Computational Disorder Analysis in Ethylene Response Factors Uncovers Binding Motifs Critical to Their Diverse Functions

 ,
,
Abstract
:
1. Introduction
2. Results and Discussion
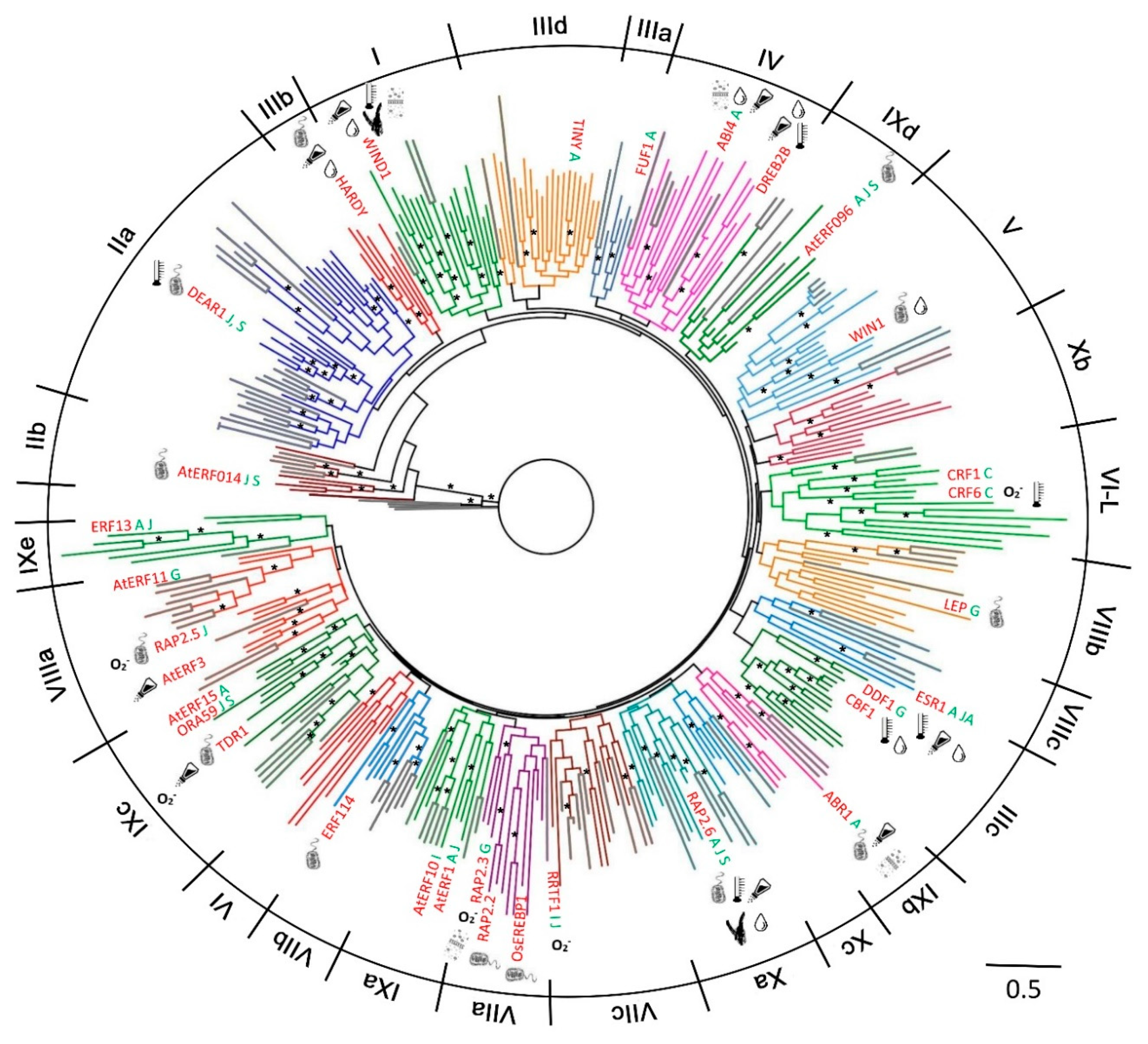
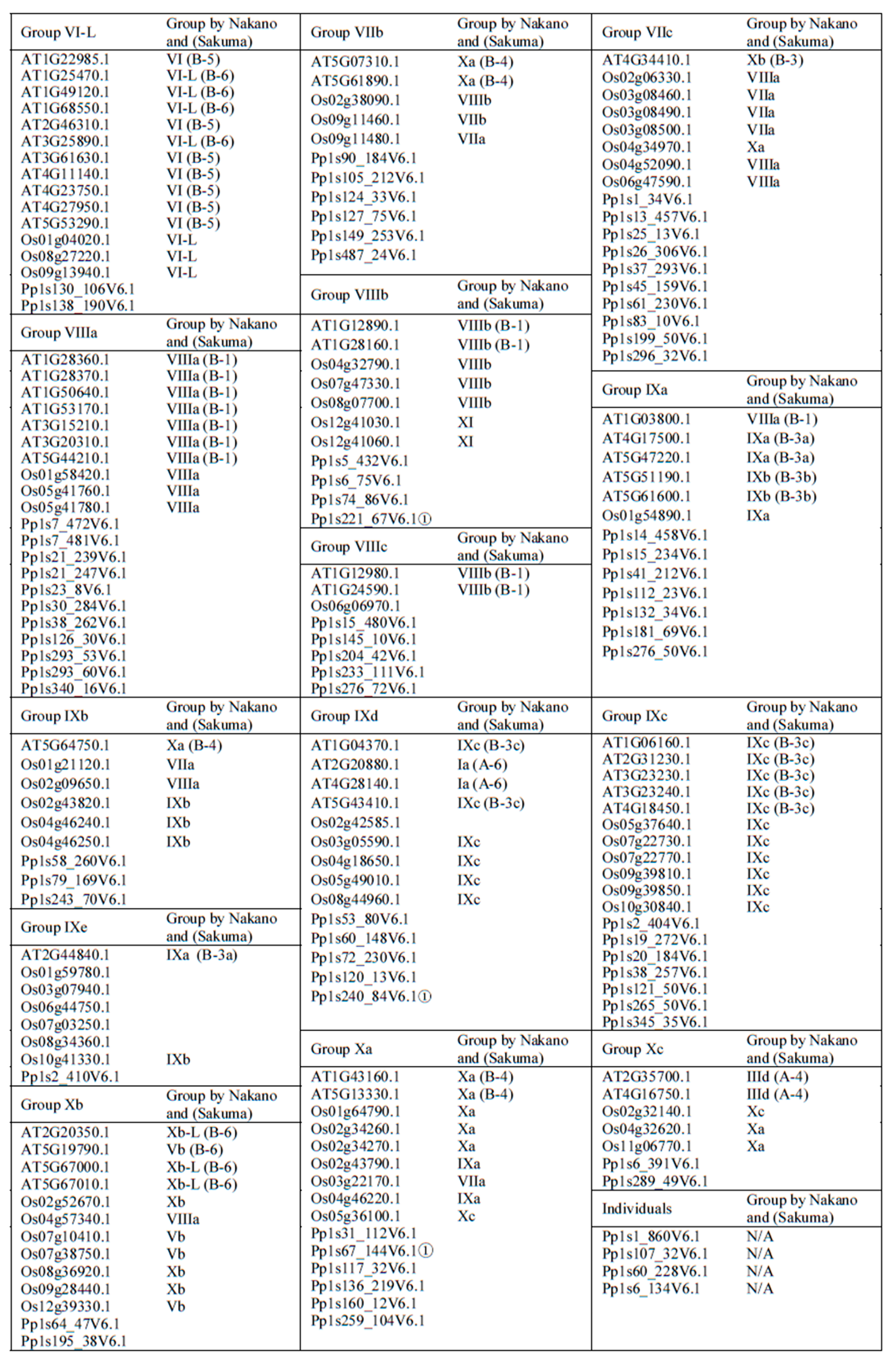
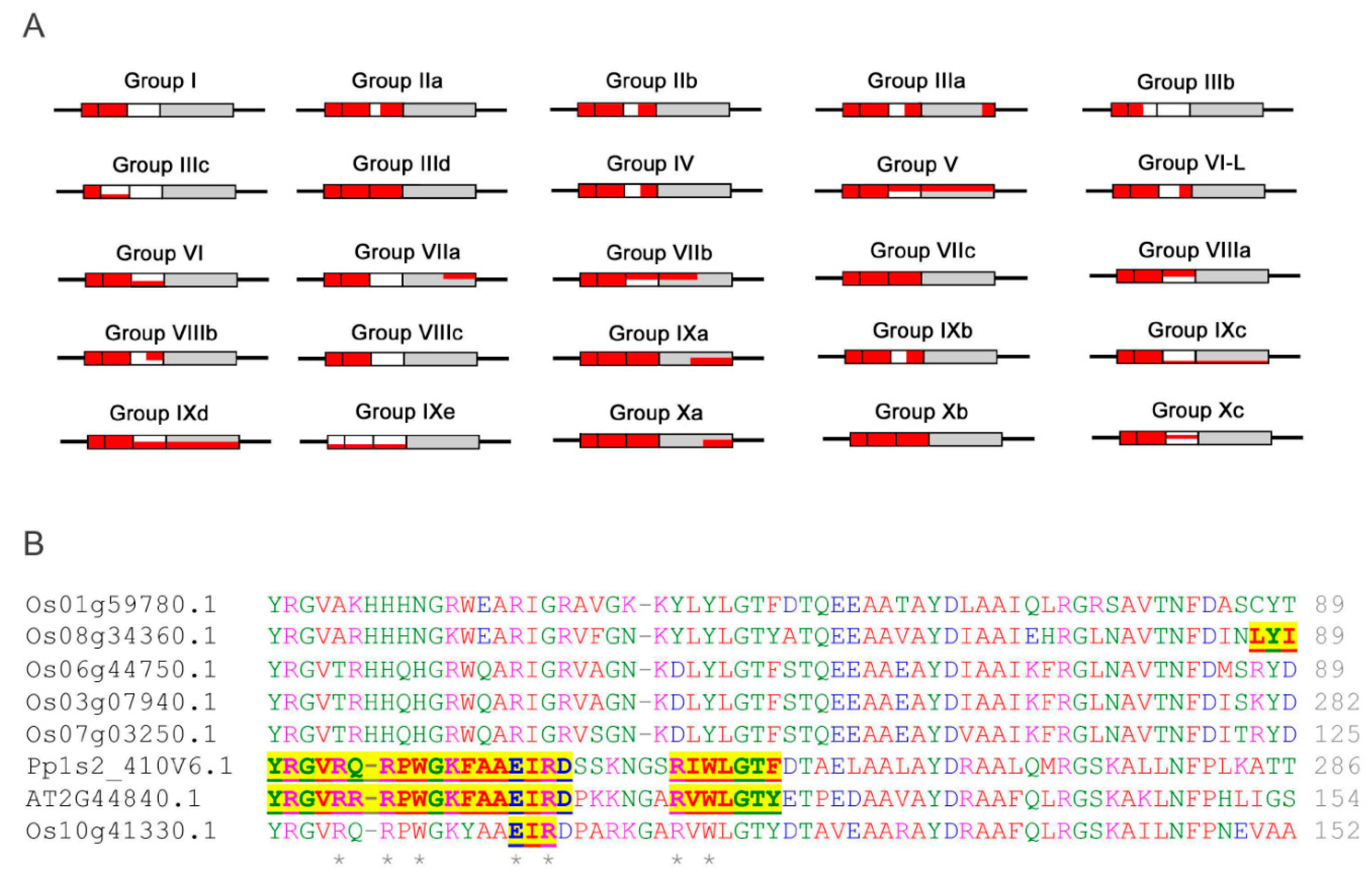
2.1. Phylogenetic Analysis and Classification of ERF Families
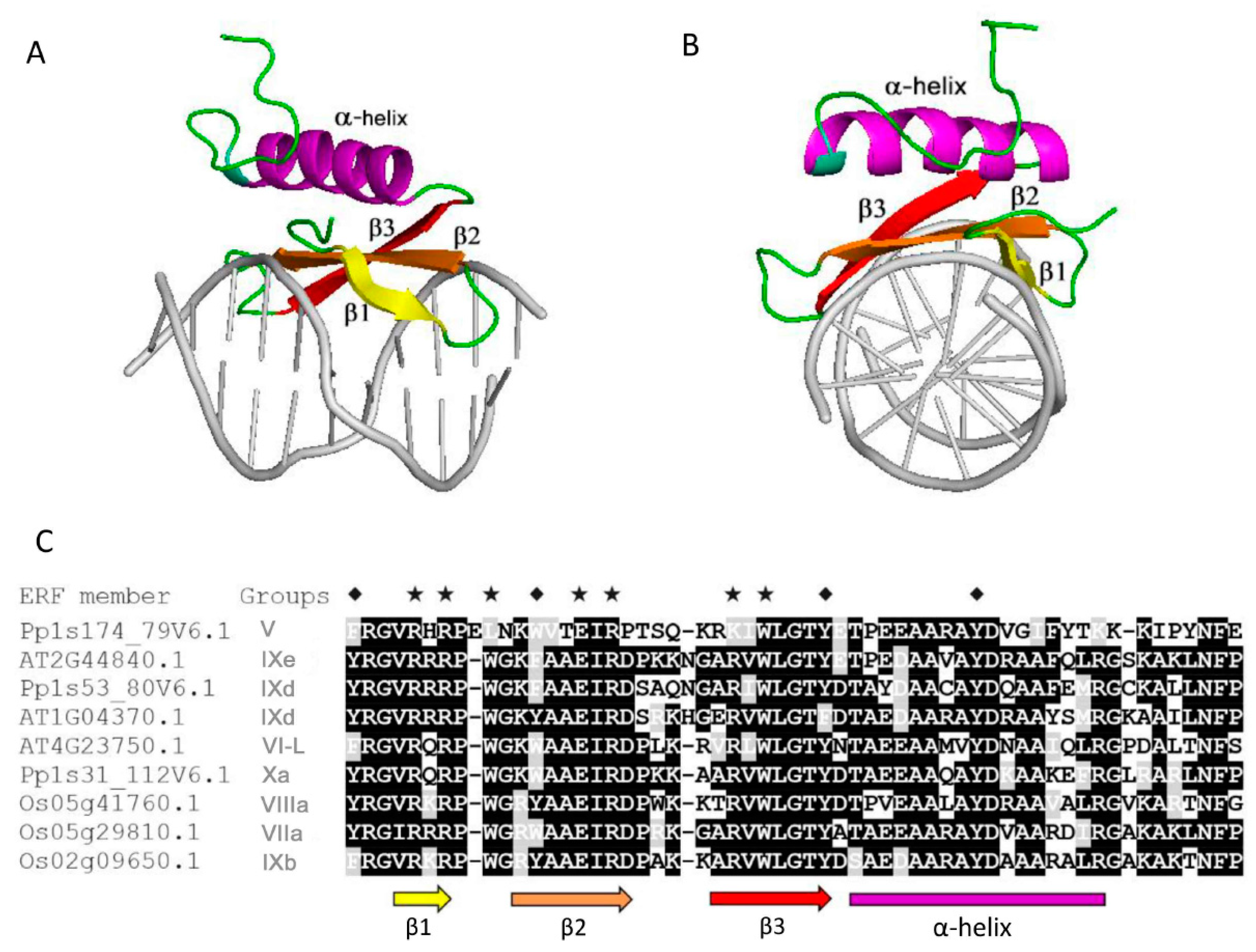
2.2. Multiple Analyses Show that the TRDs Domain of ERF Proteins are Intrinsically Disordered
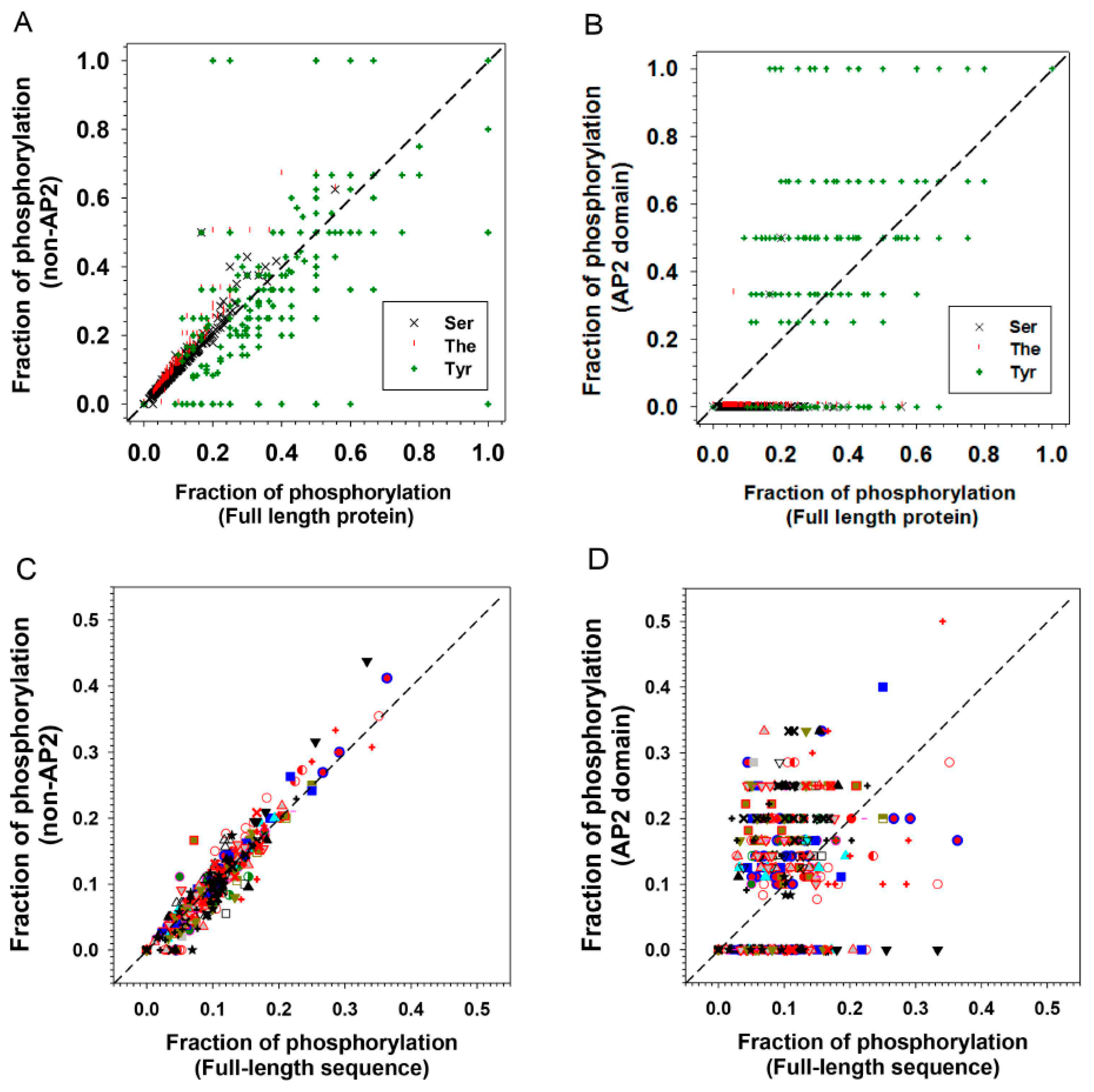
2.3. Phosphorylation/Dephosphorylation is Involved in ERF Protein Interactions
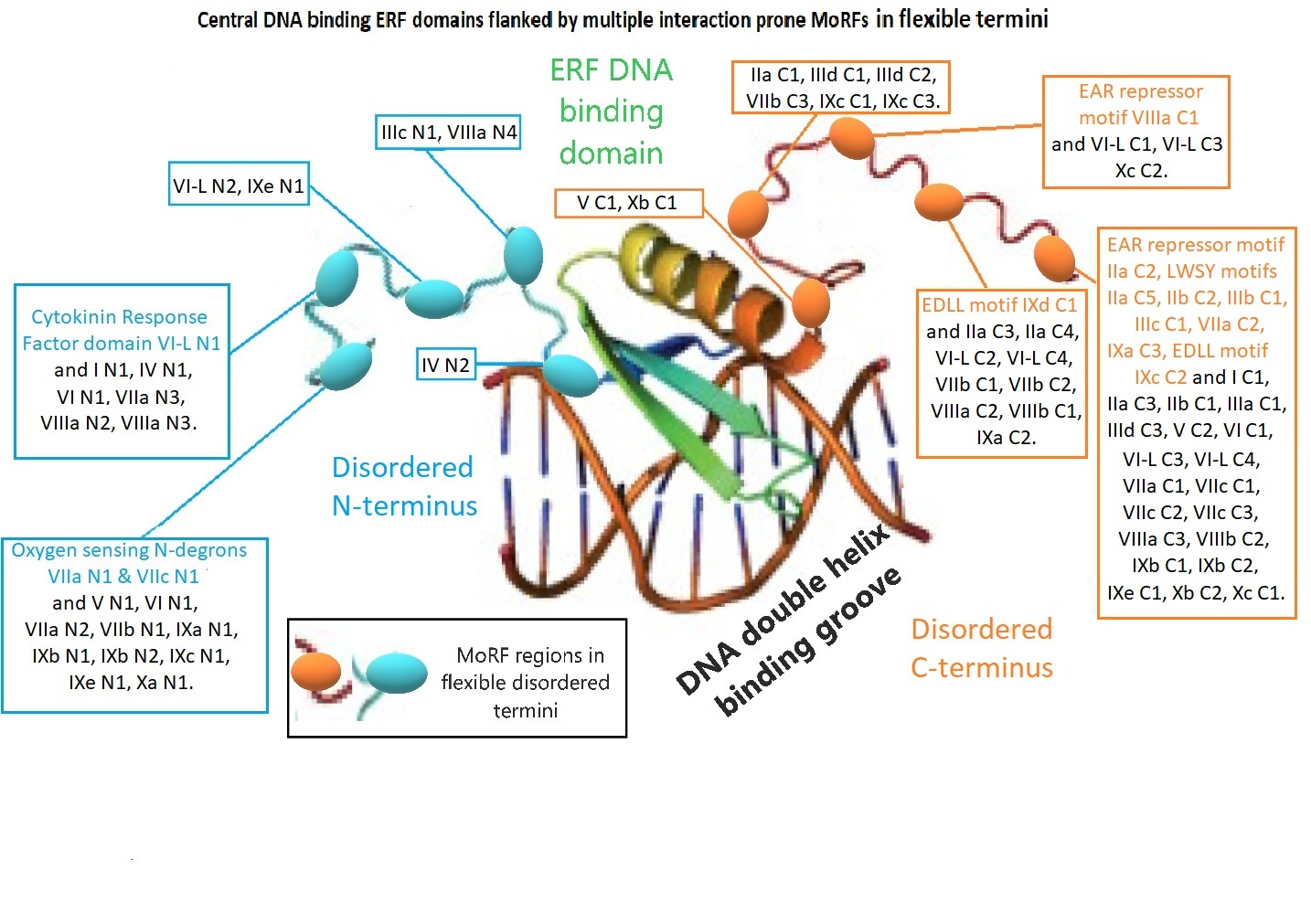
2.4. Analyses of MoRFs in ERF Groups and Their Functional Implications
2.5. Conserved MoRFs are Potentially Mobile Through Evolution
3. Materials and Methods
3.1. Sequence and Phylogenetic Analysis
3.2. Amino Acids Compositional Profile Analysis
3.3. Charge-Hydropathy (CH) and Cumulative Distribution Function (CDF) Plot
3.4. Disorder Predictions
3.5. Predictions of Phosphorylation Sites
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Muller, M.; Munne-Bosch, S. Ethylene response factors: A key regulatory hub in hormone and stress signaling. Plant Physiol. 2015, 169, 32–41. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pre, M.; Atallah, M.; Champion, A.; De Vos, M.; Pieterse, C.M.J.; Memelink, J. The AP2/ERF domain transcription factor ORA59 integrates jasmonic acid and ethylene signals in plant defense. Plant Physiol. 2008, 147, 1347–1357. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Golldack, D.; Li, C.; Mohan, H.; Probst, N. Tolerance to drought and salt stress in plants: Unraveling the signaling networks. Front. Plant Sci. 2014, 5, 151. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dey, S.; Vlot, A.C. Ethylene responsive factors in the orchestration of stress responses in monocotyledonous plants. Front. Plant Sci. 2015, 6, 640. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakano, T.; Suzuki, K.; Fujimura, T.; Shinshi, H. Genome-wide analysis of the ERF gene family in Arabidopsis and Rice. Plant Physiol. 2006, 140, 411–432. [Google Scholar] [CrossRef] [Green Version]
- Sakuma, Y.; Liu, Q.; Dubouzet, J.G.; Abe, H.; Shinozaki, K.; Yamaguchi-Shinozaki, K. DNA-binding specificity of the ERF/AP2 domain of Arabidopsis DREBS, transcription factors involved in dehydration- and cold-inducible gene expression. Biochem. Biophys. Res. Commun. 2002, 290, 998–1009. [Google Scholar] [CrossRef]
- Uversky, V.N. The mysterious unfoldome: Structureless, underappreciated, yet vital part of any given proteome. J. Biomed. Biotechnol. 2010, 2010, 568068. [Google Scholar] [CrossRef]
- Liu, J.G.; Perumal, N.B.; Oldfield, C.J.; Su, E.W.; Uversky, V.N.; Dunker, A.K. Intrinsic disorder in transcription factors. Biochemistry 2006, 45, 6873–6888. [Google Scholar] [CrossRef] [Green Version]
- Patil, A.; Nakamura, H. Disordered domains and high surface charge confer hubs with the ability to interact with multiple proteins in interaction networks. FEBS Lett. 2006, 580, 2041–2045. [Google Scholar] [CrossRef] [Green Version]
- Oldfield, C.J.; Meng, J.; Yang, J.Y.; Yang, M.Q.; Uversky, V.N.; Dunker, A.K. Flexible nets: Disorder and induced fit in the associations of p53 and 14-3-3 with their partners. BMC Genomics 2008, 9 (Suppl. S1). [Google Scholar] [CrossRef] [Green Version]
- Sun, X.; Jones, W.T.; Rikkerink, E.H.A. GRAS proteins: The versatile roles of intrinsically disordered proteins in plant signalling. Biochem. J. 2012, 442, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kjaersgaard, T.; Jensen, M.K.; Christiansen, M.W.; Gregersen, P.; Kragelund, B.B.; Skriver, K. Senescence-associated barley NAC (NAM, ATAF1,2, CUC) transcription factor interacts with radical-induced cell death 1 through a disordered regulatory domain. J. Biol. Chem. 2011, 286, 35418–35429. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, X.; Rikkerink, E.H.A.; Jones, W.T.; Uversky, V.N. Multifarious roles of intrinsic disorder in proteins illustrate its broad impact on plant biology. Plant Cell 2013, 25, 38–55. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yruela, I.; Oldfield, C.J.; Niklas, K.J.; Dunker, A.K. Evidence for a strong correlation between transcription factor protein disorder and organismic complexity. Genome Biol. Evol. 2017, 9, 1248–1265. [Google Scholar] [CrossRef]
- Niklas, K.J.; Dunker, A.K.; Yruela, I. The evolutionary origins of cell type diversification and the role of intrinsically disordered proteins. J. Exp. Bot. 2018, 69, 1437–1446. [Google Scholar] [CrossRef] [PubMed]
- Allen, M.D.; Yamasaki, K.; Ohme-Takagi, M.; Tateno, M.; Suzuki, M. A novel mode of DNA recognition by a Beta-sheet revealed by the solution structure of the GCC-box binding domain in complex with DNA. EMBO J. 1998, 17, 5484–5496. [Google Scholar] [CrossRef] [Green Version]
- Dunker, A.K.; Lawson, J.D.; Brown, C.J.; Williams, R.M.; Romero, P.; Oh, J.S.; Oldfield, C.J.; Campen, A.M.; Ratliff, C.R.; Hipps, K.W.; et al. Intrinsically disordered protein. J. Mol. Gr. Model. 2001, 19, 26–59. [Google Scholar] [CrossRef] [Green Version]
- Uversky, V.N.; Gillespie, J.R.; Fink, A.L. Why are “natively unfolded” proteins unstructured under physiologic conditions? Proteins-Struct. Funct. Genet. 2000, 41, 415–427. [Google Scholar] [CrossRef]
- Sun, X.; Jones, W.T.; Uversky, V.N. Applications of bioinformatics and experimental methods to intrinsic disorder-based protein-protein interactions. In Protein Engineering; Kaumaya, P., Ed.; InTech: Rijeka, Croatia, 2012; pp. 181–206. [Google Scholar]
- Xue, B.; Oldfield, C.J.; Dunker, A.K.; Uversky, V.N. Cdf it all: Consensus prediction of intrinsically disordered proteins based on various cumulative distribution functions. FEBS Lett. 2009, 583, 1469–1474. [Google Scholar] [CrossRef] [Green Version]
- Mohan, A.; Sullivan, W.J.; Radivojac, P.; Dunker, A.K.; Uversky, V.N. Intrinsic disorder in pathogenic and non-pathogenic microbes: Discovering and analyzing the unfoldomes of early-branching eukaryotes. Mol. Biosyst. 2008, 4, 328–340. [Google Scholar] [CrossRef]
- Yoon, M.K.; Shin, J.; Choi, G.; Choi, B.S. Intrinsically unstructured n-terminal domain of BZIP transcription factor HY5. Proteins-Struct. Funct. Bioinforma. 2006, 65, 856–866. [Google Scholar] [CrossRef] [PubMed]
- Romero, P.; Obradovic, Z.; Li, X.H.; Garner, E.C.; Brown, C.J.; Dunker, A.K. Sequence complexity of disordered protein. Proteins-Struct. Funct. Genet. 2001, 42, 38–48. [Google Scholar] [CrossRef]
- Promponas, V.J.; Enright, A.J.; Tsoka, S.; Kreil, D.P.; Leroy, C.; Hamodrakas, S.; Sander, C.; Ouzounis, C.A. Cast: An iterative algorithm for the complexity analysis of sequence tracts. Bioinformatics 2000, 16, 915–922. [Google Scholar] [CrossRef]
- Mittag, T.; Kay, L.E.; Forman-Kay, J.D. Protein dynamics and conformational disorder in molecular recognition. J. Mol. Recognit. 2010, 23, 105–116. [Google Scholar] [CrossRef] [PubMed]
- Bah, A.; Vernon, R.M.; Siddiqui, Z.; Krzeminski, M.; Muhandiram, R.; Zhao, C.; Sonenberg, N.; Kay, L.E.; Forman-Kay, J.D. Folding of an intrinsically disordered protein by phosphorylation as a regulatory switch. Nature 2015, 519, 106–109. [Google Scholar] [CrossRef]
- Chung, E.-H.; El-Kasmi, F.; He, Y.; Loehr, A.; Dangl, J.L. A plant phosphoswitch platform repeatedly targeted by type III effector proteins regulates the output of both tiers of plant immune receptors. Cell Host Microbe 2014, 16, 484–494. [Google Scholar] [CrossRef] [Green Version]
- Hussain, A.; Cao, D.N.; Peng, J.R. Identification of conserved tyrosine residues important for gibberellin sensitivity of Arabidopsis RGL2 protein. Planta 2007, 226, 475–483. [Google Scholar] [CrossRef]
- Iakoucheva, L.M.; Radivojac, P.; Brown, C.J.; O’Connor, T.R.; Sikes, J.G.; Obradovic, Z.; Dunker, A.K. The importance of intrinsic disorder for protein phosphorylation. Nucleic Acids Res. 2004, 32, 1037–1049. [Google Scholar] [CrossRef] [Green Version]
- Agarwal, P.; Agarwal, P.K.; Nair, S.; Sopory, S.K.; Reddy, M.K. Stress-inducible DREB2A transcription factor from Pennisetum glaucum is a phosphoprotein and its phosphorylation negatively regulates its DNA-binding activity. Mol. Genet. Genomics 2007, 277, 189–198. [Google Scholar] [CrossRef]
- Bethke, G.; Unthan, T.; Uhrig, J.; Poschl, Y.; Gust, A.; Scheel, D.; Lee, J. Flg22 regulates the release of an ethylene response factor substrate from MAP kinase 6 in Arabidopsis thaliana via ethylene signaling. Proc. Natl. Acad. Sci. USA 2009, 106, 8067–8072. [Google Scholar] [CrossRef] [Green Version]
- Nemoto, K.; Takemori, N.; Seki, M.; Shinozaki, K.; Sawasaki, T. Members of the plant CRK superfamily are capable of trans- and autophosphorylation of tyrosine residues. J. Biol. Chem. 2015, 290, 16665–16677. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, S.J.; Park, J.H.; Lee, M.H.; Yu, J.H.; Kim, S.Y. Isolation and functional characterization of CE1 binding proteins. BMC Plant Biol. 2010, 10, 277. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, X.; Xue, B.; Jones, W.T.; Rikkerink, E.; Dunker, A.K.; Uversky, V.N. A functionally required unfoldome from the plant kingdom: Intrinsically disordered N-terminal domains of GRAS proteins are involved in molecular recognition during plant development. Plant Mol. Biol. 2011, 77, 205–223. [Google Scholar] [CrossRef] [PubMed]
- Kranz, H.D.; Denekamp, M.; Greco, R.; Jin, H.; Leyva, A.; Meissner, R.C.; Petroni, K.; Urzainqui, A.; Bevan, M.; Martin, C.; et al. Towards functional characterisation of the members of the R2R3-MYB gene family from Arabidopsis thaliana. Plant J. 1998, 16, 263–276. [Google Scholar] [CrossRef] [PubMed]
- Eulgem, T.; Rushton, P.J.; Robatzek, S.; Somssich, I.E. The WRKY superfamily of plant transcription factors. Trends Plant Sci. 2000, 5, 199–206. [Google Scholar] [CrossRef]
- Hollenbeck, J.J.; McClain, D.L.; Oakley, M.G. The role of helix stabilizing residues in GCN4 basic region folding and DNA binding. Protein Sci. 2002, 11, 2740–2747. [Google Scholar] [CrossRef]
- Triezenberg, S.J. Structure and function of transcriptional activation domains. Curr. Opin. Genet. Dev. 1995, 5, 190–196. [Google Scholar] [CrossRef]
- Sun, X.L.; Jones, W.T.; Harvey, D.; Edwards, P.J.B.; Pascal, S.M.; Kirk, C.; Considine, T.; Sheerin, D.J.; Rakonjac, J.; Oldfield, C.J.; et al. N-terminal domains of DELLA proteins are intrinsically unstructured in the absence of interaction with GID1/gibberellic acid receptors. J. Biol. Chem. 2010, 285, 11557–11571. [Google Scholar] [CrossRef] [Green Version]
- Murase, K.; Hirano, Y.; Sun, T.P.; Hakoshima, T. Gibberellin-induced DELLA recognition by the gibberellin receptor GID1. Nature 2008, 456, 459–464. [Google Scholar] [CrossRef]
- Morohashi, K.; Minami, M.; Takase, H.; Hotta, Y.; Hiratsuka, K. Isolation and characterization of a novel Gras gene that regulates meiosis-associated gene expression. J. Biol. Chem. 2003, 278, 20865–20873. [Google Scholar] [CrossRef] [Green Version]
- Fujimoto, S.Y.; Ohta, M.; Usui, A.; Shinshi, H.; Ohme-Takagi, M. Arabidopsis ethylene-responsive element binding factors act as transcriptional activators or repressors of GCC box-mediated gene expression. Plant Cell 2000, 12, 393–404. [Google Scholar]
- Ohta, M.; Matsui, K.; Hiratsu, K.; Shinshi, H.; Ohme-Takagi, M. Repression domains of class II ERF transcriptional repressors share an essential motif for active repression. Plant Cell 2001, 13, 1959–1968. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blair, W.S.; Bogerd, H.; Cullen, B.R. Genetic-analysis indicates that the human foamy virus BEL-1 protein contains a transcription activation domain of the acidic class. J. Virol. 1994, 68, 3803–3808. [Google Scholar] [PubMed]
- Tanaka, M.; Herr, W. Reconstitution of transcriptional activation domains by reiteration of short peptide segments reveals the modular organization of a glutamine-rich activation domain. Mol. Cell. Biol. 1994, 14, 6056–6067. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tiwari, S.B.; Belachew, A.; Ma, S.F.; Young, M.; Ade, J.; Shen, Y.; Marion, C.M.; Holtan, H.E.; Bailey, A.; Stone, J.K.; et al. The EDLL motif: A potent plant transcriptional activation domain from AP2/ERF transcription factors. Plant J. 2012, 70, 855–865. [Google Scholar] [CrossRef]
- Mao, D.H.; Chen, C.Y. Colinearity and similar expression pattern of rice DREB1S reveal their functional conservation in the cold-responsive pathway. PLoS ONE 2012, 7, e47275. [Google Scholar] [CrossRef] [Green Version]
- Dubouzet, J.G.; Sakuma, Y.; Ito, Y.; Kasuga, M.; Dubouzet, E.G.; Miura, S.; Seki, M.; Shinozaki, K.; Yamaguchi-Shinozaki, K. OsDREB genes in rice, Oryza sativa l., encode transcription activators that function in drought-, high-salt- and cold-responsive gene expression. Plant J. 2003, 33, 751–763. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.B.; Triezenberg, S.J.; Thomashow, M.F.; Stockinger, E.J. Multiple hydrophobic motifs in Arabidopsis CBF1 COOH-terminus provide functional redundancy in trans-activation. Plant Mol. Biol. 2005, 58, 543–559. [Google Scholar] [CrossRef]
- Zhang, L.L.; Li, Z.J.; Li, J.F.; Wang, A.X. Ectopic overexpression of SSCBF1, a CRT/DRE-binding factor from the nightshade plant Solanum lycopersicoides, confers freezing and salt tolerance in transgenic Arabidopsis. PLoS ONE 2013, 8, e61810. [Google Scholar] [CrossRef] [Green Version]
- Tournier, B.; Sanchez-Ballesta, M.T.; Jones, B.; Pesquet, E.; Regad, F.; Latche, A.; Pech, J.C.; Bouzayen, M. New members of the tomato ERF family show specific expression pattern and diverse DNA-binding capacity to the GCC box element. FEBS Lett. 2003, 550, 149–154. [Google Scholar] [CrossRef] [Green Version]
- Gibbs, D.J.; Conde, J.V.; Berckhan, S.; Prasad, G.; Mendiondo, G.M.; Holdsworth, M.J. Group VII ethylene response factors coordinate oxygen and nitric oxide signal transduction and stress responses in plants. Plant Physiol. 2015, 169, 23–31. [Google Scholar] [CrossRef] [PubMed]
- Rashotte, A.M.; Goertzen, L.R. The CRF domain defines cytokinin response factor proteins in plants. BMC Plant Biol. 2010, 10, 74. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rashotte, A.M.; Mason, M.G.; Hutchison, C.E.; Ferreira, F.J.; Schaller, G.E.; Kieber, J.J. A subset of Arabidopsis AP2 transcription factors mediates cytokinin responses in concert with a two-component pathway. Proc. Natl. Acad. Sci. USA 2006, 103, 11081–11085. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cutcliffe, J.W.; Hellmann, E.; Heyl, A.; Rashotte, A.M. CRFs form protein-protein interactions with each other and with members of the cytokinin signalling pathway in Arabidopsis via the CRF domain. J. Exp. Bot. 2011, 62, 4995–5002. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dong, C.J.; Liu, J.Y. The Arabidopsis EAR-motif-containing protein RAP2.1 functions as an active transcriptional repressor to keep stress responses under tight control. BMC Plant Biol. 2010, 10, 47. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, J.G.; Stork, W.; Mudgett, M.B. Xanthomonas type III effector XopD desumoylates tomato transcription factor SlERF4 to suppress ethylene responses and promote pathogen growth. Cell Host Microbe 2013, 13, 143–154. [Google Scholar] [CrossRef] [Green Version]
- Vacic, V.; Uversky, V.N.; Dunker, A.K.; Lonardi, S. Composition profiler: A tool for discovery and visualization of amino acid composition differences. BMC Bioinform. 2007, 8, 211. [Google Scholar] [CrossRef] [Green Version]
- Huang, F.; Oldfield, C.; Meng, J.; Hsu, W.-L.; Xue, B.; Uversky, V.N.; Romero, P.; Dunker, A.K. Subclassifying disordered proteins by the CH-CDF plot method. Pac. Symp. Biocomput. 2012, 17, 128–139. [Google Scholar]
- Xue, B.; Dunbrack, R.L.; Williams, R.W.; Dunker, A.K.; Uversky, V.N. PONDR-FIT: A meta-predictor of intrinsically disordered amino acids. Biochim. Biophys. Acta 2010, 1804, 996–1010. [Google Scholar] [CrossRef] [Green Version]
- Petrovich, A.; Borne, A.; Uversky, V.N.; Xue, B. Identifying similar patterns of structural flexibility in proteins by disorder prediction and dynamic programming. Int. J. Mol. Sci. 2015, 16, 13829–13849. [Google Scholar] [CrossRef] [Green Version]
- Malhis, N.; Jacobson, M.; Gsponer, J. MoRFchibi SYSTEM: Software tools for the identification of MoRFs in protein sequences. Nucleic Acids Res. 2016, 44, W488–W493. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, J.J.; Thelen, J.J.; Dunker, A.K.; Xu, D. Musite, a tool for global prediction of general and kinase-specific phosphorylation sites. Mol. Cell. Proteomics 2010, 9, 2586–2600. [Google Scholar] [CrossRef] [PubMed] [Green Version]
 ; Cold/Heat
; Cold/Heat  ; Osmotic
; Osmotic  ; Oxidative
; Oxidative  ; Salt
; Salt  ; Water/Drought
; Water/Drought  ; Wounding
; Wounding  .
; Cold/Heat ; Osmotic ; Oxidative ; Salt ; Water/Drought ; Wounding .
.
; Cold/Heat ; Osmotic ; Oxidative ; Salt ; Water/Drought ; Wounding .




 , IIa
, IIa  , IIb
, IIb  , IIIa
, IIIa  , IIIb
, IIIb  , IIIc
, IIIc  , IIId
, IIId  , IV
, IV  , V
, V  , VI-L
, VI-L  , VI
, VI  , VIIa
, VIIa  , VIIb
, VIIb  , VIIc
, VIIc  , VIIIa
, VIIIa  , VIIIb
, VIIIb  , VIIIc
, VIIIc  , IXa
, IXa  , IXb
, IXb  , IXc
, IXc  , IXd
, IXd  , IXe
, IXe  , Xa
, Xa  , Xb
, Xb  , Xc
, Xc  .
, IIa , IIb , IIIa , IIIb , IIIc , IIId , IV , V , VI-L , VI , VIIa , VIIb , VIIc , VIIIa , VIIIb , VIIIc , IXa , IXb , IXc , IXd , IXe , Xa , Xb , Xc .
.
, IIa , IIb , IIIa , IIIb , IIIc , IIId , IV , V , VI-L , VI , VIIa , VIIb , VIIc , VIIIa , VIIIb , VIIIc , IXa , IXb , IXc , IXd , IXe , Xa , Xb , Xc . , IIa , IIb , IIIa , IIIb , IIIc , IIId , IV , V , VI-L , VI , VIIa , VIIb , VIIc , VIIIa , VIIIb , VIIIc , IXa , IXb , IXc , IXd , IXe , Xa , Xb , Xc .
, IIa , IIb , IIIa , IIIb , IIIc , IIId , IV , V , VI-L , VI , VIIa , VIIb , VIIc , VIIIa , VIIIb , VIIIc , IXa , IXb , IXc , IXd , IXe , Xa , Xb , Xc .
, IIa , IIb , IIIa , IIIb , IIIc , IIId , IV , V , VI-L , VI , VIIa , VIIb , VIIc , VIIIa , VIIIb , VIIIc , IXa , IXb , IXc , IXd , IXe , Xa , Xb , Xc .
, IIa , IIb , IIIa , IIIb , IIIc , IIId , IV , V , VI-L , VI , VIIa , VIIb , VIIc , VIIIa , VIIIb , VIIIc , IXa , IXb , IXc , IXd , IXe , Xa , Xb , Xc .

 , IIa
, IIa  , IIb
, IIb  , IIIa
, IIIa  , IIIb
, IIIb  , IIIc
, IIIc  , IIId
, IIId  , IV
, IV  , V
, V  , VI-L
, VI-L  , VI
, VI  , VIIa
, VIIa  , VIIb
, VIIb  , VIIc
, VIIc  , VIIIa
, VIIIa  , VIIIb
, VIIIb  , VIIIc
, VIIIc  , IXa
, IXa  , IXb
, IXb  , IXc
, IXc  , IXd
, IXd  , IXe
, IXe  , Xa
, Xa  , Xb
, Xb  , Xc
, Xc  .
, IIa , IIb , IIIa , IIIb , IIIc , IIId , IV , V , VI-L , VI , VIIa , VIIb , VIIc , VIIIa , VIIIb , VIIIc , IXa , IXb , IXc , IXd , IXe , Xa , Xb , Xc .
.
, IIa , IIb , IIIa , IIIb , IIIc , IIId , IV , V , VI-L , VI , VIIa , VIIb , VIIc , VIIIa , VIIIb , VIIIc , IXa , IXb , IXc , IXd , IXe , Xa , Xb , Xc . , IIa , IIb , IIIa , IIIb , IIIc , IIId , IV , V , VI-L , VI , VIIa , VIIb , VIIc , VIIIa , VIIIb , VIIIc , IXa , IXb , IXc , IXd , IXe , Xa , Xb , Xc .
, IIa , IIb , IIIa , IIIb , IIIc , IIId , IV , V , VI-L , VI , VIIa , VIIb , VIIc , VIIIa , VIIIb , VIIIc , IXa , IXb , IXc , IXd , IXe , Xa , Xb , Xc .
, IIa , IIb , IIIa , IIIb , IIIc , IIId , IV , V , VI-L , VI , VIIa , VIIb , VIIc , VIIIa , VIIIb , VIIIc , IXa , IXb , IXc , IXd , IXe , Xa , Xb , Xc .
, IIa , IIb , IIIa , IIIb , IIIc , IIId , IV , V , VI-L , VI , VIIa , VIIb , VIIc , VIIIa , VIIIb , VIIIc , IXa , IXb , IXc , IXd , IXe , Xa , Xb , Xc .



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| S | T | Y | All (S + T + Y) | |
|---|---|---|---|---|
| AP2 domain only | 0.28% | 0.10% | 34.29% | 11.15% |
| Non-AP2 domain | 8.59% | 5.93% | 24.85% | 9.67% |
| Full-length | 8.09% | 4.92% | 30.32% | 9.97% |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sun, X.; Malhis, N.; Zhao, B.; Xue, B.; Gsponer, J.; Rikkerink, E.H.A. Computational Disorder Analysis in Ethylene Response Factors Uncovers Binding Motifs Critical to Their Diverse Functions. Int. J. Mol. Sci. 2020, 21, 74. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21010074
Sun X, Malhis N, Zhao B, Xue B, Gsponer J, Rikkerink EHA. Computational Disorder Analysis in Ethylene Response Factors Uncovers Binding Motifs Critical to Their Diverse Functions. International Journal of Molecular Sciences. 2020; 21(1):74. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21010074
Chicago/Turabian StyleSun, Xiaolin, Nawar Malhis, Bi Zhao, Bin Xue, Joerg Gsponer, and Erik H. A. Rikkerink. 2020. "Computational Disorder Analysis in Ethylene Response Factors Uncovers Binding Motifs Critical to Their Diverse Functions" International Journal of Molecular Sciences 21, no. 1: 74. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21010074