Proteomics Analysis of Early Developmental Stages of Zebrafish Embryos


 , and
, and
Abstract
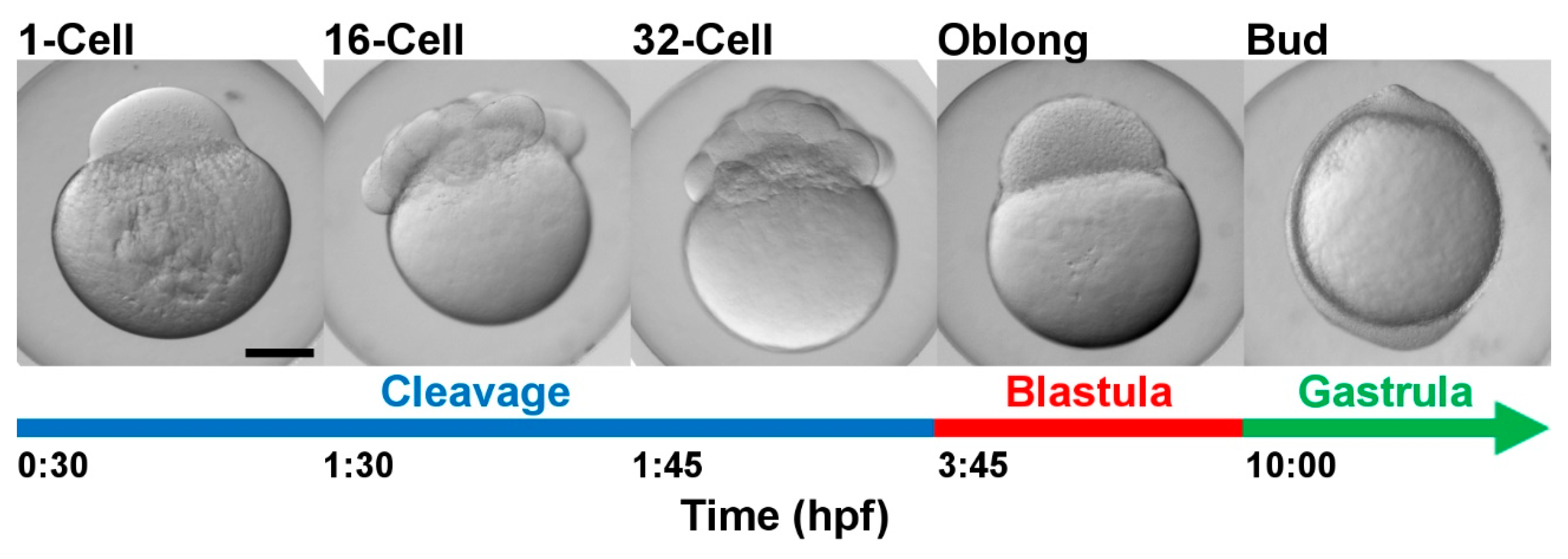
:1. Introduction
2. Results
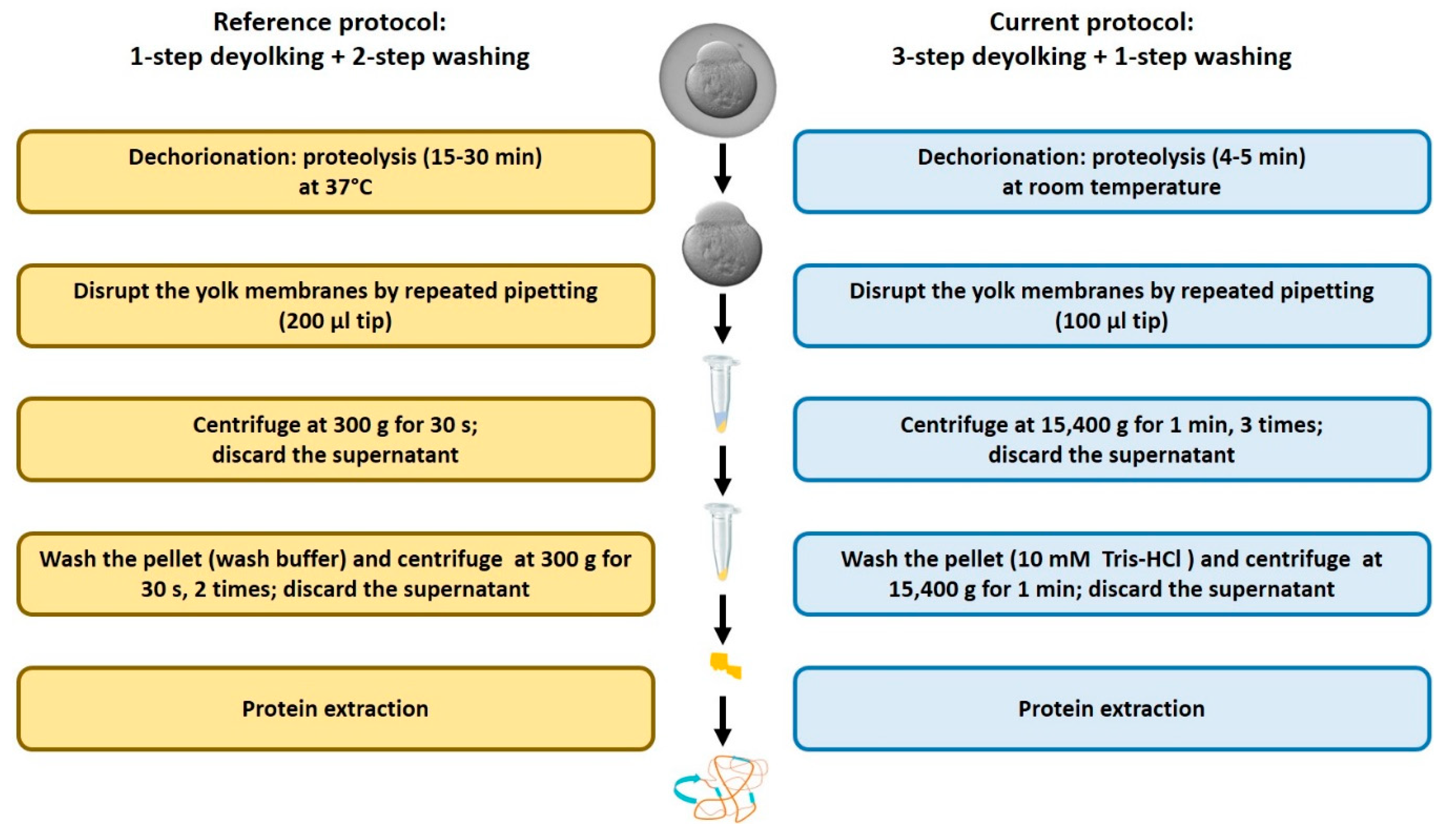
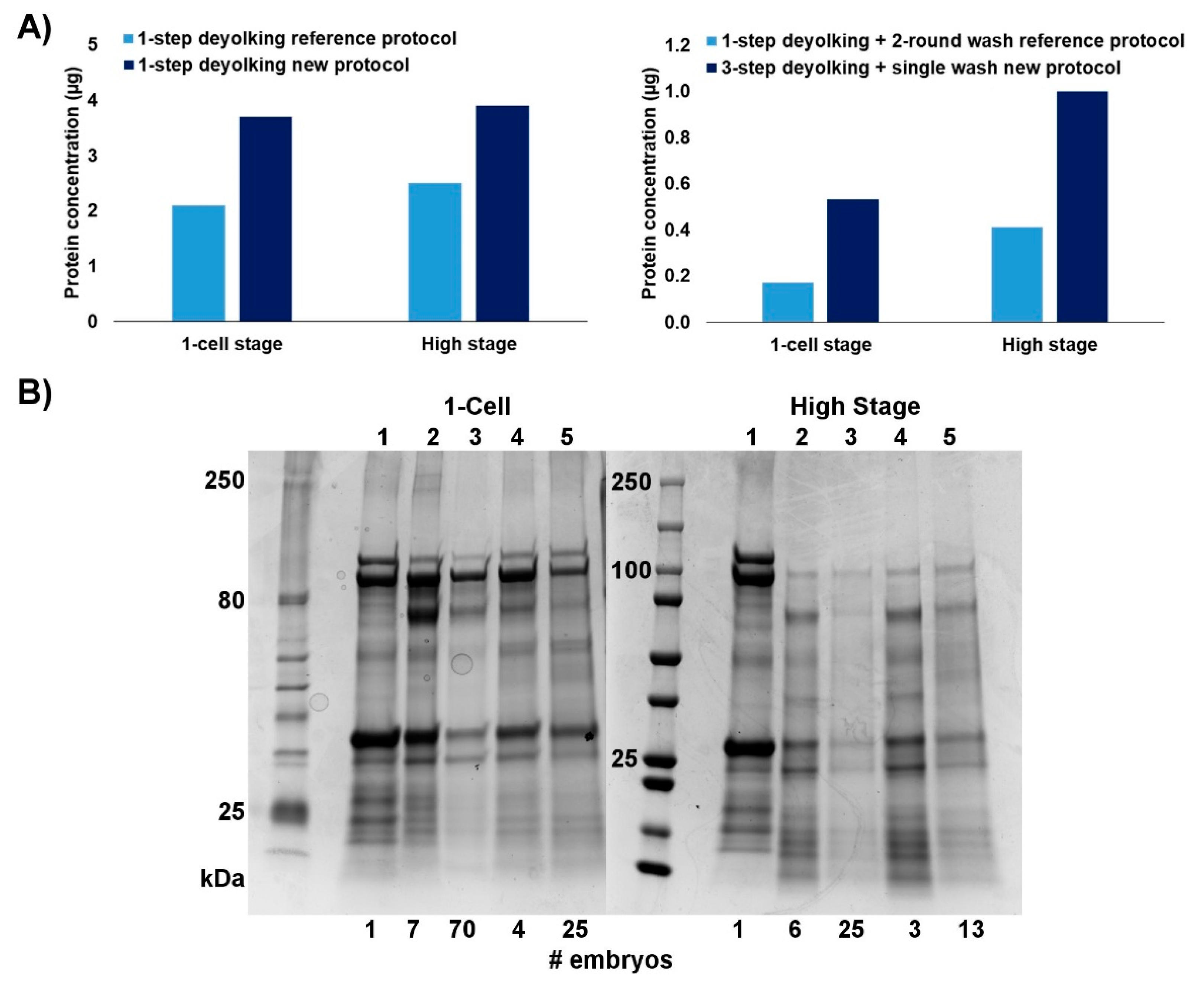
2.1. Efficiency of the New Extraction Protocol
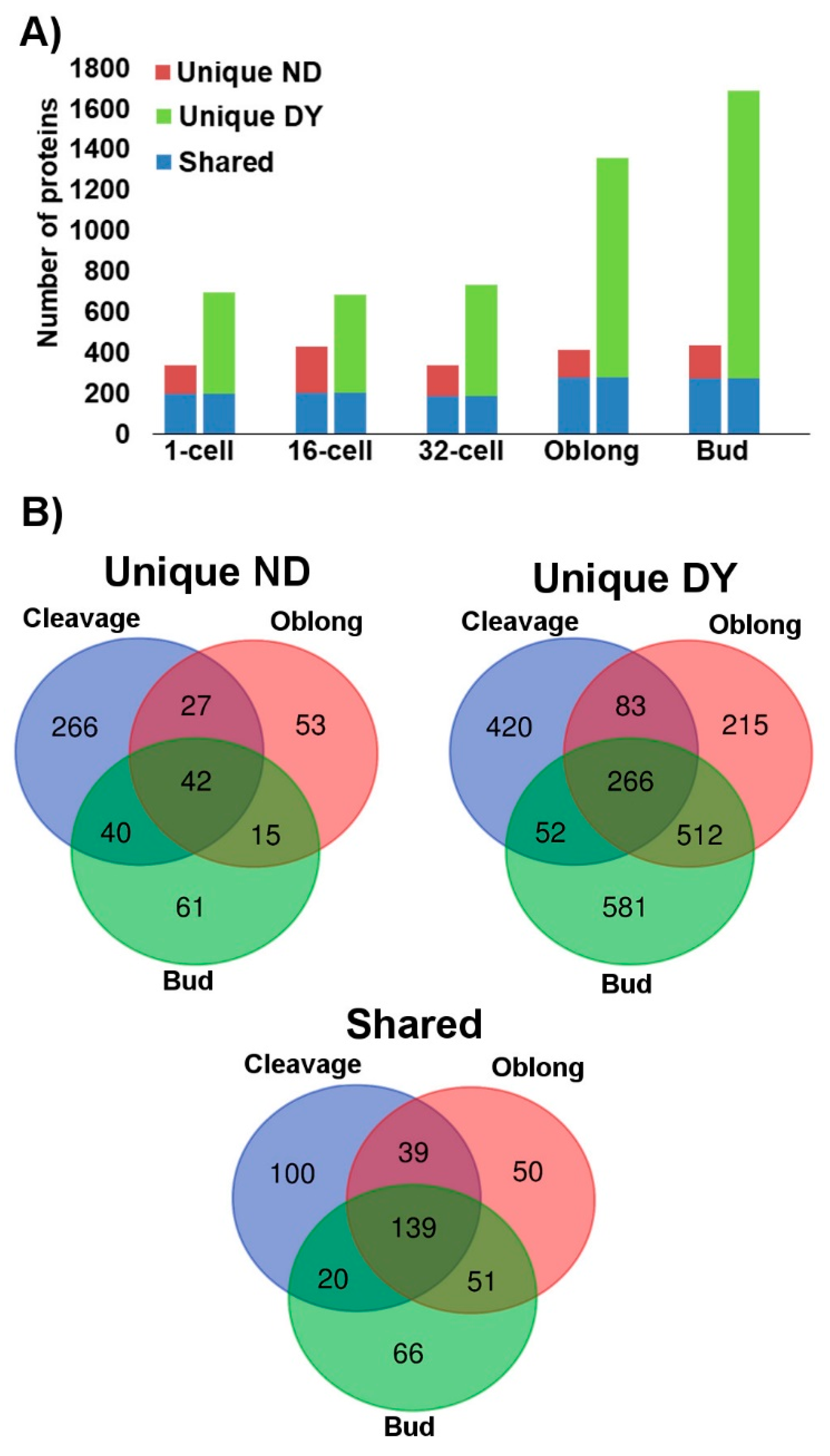
2.2. Proteome in Deyolked Versus Non-Deyolked Samples
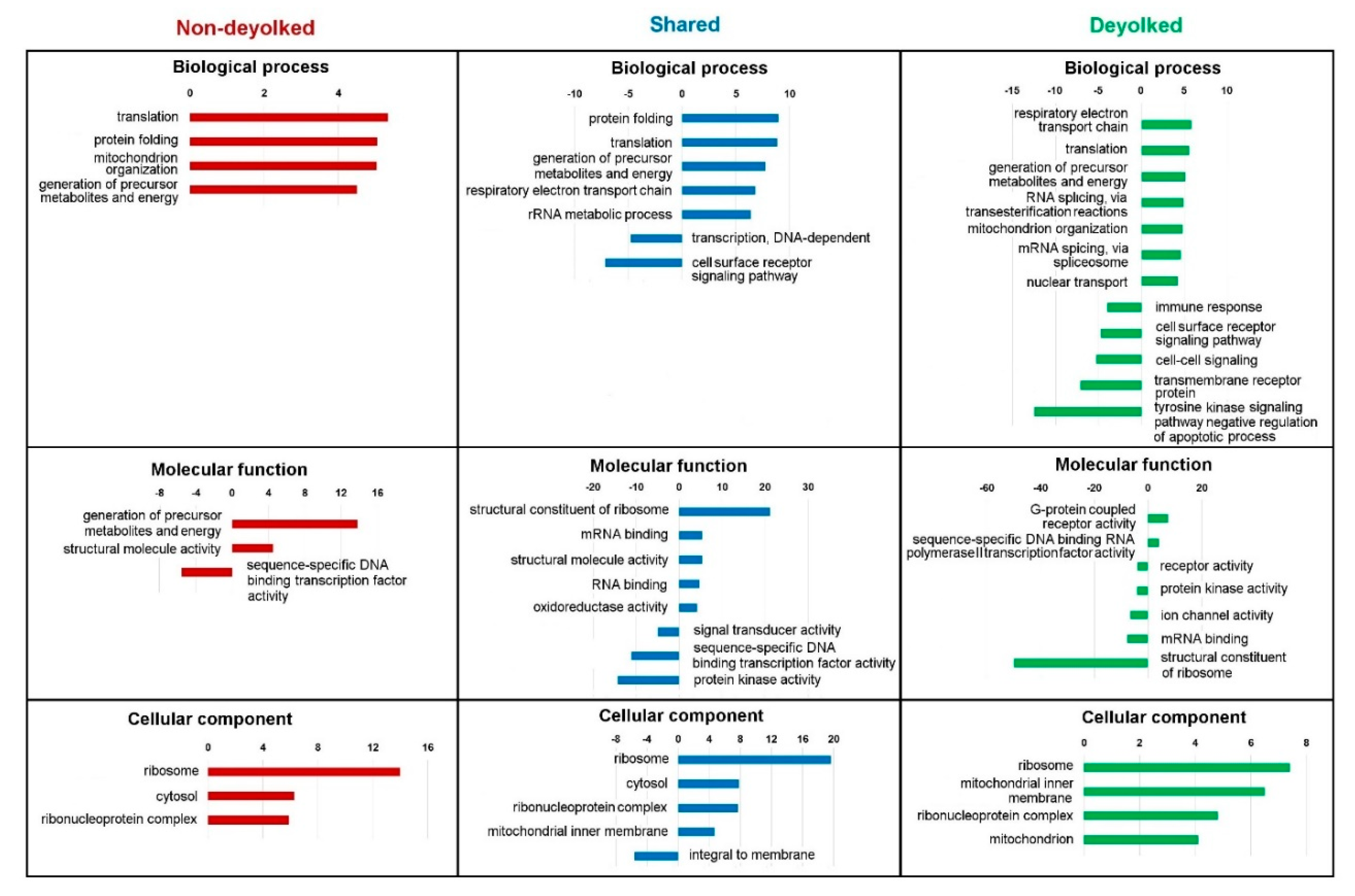
2.3. Functional Annotations of the Proteome
3. Discussion
4. Materials and Methods
4.1. Fish
4.2. Sample Collection
4.3. Dechorionation and Deyolking
4.4. Protein Extraction
4.5. Polyacrylamide Gel Electrophoresis
4.6. Tube-Gel Digestion and Sample Clean up
4.7. 1D LC–MS/MS Analysis
4.8. Protein Identification and Quantification
4.9. KEGG and Gene Ontology (GO) Functional Pathways Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| ND | Non-deyolked |
| DY | Deyolked |
| SH | Shared |
| iTRAQ | Isobaric tag for relative and absolute quantitation |
| LC–MS/MS | Liquid chromatography–tandem mass spectrometry |
| 1D LC–MS/MS | One-dimension liquid chromatography–tandem mass spectrometry |
| MZT | Maternal-to-zygotic transition |
| PBS | Phosphate-buffered saline |
| SDS | Sodium dodecyl sulfate |
| TCEP | Tris-(2-carboxyethyl) phosphine |
| MMTS | Methyl methane-thiosulfonate |
| TEAB | Triethylammonium bicarbonate |
| ACN | Acetonitrile |
| FDR | False discovery rate |
| COG | Clusters of orthologous groups |
| GO | Gene Ontology |
| KEGG | Kyoto Encyclopedia of Genes and Genomes |
References
- Amsterdam, A.; Hopkins, N. Mutagenesis strategies in zebrafish for identifying genes involved in development and disease. Trends Genet. 2006, 22, 473–478. [Google Scholar] [CrossRef] [PubMed]
- Brownlie, A.; Donovan, A.; Pratt, S.J.; Paw, B.H.; Oates, A.C.; Brugnara, C.; Witkowska, H.E.; Sassa, S.; Zon, L.I. Positional cloning of the zebrafish sauternes gene: A model for congenital sideroblastic anaemia. Nat. Genet. 1998, 20, 244. [Google Scholar] [CrossRef] [PubMed]
- Grosser, T.; Yusuff, S.; Cheskis, E.; Pack, M.A.; FitzGerald, G.A. Developmental expression of functional cyclooxygenases in zebrafish. Proc. Natl. Acad. Sci. USA. 2002, 99, 8418–8423. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gündel, U.; Benndorf, D.; von Bergen, M.; Altenburger, R.; Küster, E. Vitellogenin cleavage products as indicators for toxic stress in zebra fish embryos: A proteomic approach. Proteom. 2007, 7, 4541–4554. [Google Scholar] [CrossRef] [PubMed]
- Hanisch, K.; Küster, E.; Altenburger, R.; Gündel, U. Proteomic signatures of the zebrafish (Danio rerio) embryo: Sensitivity and specificity in toxicity assessment of chemicals. Int. J. Proteom. 2010. [CrossRef] [PubMed] [Green Version]
- Howe, K.; Clark, M.D.; Torroja, C.F.; Torrance, J.; Berthelot, C.; Muffato, M.; Collins, J.E.; Humphray, S.; McLaren, K.; Matthews, L. The zebrafish reference genome sequence and its relationship to the human genome. Nature 2013, 496, 498–503. [Google Scholar] [CrossRef] [Green Version]
- Aanes, H.; Winata, C.L.; Lin, C.H.; Chen, J.P.; Srinivasan, K.G.; Lee, S.G.; Lim, A.Y.; Hajan, H.S.; Collas, P.; Bourque, G. Zebrafish mRNA sequencing deciphers novelties in transcriptome dynamics during maternal to zygotic transition. Genome. Res. 2011, 21, 1328–1338. [Google Scholar] [CrossRef] [Green Version]
- Heyn, P.; Kircher, M.; Dahl, A.; Kelso, J.; Tomancak, P.; Kalinka, A.T.; Neugebauer, K.M. The earliest transcribed zygotic genes are short, newly evolved, and different across species. Cell Rep. 2014, 6, 285–292. [Google Scholar] [CrossRef] [Green Version]
- Mehjabin, R.; Xiong, L.; Huang, R.; Yang, C.; Chen, G.; He, L.; Liao, L.; Zhu, Z.; Wang, Y. Full-Length Transcriptome Sequencing and the Discovery of New Transcripts in the Unfertilized Eggs of Zebrafish (Danio rerio). G3 2019. [Google Scholar] [CrossRef] [Green Version]
- Nudelman, G.; Frasca, A.; Kent, B.; Sadler, K.C.; Sealfon, S.C.; Walsh, M.J.; Zaslavsky, E. High resolution annotation of zebrafish transcriptome using long-read sequencing. Genome. Res. 2018, 28, 1415–1425. [Google Scholar] [CrossRef] [Green Version]
- White, R.J.; Collins, J.E.; Sealy, I.M.; Wali, N.; Dooley, C.M.; Digby, Z.; Stemple, D.L.; Murphy, D.N.; Billis, K.; Hourlier, T. A high-resolution mRNA expression time course of embryonic development in zebrafish. Elife 2017. [Google Scholar] [CrossRef] [PubMed]
- De Souza, A.G.; Mac Cormack, T.J.; Wang, N.; Li, L.; Goss, G.G. Large-scale proteome profile of the zebrafish (Danio rerio) gill for physiological and biomarker discovery studies. Zebrafish 2009, 6, 229–238. [Google Scholar] [CrossRef] [PubMed]
- Groh, K.J.; Nesatyy, V.J.; Segner, H.; Eggen, R.I.; Suter, M.J.-F. Global proteomics analysis of testis and ovary in adult zebrafish (Danio rerio). Fish Physiol. Biochem. 2011, 37, 619–647. [Google Scholar] [CrossRef] [Green Version]
- Singh, S.K.; Sundaram, C.S.; Shanbhag, S.; Idris, M.M. Proteomic profile of zebrafish brain based on two-dimensional gel electrophoresis matrix-assisted laser desorption/ionization MS/MS analysis. Zebrafish 2010, 7, 169–177. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Lanham, K.A.; Peterson, R.E.; Heideman, W.; Li, L. Characterization of the adult zebrafish cardiac proteome using online pH gradient strong cation exchange-RP 2D LC coupled with ESI MS/MS. J. Sep. Sci. 2010, 33, 1462–1471. [Google Scholar] [CrossRef] [PubMed]
- Link, V.; Shevchenko, A.; Heisenberg, C.-P. Proteomics of early zebrafish embryos. Bmc. Dev. Biol. 2006. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, W.; Yin, X.; Liu, X.; Yan, G.; Yang, P. Response of peptide intensity to concentration in ESI-MS-based proteome. Sci. China Chem. 2014, 57, 686–694. [Google Scholar] [CrossRef]
- Vaidyanathan, S.; Kell, D.B.; Goodacre, R. Selective detection of proteins in mixtures using electrospray ionization mass spectrometry: Influence of instrumental settings and implications for proteomics. Anal. Chem. 2004, 76, 5024–5032. [Google Scholar] [CrossRef]
- Lucitt, M.B.; Price, T.S.; Pizarro, A.; Wu, W.; Yocum, A.K.; Seiler, C.; Pack, M.A.; Blair, I.A.; FitzGerald, G.A.; Grosser, T. Analysis of the zebrafish proteome during embryonic development. Mol. Cell Proteom. 2008, 7, 981–994. [Google Scholar] [CrossRef] [Green Version]
- Rahlouni, F.; Szarka, S.; Shulaev, V.; Prokai, L. A Survey of the Impact of Deyolking on Biological Processes Covered by Shotgun Proteomic Analyses of Zebrafish Embryos. Zebrafish 2015, 12, 398–407. [Google Scholar] [CrossRef] [Green Version]
- Tay, T.L.; Lin, Q.; Seow, T.K.; Tan, K.H.; Hew, C.L.; Gong, Z. Proteomic analysis of protein profiles during early development of the zebrafish, Danio rerio. Proteomics 2006, 6, 3176–3188. [Google Scholar] [CrossRef] [PubMed]
- Shaik, A.A.; Wee, S.; Li, R.H.X.; Li, Z.; Carney, T.J.; Mathavan, S.; Gunaratne, J. Functional mapping of the zebrafish early embryo proteome and transcriptome. J. Proteome Res. 2014, 13, 5536–5550. [Google Scholar] [CrossRef] [PubMed]
- Lößner, C.; Wee, S.; Ler, S.G.; Li, R.H.; Carney, T.; Blackstock, W.; Gunaratne, J. Expanding the zebrafish embryo proteome using multiple fractionation approaches and tandem mass spectrometry. Proteomics 2012, 12, 1879–1882. [Google Scholar] [CrossRef] [PubMed]
- Tadros, W.; Lipshitz, H.D. The maternal-to-zygotic transition: A play in two acts. Development 2009, 136, 3033–3042. [Google Scholar] [CrossRef] [Green Version]
- Gong, Y.; Mo, C.; Fraser, S.E. Planar cell polarity signalling controls cell division orientation during zebrafish gastrulation. Nature 2004, 430, 689. [Google Scholar] [CrossRef]
- Sugiyama, M.; Sakaue-Sawano, A.; Iimura, T.; Fukami, K.; Kitaguchi, T.; Kawakami, K.; Okamoto, H.; Higashijima, S.-i.; Miyawaki, A. Illuminating cell-cycle progression in the developing zebrafish embryo. Proc. Natl. Acad. Sci. USA 2009, 106, 20812–20817. [Google Scholar] [CrossRef] [Green Version]
- Artuso, L.; Romano, A.; Verri, T.; Domenichini, A.; Argenton, F.; Santorelli, F.M.; Petruzzella, V. Mitochondrial DNA metabolism in early development of zebrafish (Danio rerio). Biochim. Biophys. Acta. 2012, 1817, 1002–1011. [Google Scholar] [CrossRef]
- Sun, J.; Yan, L.; Shen, W.; Meng, A. Maternal Ybx1 safeguards zebrafish oocyte maturation and maternal-to-zygotic transition by repressing global translation. Development 2018. [Google Scholar] [CrossRef] [Green Version]
- Lee, M.T.; Bonneau, A.R.; Giraldez, A.J. Zygotic genome activation during the maternal-to-zygotic transition. Annu. Rev. Cell Dev. Biol. 2014, 30, 581–613. [Google Scholar] [CrossRef] [Green Version]
- Lubzens, E.; Bobe, J.; Young, G.; Sullivan, C.V. Maternal investment in fish oocytes and eggs: The molecular cargo and its contributions to fertility and early development. Aquaculture 2017, 472, 107–143. [Google Scholar] [CrossRef] [Green Version]
- Westerfield, M. A guide for the laboratory use of zebrafish (Danio rerio). The Zebrafish Book 2000, 4. [Google Scholar]
- Kimmel, C.B.; Ballard, W.W.; Kimmel, S.R.; Ullmann, B.; Schilling, T.F. Stages of embryonic development of the zebrafish. Dev. Dyn. 1995, 203, 253–310. [Google Scholar] [CrossRef] [PubMed]
- Suriyanarayanan, T.; Qingsong, L.; Kwang, L.T.; Mun, L.Y.; Seneviratne, C.J. Quantitative proteomics of strong and weak biofilm formers of Enterococcus faecalis reveals novel regulators of biofilm formation. Mol. Cell Proteom. 2018, 17, 643–654. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moriya, Y.; Itoh, M.; Okuda, S.; Yoshizawa, A.C.; Kanehisa, M. KAAS: An automatic genome annotation and pathway reconstruction server. Nucleic. Acids Res. 2007, 35, W182–W185. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kanehisa, M.; Sato, Y.; Kawashima, M.; Furumichi, M.; Tanabe, M. KEGG as a reference resource for gene and protein annotation. Nucleic. Acids Res. 2015, 44, D457–D462. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mi, H.; Huang, X.; Muruganujan, A.; Tang, H.; Mills, C.; Kang, D.; Thomas, P.D. PANTHER version 11: Expanded annotation data from Gene Ontology and Reactome pathways, and data analysis tool enhancements. Nucleic. Acids Res. 2016, 45, D183–D189. [Google Scholar] [CrossRef] [Green Version]
- Zerbino, D.R.; Achuthan, P.; Akanni, W.; Amode, M.R.; Barrell, D.; Bhai, J.; Billis, K.; Cummins, C.; Gall, A.; Girón, C.G.; et al. Ensembl 2018. Nucleic. Acids Res. 2017, 46, D754–D761. [Google Scholar] [CrossRef]
- Apweiler, R.; Bairoch, A.; Wu, C.H.; Barker, W.C.; Boeckmann, B.; Ferro, S.; Gasteiger, E.; Huang, H.; Lopez, R.; Magrane, M. UniProt: The universal protein knowledgebase. Nucleic. Acids Res. 2004, 32, D115–D119. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Developmental Stage | Number of Embryos | Total Sample Volume (µL) | Amount of Extracted Protein (µg) | ||
|---|---|---|---|---|---|
| Total | Per µL | Per Embryo | |||
| Non-deyolked | |||||
| 1-cell | 28 | 119 | 395.08 | 3.32 | 14.11 |
| 16-cell | 20 | 88 | 327.36 | 3.72 | 16.37 |
| 32-cell | 40 | 170 | 697.00 | 4.10 | 17.42 |
| Oblong | 20 | 120 | 478.80 | 3.99 | 23.94 |
| Bud | 20 | 92 | 524.40 | 5.70 | 26.22 |
| Deyolked | |||||
| 1-cell | 575 | 94 | 304.56 | 3.24 | 0.53 |
| 16-cell | 300 | 58 | 191.41 | 3.30 | 0.63 |
| 32-cell | 400 | 99 | 277.22 | 2.81 | 0.69 |
| Oblong | 225 | 42 | 246.54 | 5.87 | 1.09 |
| Bud | 250 | 70 | 413.70 | 5.91 | 1.65 |
| Pathway Name | ND-Cleavage Stage-Unique | ND- Oblong & Bud Stages Unique | ND-Common in All Stages | DY-Cleavage Stage Unique | DY-Oblong & Bud Stages Unique | DY-Common in All Stages | Shared Proteins |
|---|---|---|---|---|---|---|---|
| map01100 Metabolic pathways | 22 | 23 | 6 | 66 | 62 | 87 | 69 |
| map03010 Ribosome | 27 | 11 | 2 | 21 | 19 | 11 | 52 |
| map01110 Biosynthesis of secondary metabolites | 12 | 11 | 3 | 21 | 25 | 27 | 32 |
| map04714 Thermogenesis | 2 | 6 | 1 | 28 | 6 | 37 | 20 |
| map01200 Carbon metabolism | 6 | 6 | 1 | 10 | 12 | 21 | 23 |
| map04141 Protein processing in endoplasmic reticulum | 4 | 0 | 0 | 5 | 14 | 11 | 20 |
| map03050 Proteasome | 14 | 4 | 1 | 1 | 15 | 0 | 14 |
| map00010 Glycolysis / Gluconeogenesis | 6 | 3 | 0 | 2 | 6 | 6 | 12 |
| map00071 Fatty acid degradation | 3 | 1 | 0 | 9 | 1 | 11 | 9 |
| map01212 Fatty acid metabolism | 8 | 3 | 0 | 7 | 0 | 10 | 8 |
| map04530 Tight junction | 5 | 5 | 2 | 6 | 13 | 2 | 7 |
| map03013 RNA transport | 7 | 0 | 0 | 3 | 22 | 1 | 6 |
| map04110 Cell cycle | 9 | 2 | 0 | 3 | 12 | 1 | 5 |
| map04810 Regulation of actin cytoskeleton | 5 | 1 | 1 | 6 | 12 | 1 | 4 |
| map04144 Endocytosis | 3 | 0 | 0 | 3 | 10 | 6 | 4 |
| map00230 Purine metabolism | 5 | 5 | 1 | 2 | 11 | 1 | 4 |
| map03018 RNA degradation | 3 | 0 | 0 | 0 | 8 | 1 | 4 |
| map04210 Apoptosis | 0 | 4 | 1 | 4 | 5 | 1 | 4 |
| map00970 Aminoacyl-tRNA biosynthesis | 2 | 1 | 1 | 8 | 12 | 1 | 3 |
| map03030 DNA replication | 4 | 3 | 0 | 0 | 14 | 0 | 3 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Purushothaman, K.; Das, P.P.; Presslauer, C.; Lim, T.K.; Johansen, S.D.; Lin, Q.; Babiak, I. Proteomics Analysis of Early Developmental Stages of Zebrafish Embryos. Int. J. Mol. Sci. 2019, 20, 6359. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20246359
Purushothaman K, Das PP, Presslauer C, Lim TK, Johansen SD, Lin Q, Babiak I. Proteomics Analysis of Early Developmental Stages of Zebrafish Embryos. International Journal of Molecular Sciences. 2019; 20(24):6359. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20246359
Chicago/Turabian StylePurushothaman, Kathiresan, Prem Prakash Das, Christopher Presslauer, Teck Kwang Lim, Steinar D. Johansen, Qingsong Lin, and Igor Babiak. 2019. "Proteomics Analysis of Early Developmental Stages of Zebrafish Embryos" International Journal of Molecular Sciences 20, no. 24: 6359. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms20246359