Based Statistics


Abstract
:1. Introduction
2. Results and Discussion
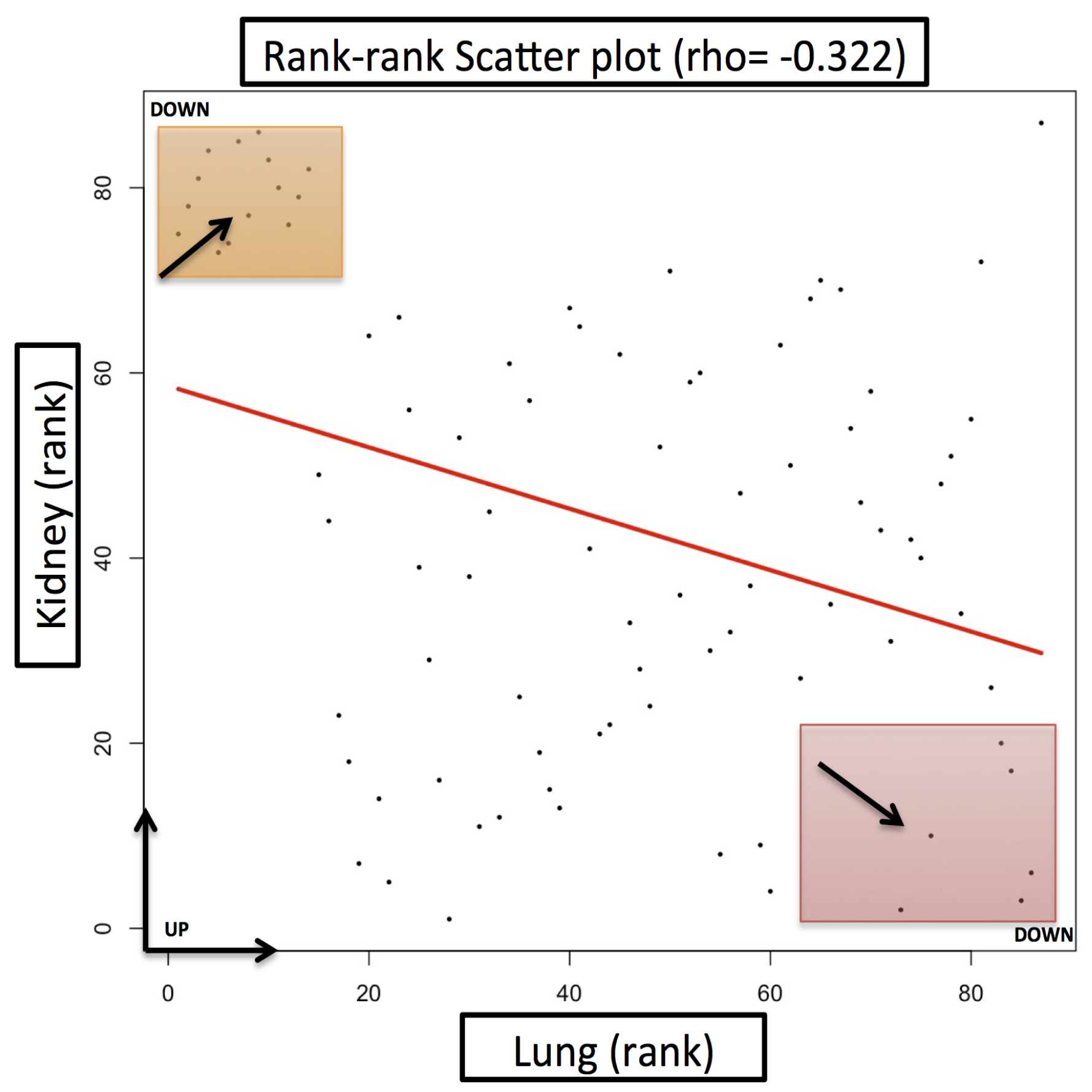
Case Study
3. Method and Implementation
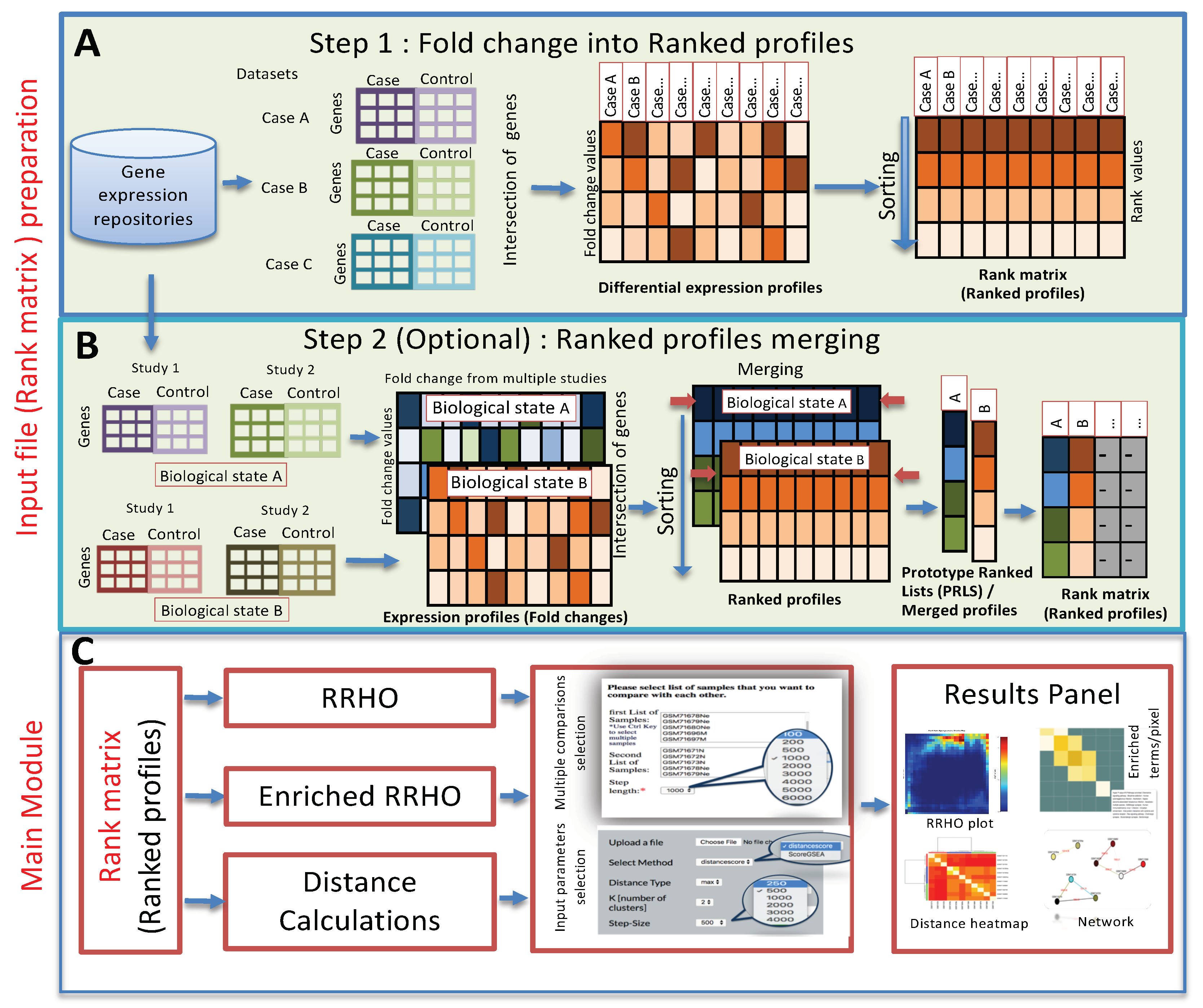
3.1. Method
3.2. Web Server
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| GSEA | Gene set enrichment analysis |
| KIRC | Kidney renal clear cell carcinoma |
| LUAD | Lung adenocarcinoma |
| LUSC | Lung squamous cell carcinoma |
| PRL | Prototype ranked list |
| RRHO | Rank–rank hyper-geometric overlaps |
References
- Hu, G.; Agarwal, P. Human disease-drug network based on genomic expression profiles. PLoS ONE 2009, 4, e6536. [Google Scholar] [CrossRef] [PubMed]
- Iorio, F.; Bosotti, R.; Scacheri, E.; Belcastro, V.; Mithbaokar, P.; Ferriero, R.; Murino, L.; Tagliaferri, R.; Brunetti-Pierri, N.; Isacchi, A.; et al. Discovery of drug mode of action and drug repositioning from transcriptional responses. Proc. Natl. Acad. Sci. USA 2010, 107, 14621–14626. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: A knowledge based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tripathi, K.P.; Chavan, S.G.; Parashuraman, S.; Piccirillo, M.; Magliocca, S.; Guarracino, M.R. Comparison of gene expression signature using rank based statistical inference. In Proceedings of the International Meeting on Computational Intelligence Methods for Bioinformatics and Biostatistics, Naples, Italy, 10–12 September 2015; pp. 28–41. [Google Scholar]
- Chavan, S.G.; Tripathi, K.P.; Piccirilo, M.; Roy, P.D.; Guarracino, M.; Luini, A.; Parashuraman, S. Dissecting the Functions of the Secretory Pathway by Transcriptional Profiling. In Dynamics of Mathematical Models in Biology; Springer: Berlin, Germany, 2016; pp. 79–87. [Google Scholar]
- Yang, X.; Bentink, S.; Scheid, S.; Spang, R. Similarities of ordered gene lists. J. Bioinform. Comput. Biol. 2006, 4, 693–708. [Google Scholar] [CrossRef] [PubMed]
- Khan, H.A. ArraySolver: An algorithm for color-coded graphical display and Wilcoxon signed-rank statistics for comparing microarray gene expression data. Comp. Funct. Genom. 2004, 5, 39–47. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eden, E.; Navon, R.; Steinfeld, I.; Lipson, D.; Yakhini, Z. GOrilla: A tool for discovery and visualization of enriched GO terms in ranked gene lists. BMC Bioinform. 2009, 10, 48. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Plaisier, S.B.; Taschereau, R.; Wong, J.A.; Graeber, T.G. Rank–rank hypergeometric overlap: identification of statistically significant overlap between gene-expression signatures. Nucleic Acids Res. 2010, 38, e169. [Google Scholar] [CrossRef] [Green Version]
- Carrella, D.; Napolitano, F.; Rispoli, R.; Miglietta, M.; Carissimo, A.; Cutillo, L.; Sirci, F.; Gregoretti, F.; Di Bernardo, D. Mantra 2.0: An online collaborative resource for drug mode of action and repurposing by network analysis. Bioinformatics 2014, 30, 1787–1788. [Google Scholar] [CrossRef] [Green Version]
- Furge, K.; Dykema, K. PGSEA: Parametric Gene Set Enrichment Analysis, R package version 1.2; Rpackage: Auckland, New Zealand, 2006. [Google Scholar]
- Li, F.; Cao, Y.; Han, L.; Cui, X.; Xie, D.; Wang, S.; Bo, X. GeneExpressionSignature: An R package for discovering functional connections using gene expression signatures. Omics J. Integr. Biol. 2013, 17, 116–118. [Google Scholar] [CrossRef]
- Slonim, D.K.; Yanai, I. Getting started in gene expression microarray analysis. PLoS Comput. Biol. 2009, 5, e1000543. [Google Scholar] [CrossRef]
- Costa-Silva, J.; Domingues, D.; Lopes, F.M. RNA-Seq differential expression analysis: An extended review and a software tool. PLoS ONE 2017, 12, e0190152. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eijssen, L.M.; Jaillard, M.; Adriaens, M.E.; Gaj, S.; de Groot, P.J.; Müller, M.; Evelo, C.T. User-friendly solutions for microarray quality control and pre-processing on ArrayAnalysis. org. Nucleic Acids Res. 2013, 41, W71–W76. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kanehisa, M.; Goto, S. KEGG: kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000, 28, 27–30. [Google Scholar] [CrossRef] [PubMed]
- Jones, M.H.; Virtanen, C.; Honjoh, D.; Miyoshi, T.; Satoh, Y.; Okumura, S.; Nakagawa, K.; Nomura, H.; Ishikawa, Y. Two prognostically significant subtypes of high-grade lung neuroendocrine tumors independent of small-cell and large-cell neuroendocrine carcinomas identified by gene expression profiles. Lancet 2004, 363, 775–781. [Google Scholar] [CrossRef]
- Fujiwara, T.; Hiramatsu, M.; Isagawa, T.; Ninomiya, H.; Inamura, K.; Ishikawa, S.; Ushijima, M.; Matsuura, M.; Jones, M.H.; Shimane, M.; et al. ASCL1-coexpression profiling but not single gene expression profiling defines lung adenocarcinomas of neuroendocrine nature with poor prognosis. Lung Cancer 2012, 75, 119–125. [Google Scholar] [CrossRef]
- Lenburg, M.E.; Liou, L.S.; Gerry, N.P.; Frampton, G.M.; Cohen, H.T.; Christman, M.F. Previously unidentified changes in renal cell carcinoma gene expression identified by parametric analysis of microarray data. BMC Cancer 2003, 3, 31. [Google Scholar] [CrossRef] [Green Version]
- Wachi, S.; Yoneda, K.; Wu, R. Interactome-transcriptome analysis reveals the high centrality of genes differentially expressed in lung cancer tissues. Bioinformatics 2005, 21, 4205–4208. [Google Scholar] [CrossRef] [Green Version]
- Smyth, G.K. Linear models and empirical bayes methods for assessing differential expression in microarray experiments. Stat. Appl. Genet. Mol. Biol. 2004, 3, 1–25. [Google Scholar] [CrossRef]
- Davis, S.; Meltzer, P.S. GEOquery: A bridge between the Gene Expression Omnibus (GEO) and BioConductor. Bioinformatics 2007, 23, 1846–1847. [Google Scholar] [CrossRef] [Green Version]
- Bindea, G.; Mlecnik, B.; Hackl, H.; Charoentong, P.; Tosolini, M.; Kirilovsky, A.; Fridman, W.H.; Pagès, F.; Trajanoski, Z.; Galon, J. ClueGO: A Cytoscape plug-in to decipher functionally grouped gene ontology and pathway annotation networks. Bioinformatics 2009, 25, 1091–1093. [Google Scholar] [CrossRef] [Green Version]
- Tang, Z.; Li, C.; Kang, B.; Gao, G.; Li, C.; Zhang, Z. GEPIA: A web server for cancer and normal gene expression profiling and interactive analyses. Nucleic Acids Res. 2017, 45, W98–W102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kruskal, J.B. On the shortest spanning subtree of a graph and the traveling salesman problem. Proc. Am. Math. Soc. 1956, 7, 48–50. [Google Scholar] [CrossRef]
- Diaconis, P.; Graham, R.L. Spearman’s footrule as a measure of disarray. J. R. Stat. Soc. Ser. B (Methodol.) 1977, 39, 262–268. [Google Scholar] [CrossRef]
- Chamberlin, J.R.; Courant, P.N. Representative deliberations and representative decisions: Proportional representation and the Borda rule. Am. Political Sci. Rev. 1983, 77, 718–733. [Google Scholar] [CrossRef]
- Yu, G.; Wang, L.G.; Han, Y.; He, Q.Y. clusterProfiler: An R package for comparing biological themes among gene clusters. Omics J. Integr. Biol. 2012, 16, 284–287. [Google Scholar] [CrossRef]
- Frey, B.J.; Dueck, D. Clustering by passing messages between data points. Science 2007, 315, 972–976. [Google Scholar] [CrossRef] [Green Version]
- Bodenhofer, U.; Kothmeier, A.; Hochreiter, S. APCluster: An R package for affinity propagation clustering. Bioinformatics 2011, 27, 2463–2464. [Google Scholar] [CrossRef]
- Franz, M.; Lopes, C.T.; Huck, G.; Dong, Y.; Sumer, O.; Bader, G.D. Cytoscape. js: A graph theory library for visualization and analysis. Bioinformatics 2015, 32, 309–311. [Google Scholar]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene Name | Fold Change (FC) (Lung Cancer) | Fold Change (FC) (Kidney Cancer) | Description |
|---|---|---|---|
| ALDOB | 1.13 | −7.01 | aldolase, fructose-bisphosphate B |
| TFAP2B | 1.17 | −4.16 | transcription factor AP-2 beta |
| AZGP1 | 1.23 | −3.95 | alpha-2-glycoprotein 1, zinc-binding |
| PC | 1.05 | −2.39 | pyruvate carboxylase |
| PPM1H | 1.01 | −2.37 | protein phosphatase, Mg2+/Mn2+ dependent 1H |
| GGH | 1.24 | −2.35 | gamma-glutamyl hydrolase |
| FOXI1 | 1.05 | −2.26 | forkhead box I1 |
| MYCN | 1.02 | −2.10 | v-myc avian myelocytomatosis viral oncogene neuroblastoma derived homolog |
| UCHL1 | 1.47 | −1.70 | ubiquitin C-terminal hydrolase L1 |
| TUBB2A | 1.15 | −1.52 | tubulin beta 2A class IIa |
| PPIF | 1.03 | −1.32 | peptidylprolyl isomerase F |
| SPP1 | 2.29 | −1.15 | secreted phosphoprotein 1 |
| PFN2 | 1.2 | −1.06 | profilin 2 |
| PDHA1 | 1.21 | −1.02 | pyruvate dehydrogenase (lipoamide) alpha 1 |
| CALCRL | −2.04 | 1.00 | calcitonin receptor like receptor |
| CDH5 | −2.08 | 1.71 | cadherin 5 |
| CAV2 | −2.1 | 1.86 | caveolin 2 |
| PMP22 | −2.11 | 1.97 | peripheral myelin protein 22 |
| FHL1 | −2.52 | 3.09 | four and a half LIM domains 1 |
| CAV1 | −3.39 | 2.92 | caveolin 1 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Thind, A.S.; Tripathi, K.P.; Guarracino, M.R. RankerGUI: A Computational Framework to Compare Differential Gene Expression Profiles Using Rank Based Statistics. Int. J. Mol. Sci. 2019, 20, 6098. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20236098
Thind AS, Tripathi KP, Guarracino MR. RankerGUI: A Computational Framework to Compare Differential Gene Expression Profiles Using Rank Based Statistics. International Journal of Molecular Sciences. 2019; 20(23):6098. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20236098
Chicago/Turabian StyleThind, Amarinder Singh, Kumar Parijat Tripathi, and Mario Rosario Guarracino. 2019. "RankerGUI: A Computational Framework to Compare Differential Gene Expression Profiles Using Rank Based Statistics" International Journal of Molecular Sciences 20, no. 23: 6098. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20236098