Cardiosomal microRNAs Are Essential in Post-Infarction Myofibroblast Phenoconversion




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
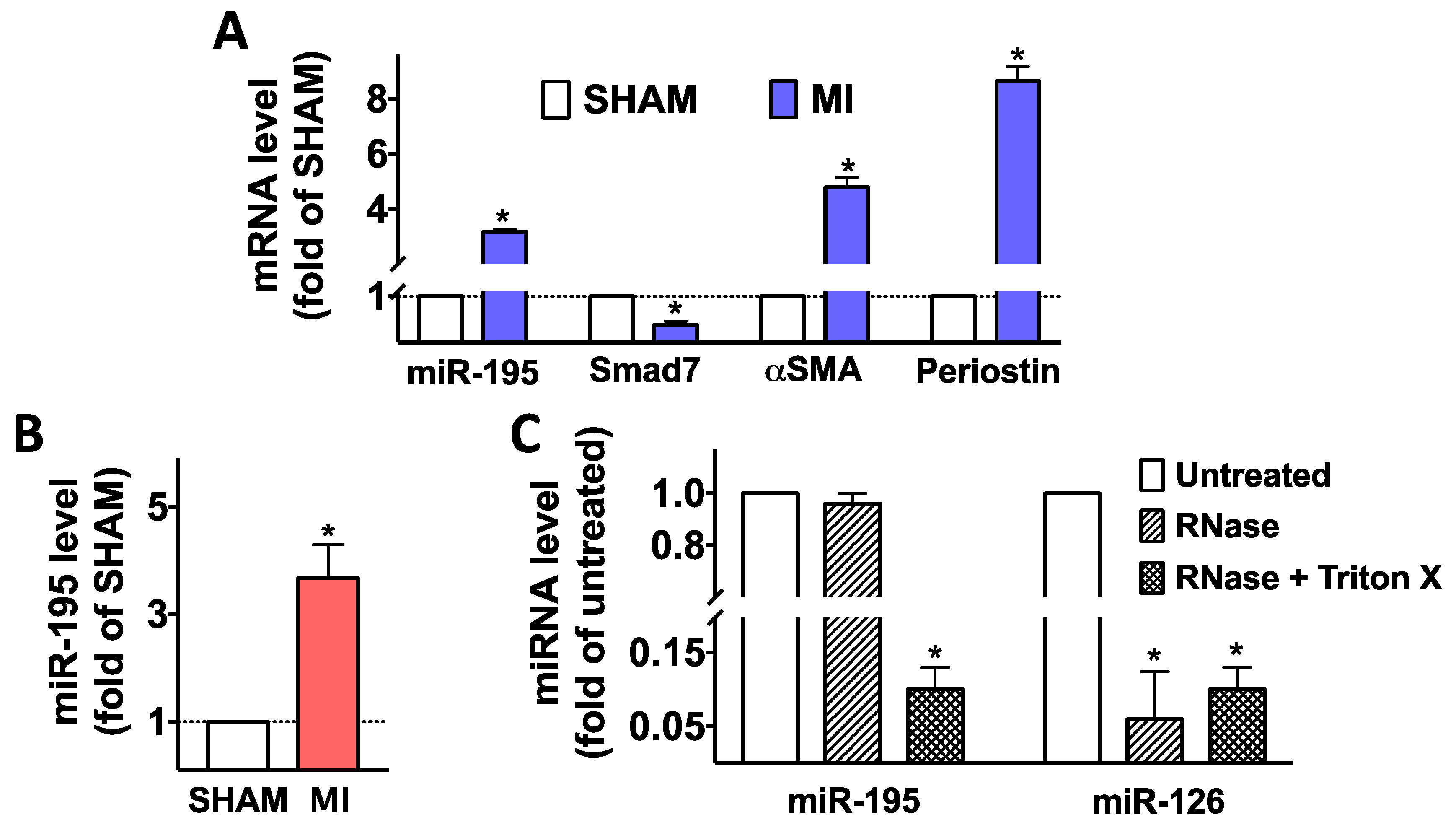
2.1. Cardiac Ischemic Injury Causes Upregulation of miR-195 in Myofibroblasts
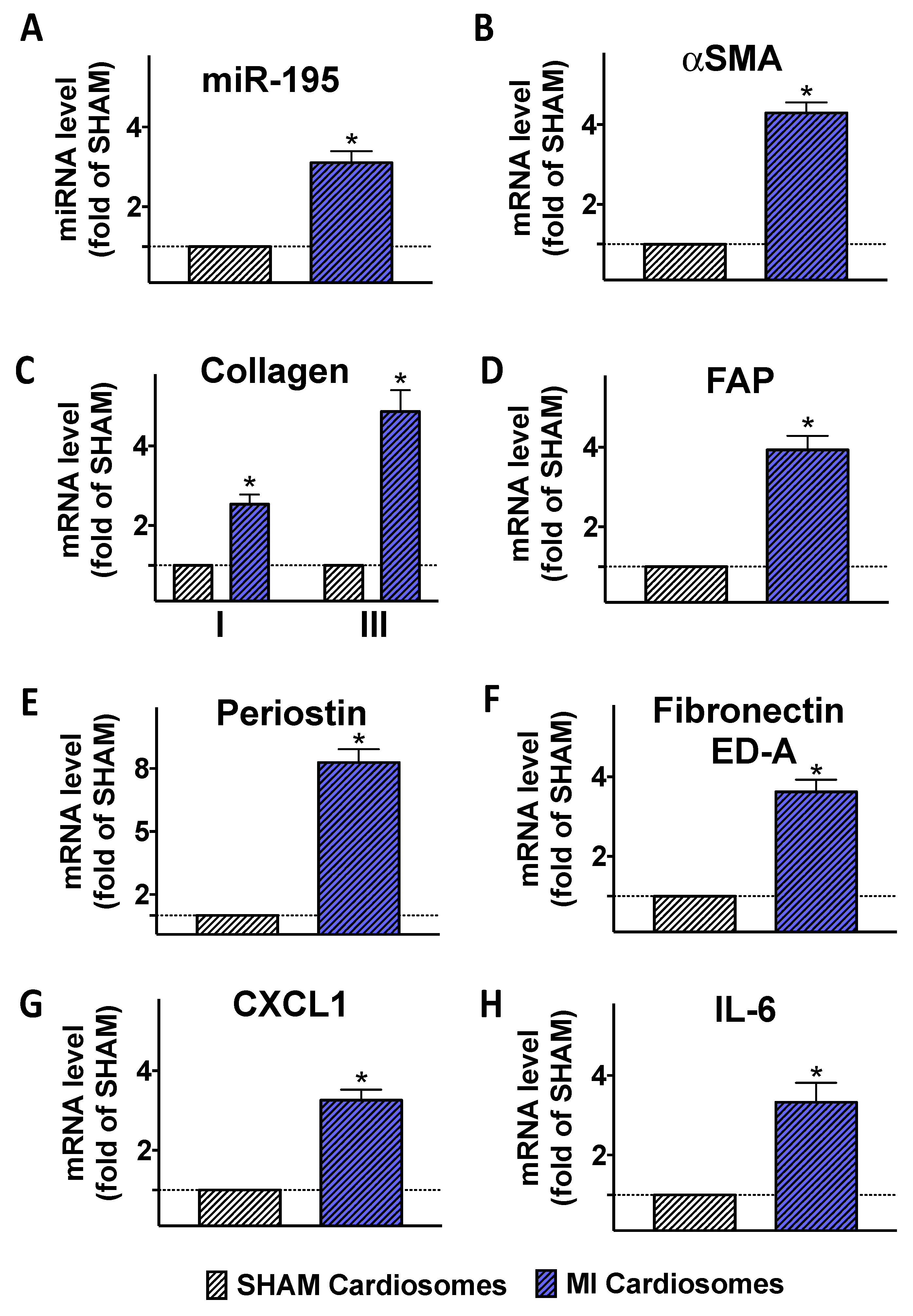
2.2. Fibroblasts Are Activated Following the Incubation with Cardiosomes Isolated from Ischemic Cardiomyocytes
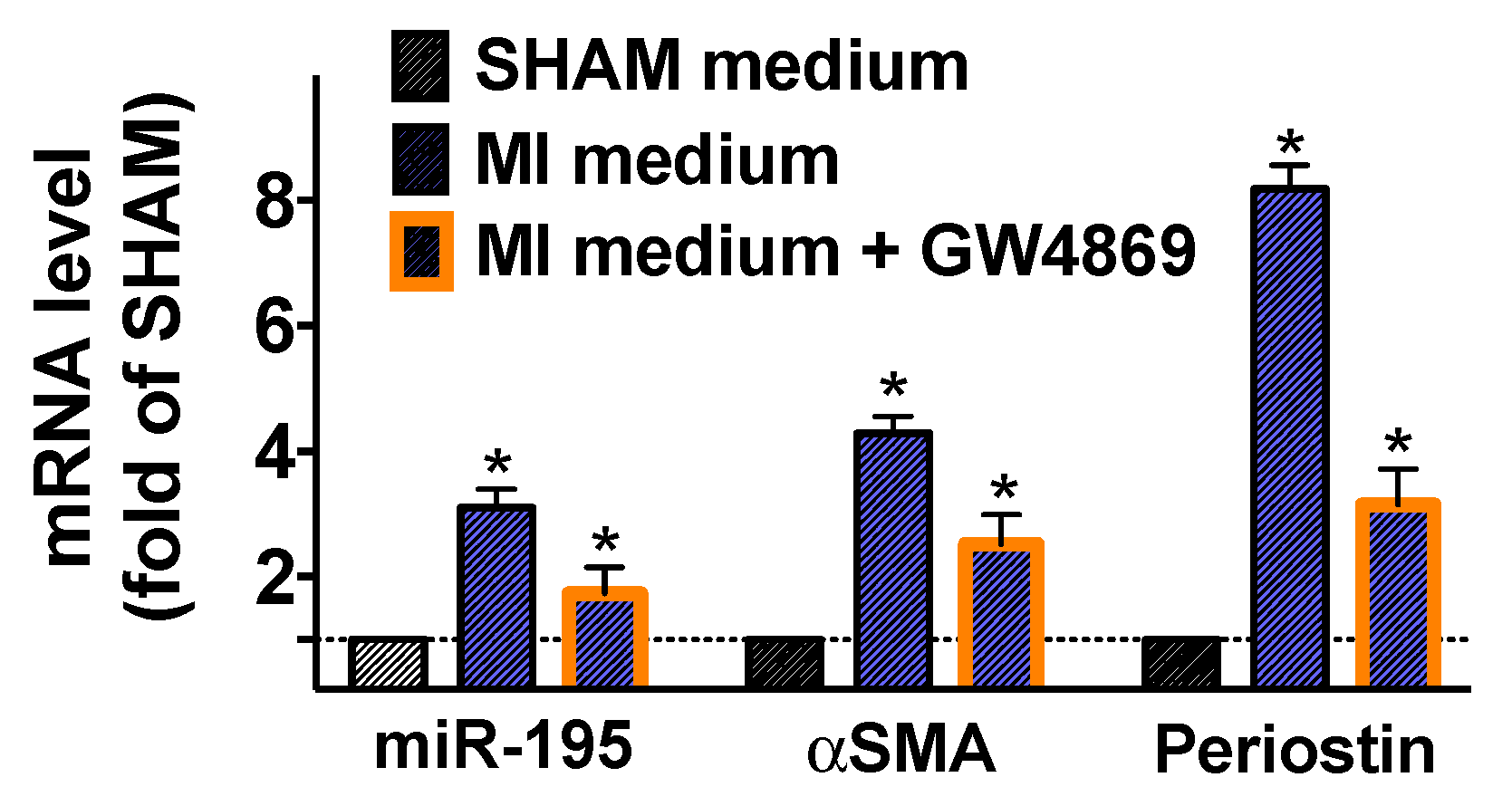
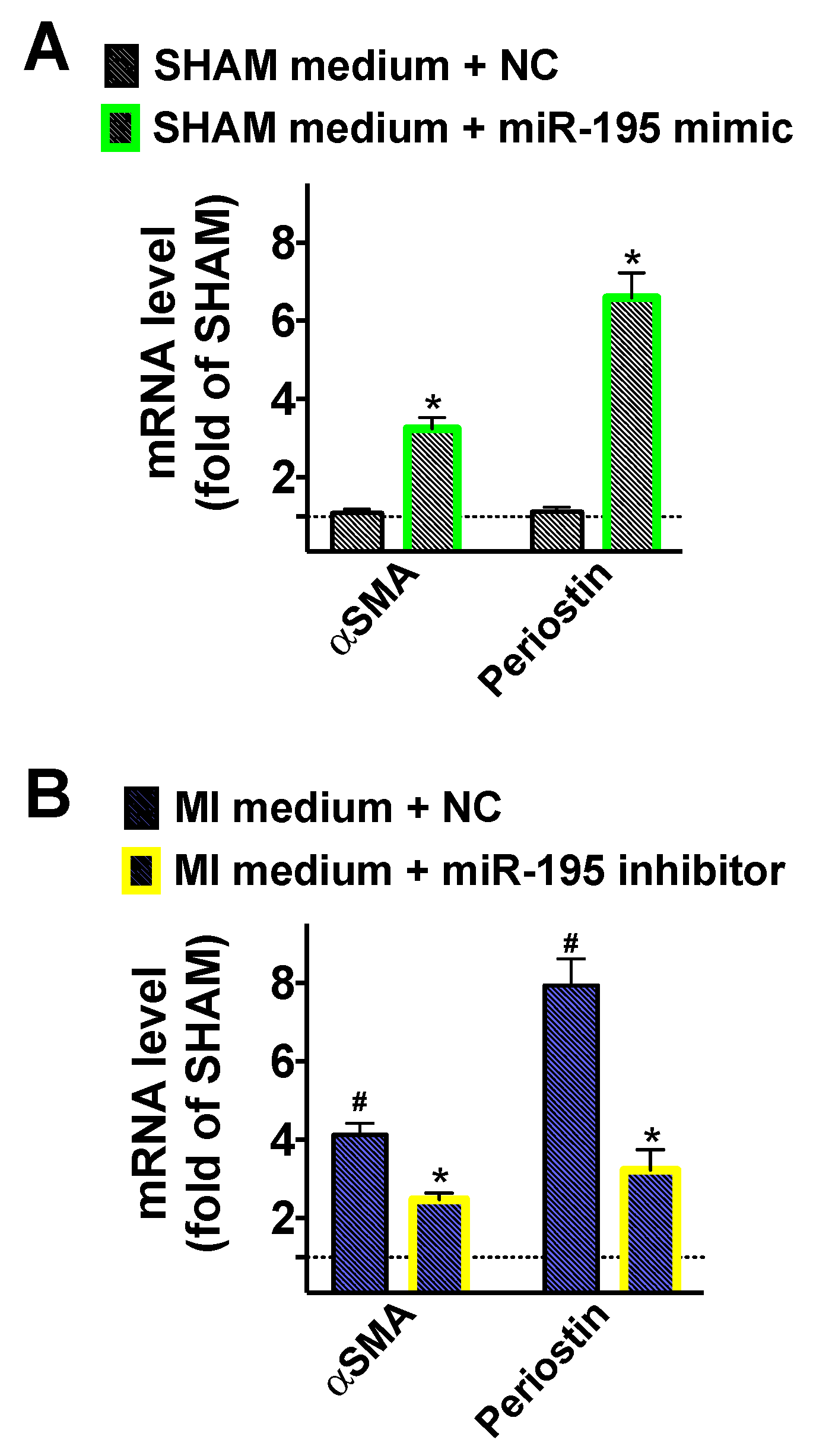
2.3. Fibroblasts Are Activated when Cultured in Conditioned Medium of Post-MI Cardiomyocytes
3. Discussion
4. Materials and Methods
4.1. Animal Studies
4.2. Isolation of Cardiomyocytes and Fibroblasts
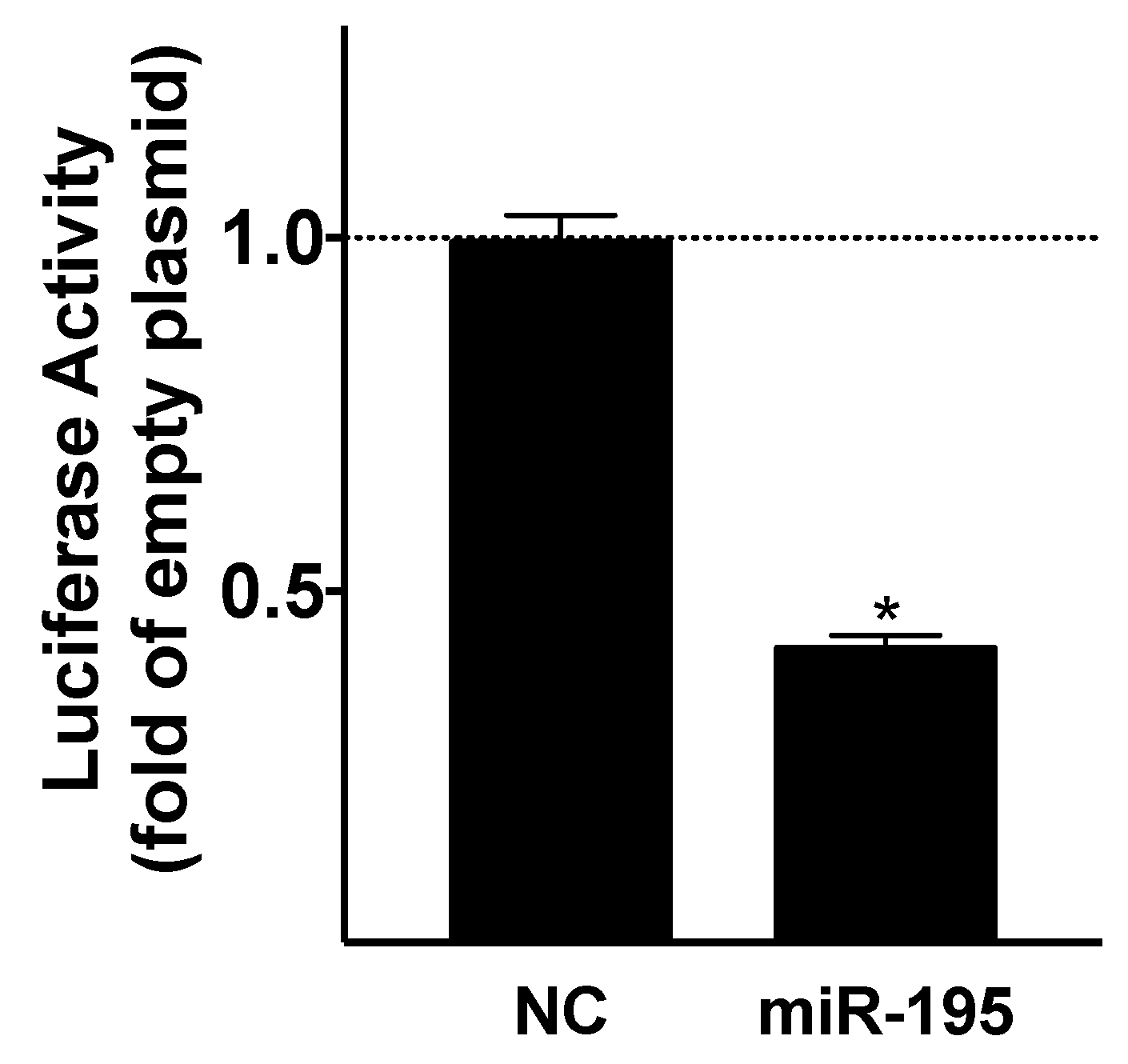
4.3. Luciferase Assay
4.4. Exosome Isolation
4.5. Quantitative Real-Time PCR (RT-qPCR) and Normalization
4.6. Bioinformatic Analyses
4.7. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| α-SMA | α-smooth muscle actin |
| CXCL1 | chemokine (C-X-C motif) ligand 1 |
| FAP | fibroblast activation protein |
| Fibronectin ED-A | fibronectin containing extra domain A |
| GAPDH | glyceraldehyde 3-phosphate dehydrogenase |
| IL-6 | interleukin-6 |
| MI | myocardial infarction |
| miRs (miRNAs) | microRNAs |
| Smad7 | SMAD family member |
References
- Russo, I.; Cavalera, M.; Huang, S.; Su, Y.; Hanna, A.; Chen, B.; Shinde, A.V.; Conway, S.J.; Graff, J.M.; Frangogiannis, N.G. Protective Effects of Activated Myofibroblasts in the Pressure-Overloaded Myocardium Are Mediated Through Smad-Dependent Activation of a Matrix-Preserving Program. Circ. Res. 2019, 8, 1214–1227. [Google Scholar] [CrossRef] [PubMed]
- Bhandary, B.; Meng, Q.; James, J.; Osinska, H.; Gulick, J.; Valiente-Alandi, I.; Sargent, M.A.; Bhuiyan, M.S.; Blaxall, B.C.; Molkentin, J.D.; et al. Cardiac Fibrosis in Proteotoxic Cardiac Disease is Dependent Upon Myofibroblast TGF -beta Signaling. J. Am. Heart Assoc. 2018, 7, e010013. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Turner, N.A. Inflammatory and fibrotic responses of cardiac fibroblasts to myocardial damage associated molecular patterns (DAMPs). J. Mol. Cell. Cardiol. 2016, 94, 189–200. [Google Scholar] [CrossRef] [PubMed]
- Kanisicak, O.; Khalil, H.; Ivey, M.J.; Karch, J.; Maliken, B.D.; Correll, R.N.; Brody, M.J.; SC, J.L.; Aronow, B.J.; Tallquist, M.D.; et al. Genetic lineage tracing defines myofibroblast origin and function in the injured heart. Nat. Commun. 2016, 7, 12260. [Google Scholar] [CrossRef] [Green Version]
- Molkentin, J.D.; Bugg, D.; Ghearing, N.; Dorn, L.E.; Kim, P.; Sargent, M.A.; Gunaje, J.; Otsu, K.; Davis, J. Fibroblast-Specific Genetic Manipulation of p38 Mitogen-Activated Protein Kinase In Vivo Reveals Its Central Regulatory Role in Fibrosis. Circulation 2017, 136, 549–561. [Google Scholar] [CrossRef]
- Nakaya, M.; Watari, K.; Tajima, M.; Nakaya, T.; Matsuda, S.; Ohara, H.; Nishihara, H.; Yamaguchi, H.; Hashimoto, A.; Nishida, M.; et al. Cardiac myofibroblast engulfment of dead cells facilitates recovery after myocardial infarction. J. Clin. Investig. 2017, 127, 383–401. [Google Scholar] [CrossRef]
- Valiente-Alandi, I.; Potter, S.J.; Salvador, A.M.; Schafer, A.E.; Schips, T.; Carrillo-Salinas, F.; Gibson, A.M.; Nieman, M.L.; Perkins, C.; Sargent, M.A.; et al. Inhibiting Fibronectin Attenuates Fibrosis and Improves Cardiac Function in a Model of Heart Failure. Circulation 2018, 138, 1236–1252. [Google Scholar] [CrossRef]
- Cheng, M.; Yang, J.; Zhao, X.; Zhang, E.; Zeng, Q.; Yu, Y.; Yang, L.; Wu, B.; Yi, G.; Mao, X.; et al. Circulating myocardial microRNAs from infarcted hearts are carried in exosomes and mobilise bone marrow progenitor cells. Nat. Commun. 2019, 10, 959. [Google Scholar] [CrossRef]
- Wronska, A.; Kurkowska-Jastrzebska, I.; Santulli, G. Application of microRNAs in diagnosis and treatment of cardiovascular disease. Acta Physiol. 2015, 213, 60–83. [Google Scholar] [CrossRef] [Green Version]
- Santulli, G. MicroRNAs and Endothelial (Dys) Function. J. Cell Physiol. 2016, 231, 1638–1644. [Google Scholar] [CrossRef] [Green Version]
- El Harane, N.; Kervadec, A.; Bellamy, V.; Pidial, L.; Neametalla, H.J.; Perier, M.C.; Lima Correa, B.; Thiebault, L.; Cagnard, N.; Duche, A.; et al. Acellular therapeutic approach for heart failure: In vitro production of extracellular vesicles from human cardiovascular progenitors. Eur. Heart J. 2018, 39, 1835–1847. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Santulli, G.; Iaccarino, G.; De Luca, N.; Trimarco, B.; Condorelli, G. Atrial fibrillation and microRNAs. Front. Physiol. 2014, 5, 15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Santulli, G. MicroRNA: From Molecular Biology to Clinical Practice; Springer Nature: New York, NY, USA, 2016. [Google Scholar]
- Elia, L.; Kunderfranco, P.; Carullo, P.; Vacchiano, M.; Farina, F.M.; Hall, I.F.; Mantero, S.; Panico, C.; Papait, R.; Condorelli, G.; et al. UHRF1 epigenetically orchestrates smooth muscle cell plasticity in arterial disease. J. Clin. Investig. 2018, 128, 2473–2486. [Google Scholar] [CrossRef] [PubMed]
- Wong, L.L.; Wang, J.; Liew, O.W.; Richards, A.M.; Chen, Y.T. MicroRNA and Heart Failure. Int. J. Mol. Sci. 2016, 17, 502. [Google Scholar] [CrossRef]
- Bang, C.; Batkai, S.; Dangwal, S.; Gupta, S.K.; Foinquinos, A.; Holzmann, A.; Just, A.; Remke, J.; Zimmer, K.; Zeug, A.; et al. Cardiac fibroblast-derived microRNA passenger strand-enriched exosomes mediate cardiomyocyte hypertrophy. J. Clin. Investig. 2014, 124, 2136–2146. [Google Scholar] [CrossRef]
- Hinz, B.; Phan, S.H.; Thannickal, V.J.; Prunotto, M.; Desmouliere, A.; Varga, J.; De Wever, O.; Mareel, M.; Gabbiani, G. Recent developments in myofibroblast biology: Paradigms for connective tissue remodeling. Am. J. Pathol. 2012, 180, 1340–1355. [Google Scholar] [CrossRef]
- Feinberg, M.W.; Moore, K.J. MicroRNA Regulation of Atherosclerosis. Circ. Res. 2016, 118, 703–720. [Google Scholar] [CrossRef] [Green Version]
- Sassi, Y.; Avramopoulos, P.; Ramanujam, D.; Gruter, L.; Werfel, S.; Giosele, S.; Brunner, A.D.; Esfandyari, D.; Papadopoulou, A.S.; De Strooper, B.; et al. Cardiac myocyte miR-29 promotes pathological remodeling of the heart by activating Wnt signaling. Nat. Commun. 2017, 8, 1614. [Google Scholar] [CrossRef]
- Hayashi, T.; Hoffman, M.P. Exosomal microRNA communication between tissues during organogenesis. RNA Biol. 2017, 14, 1683–1689. [Google Scholar] [CrossRef] [Green Version]
- de Couto, G.; Gallet, R.; Cambier, L.; Jaghatspanyan, E.; Makkar, N.; Dawkins, J.F.; Berman, B.P.; Marban, E. Exosomal MicroRNA Transfer Into Macrophages Mediates Cellular Postconditioning. Circulation 2017, 136, 200–214. [Google Scholar] [CrossRef]
- Santulli, G. Exosomal microRNA: The revolutionary endogenous Innerspace nanotechnology. Sci. Transl. Med. 2018, 10, eaav9141. [Google Scholar] [CrossRef] [PubMed]
- Waldenstrom, A.; Genneback, N.; Hellman, U.; Ronquist, G. Cardiomyocyte microvesicles contain DNA/RNA and convey biological messages to target cells. PLoS ONE 2012, 7, e34653. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- D’Alessandra, Y.; Devanna, P.; Limana, F.; Straino, S.; Di Carlo, A.; Brambilla, P.G.; Rubino, M.; Carena, M.C.; Spazzafumo, L.; De Simone, M.; et al. Circulating microRNAs are new and sensitive biomarkers of myocardial infarction. Eur. Heart J. 2010, 31, 2765–2773. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, Y.; Wang, X.; Yang, J.; Duan, X.; Yao, Y.; Shi, X.; Chen, Z.; Fan, Z.; Liu, X.; Qin, S.; et al. A translational study of urine miRNAs in acute myocardial infarction. J. Mol. Cell. Cardiol. 2012, 53, 668–676. [Google Scholar] [CrossRef] [Green Version]
- Nazari-Shafti, T.Z.; Stamm, C.; Falk, V.; Emmert, M.Y. Exosomes for Cardioprotection: Are We Ready for Clinical Translation? Eur. Heart J. 2019, 40, 953–956. [Google Scholar] [CrossRef]
- Patil, M.; Henderson, J.; Luong, H.; Annamalai, D.; Sreejit, G.; Krishnamurthy, P. The Art of Intercellular Wireless Communications: Exosomes in Heart Disease and Therapy. Front. Cell Dev. Biol. 2019, 7, 315. [Google Scholar] [CrossRef] [Green Version]
- Moghaddam, A.S.; Afshari, J.T.; Esmaeili, S.A.; Saburi, E.; Joneidi, Z.; Momtazi-Borojeni, A.A. Cardioprotective microRNAs: Lessons from stem cell-derived exosomal microRNAs to treat cardiovascular disease. Atherosclerosis 2019, 285, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Trovato, E.; Di Felice, V.; Barone, R. Extracellular Vesicles: Delivery Vehicles of Myokines. Front. Physiol. 2019, 10, 522. [Google Scholar] [CrossRef] [Green Version]
- Nagaraju, C.K.; Robinson, E.L.; Abdesselem, M.; Trenson, S.; Dries, E.; Gilbert, G.; Janssens, S.; Van Cleemput, J.; Rega, F.; Meyns, B.; et al. Myofibroblast Phenotype and Reversibility of Fibrosis in Patients with End-Stage Heart Failure. J. Am. Coll. Cardiol. 2019, 73, 2267–2282. [Google Scholar] [CrossRef]
- Aguado, B.A.; Schuetze, K.B.; Grim, J.C.; Walker, C.J.; Cox, A.C.; Ceccato, T.L.; Tan, A.C.; Sucharov, C.C.; Leinwand, L.A.; Taylor, M.R.G.; et al. Transcatheter aortic valve replacements alter circulating serum factors to mediate myofibroblast deactivation. Sci. Transl. Med. 2019, 11, eaav3233. [Google Scholar] [CrossRef]
- Emelyanova, L.; Sra, A.; Schmuck, E.G.; Raval, A.N.; Downey, F.X.; Jahangir, A.; Rizvi, F.; Ross, G.R. Impact of statins on cellular respiration and de-differentiation of myofibroblasts in human failing hearts. ESC Heart Fail. 2019, 6, 1027–1040. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, T.; Dong, Y.H.; Du, W.; Shi, C.Y.; Wang, K.; Tariq, M.A.; Wang, J.X.; Li, P.F. The Role of MicroRNAs in Myocardial Infarction: From Molecular Mechanism to Clinical Application. Int. J. Mol. Sci. 2017, 18, 745. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Santulli, G.; Xie, W.; Reiken, S.R.; Marks, A.R. Mitochondrial calcium overload is a key determinant in heart failure. Proc. Natl. Acad. Sci. USA 2015, 112, 11389–11394. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Menck, K.; Sonmezer, C.; Worst, T.S.; Schulz, M.; Dihazi, G.H.; Streit, F.; Erdmann, G.; Kling, S.; Boutros, M.; Binder, C.; et al. Neutral sphingomyelinases control extracellular vesicles budding from the plasma membrane. J. Extracell. Vesicles 2017, 6, 1378056. [Google Scholar] [CrossRef]
- Kakkar, R.; Lee, R.T. Intramyocardial fibroblast myocyte communication. Circ. Res. 2010, 106, 47–57. [Google Scholar] [CrossRef] [Green Version]
- Cartledge, J.E.; Kane, C.; Dias, P.; Tesfom, M.; Clarke, L.; McKee, B.; Al Ayoubi, S.; Chester, A.; Yacoub, M.H.; Camelliti, P.; et al. Functional crosstalk between cardiac fibroblasts and adult cardiomyocytes by soluble mediators. Cardiovasc. Res. 2015, 105, 260–270. [Google Scholar] [CrossRef]
- Salvarani, N.; Maguy, A.; De Simone, S.A.; Miragoli, M.; Jousset, F.; Rohr, S. TGF-beta1 (Transforming Growth Factor-beta1) Plays a Pivotal Role in Cardiac Myofibroblast Arrhythmogenicity. Circ. Arrhythm. Electrophysiol. 2017, 10, e004567. [Google Scholar] [CrossRef]
- Lai, K.B.; Sanderson, J.E.; Izzat, M.B.; Yu, C.M. Micro-RNA and mRNA myocardial tissue expression in biopsy specimen from patients with heart failure. Int. J. Cardiol. 2015, 199, 79–83. [Google Scholar] [CrossRef]
- Santulli, G. The Adrenergic System in Cardiovascular Metabolism and Aging. In The Cardiovascular Adrenergic System; Lymperopoulos, A., Ed.; Springer: Cham, Switzerland, 2015; pp. 97–116. [Google Scholar]
- Lymperopoulos, A.; Rengo, G.; Koch, W.J. Adrenergic nervous system in heart failure: Pathophysiology and therapy. Circ. Res. 2016, 119, e38. [Google Scholar] [CrossRef]
- Santulli, G.; Campanile, A.; Spinelli, L.; Assante di Panzillo, E.; Ciccarelli, M.; Trimarco, B.; Iaccarino, G. G protein-coupled receptor kinase 2 in patients with acute myocardial infarction. Am. J. Cardiol. 2011, 107, 1125–1130. [Google Scholar] [CrossRef]
- Ciccarelli, M.; Sorriento, D.; Coscioni, E.; Iaccarino, G.; Santulli, G. Adrenergic Receptors. In Endocrinology of the Heart in Health and Disease; Schisler, J., Lang, C., Willis, M., Eds.; Academic Press: London, UK, 2017; pp. 285–315. [Google Scholar]
- Santulli, G. The lymphatic border patrol outwits inflammatory cells in myocardial infarction. Sci. Transl. Med. 2018, 10, eaau7379. [Google Scholar] [CrossRef] [PubMed]
- Capote, L.A.; Mendez Perez, R.; Lymperopoulos, A. GPCR signaling and cardiac function. Eur. J. Pharmacol. 2015, 763, 143–148. [Google Scholar] [CrossRef] [PubMed]
- Santulli, G. Adrenal signaling in heart failure: Something more than a distant ship’s smoke on the horizon. Hypertension 2014, 63, 215–216. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- You, X.Y.; Huang, J.H.; Liu, B.; Liu, S.J.; Zhong, Y.; Liu, S.M. HMGA1 is a new target of miR-195 involving isoprenaline-induced cardiomyocyte hypertrophy. Biochemistry 2014, 79, 538–544. [Google Scholar] [CrossRef]
- Okamoto, M.; Fukushima, Y.; Kouwaki, T.; Daito, T.; Kohara, M.; Kida, H.; Oshiumi, H. MicroRNA-451a in extracellular, blood-resident vesicles attenuates macrophage and dendritic cell responses to influenza whole-virus vaccine. J. Biol. Chem. 2018, 293, 18585–18600. [Google Scholar] [CrossRef] [Green Version]
- Buschmann, D.; Kirchner, B.; Hermann, S.; Marte, M.; Wurmser, C.; Brandes, F.; Kotschote, S.; Bonin, M.; Steinlein, O.K.; Pfaffl, M.W.; et al. Evaluation of serum extracellular vesicle isolation methods for profiling miRNAs by next-generation sequencing. J. Extracell. Vesicles 2018, 7, 1481321. [Google Scholar] [CrossRef]
- Stahl, P.D.; Raposo, G. Exosomes and extracellular vesicles: The path forward. Essays Biochem. 2018, 62, 119–124. [Google Scholar] [CrossRef]
- Huang-Doran, I.; Zhang, C.Y.; Vidal-Puig, A. Extracellular Vesicles: Novel Mediators of Cell Communication In Metabolic Disease. Trends Endocrinol. Metab. 2017, 28, 3–18. [Google Scholar] [CrossRef]
- Thery, C.; Ostrowski, M.; Segura, E. Membrane vesicles as conveyors of immune responses. Nat. Rev. Immunol. 2009, 9, 581–593. [Google Scholar] [CrossRef]
- Harding, C.; Heuser, J.; Stahl, P. Receptor-mediated endocytosis of transferrin and recycling of the transferrin receptor in rat reticulocytes. J. Cell Biol. 1983, 97, 329–339. [Google Scholar] [CrossRef]
- Burnley-Hall, N.; Abdul, F.; Androshchuk, V.; Morris, K.; Ossei-Gerning, N.; Anderson, R.; Rees, D.A.; James, P.E. Dietary Nitrate Supplementation Reduces Circulating Platelet-Derived Extracellular Vesicles in Coronary Artery Disease Patients on Clopidogrel Therapy: A Randomised, Double-Blind, Placebo-Controlled Study. Thromb. Haemost. 2018, 118, 112–122. [Google Scholar] [CrossRef] [PubMed]
- Chistiakov, D.A.; Orekhov, A.N.; Bobryshev, Y.V. Cardiac Extracellular Vesicles in Normal and Infarcted Heart. Int. J. Mol. Sci. 2016, 17, 63. [Google Scholar] [CrossRef] [PubMed]
- Ong, S.G.; Lee, W.H.; Huang, M.; Dey, D.; Kodo, K.; Sanchez-Freire, V.; Gold, J.D.; Wu, J.C. Cross talk of combined gene and cell therapy in ischemic heart disease: role of exosomal microRNA transfer. Circulation 2014, 130, S60–S69. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sahoo, S.; Losordo, D.W. Exosomes and cardiac repair after myocardial infarction. Circ. Res. 2014, 114, 333–344. [Google Scholar] [CrossRef]
- Ciullo, A.; Biemmi, V.; Milano, G.; Bolis, S.; Cervio, E.; Fertig, E.T.; Gherghiceanu, M.; Moccetti, T.; Camici, G.G.; Vassalli, G.; et al. Exosomal Expression of CXCR4 Targets Cardioprotective Vesicles to Myocardial Infarction and Improves Outcome after Systemic Administration. Int. J. Mol. Sci. 2019, 20, 468. [Google Scholar] [CrossRef] [Green Version]
- Boulanger, C.M.; Loyer, X.; Rautou, P.E.; Amabile, N. Extracellular vesicles in coronary artery disease. Nat. Rev. Cardiol. 2017, 14, 259–272. [Google Scholar] [CrossRef]
- Spannbauer, A.; Traxler, D.; Lukovic, D.; Zlabinger, K.; Winkler, J.; Gugerell, A.; Ferdinandy, P.; Hausenloy, D.J.; Pavo, N.; Emmert, M.Y.; et al. Effect of Ischemic Preconditioning and Postconditioning on Exosome-Rich Fraction microRNA Levels, in Relation with Electrophysiological Parameters and Ventricular Arrhythmia in Experimental Closed-Chest Reperfused Myocardial Infarction. Int. J. Mol. Sci. 2019, 20, 2140. [Google Scholar] [CrossRef] [Green Version]
- Gollmann-Tepekoylu, C.; Polzl, L.; Graber, M.; Hirsch, J.; Nagele, F.; Lobenwein, D.; Hess, M.W.; Blumer, M.J.; Kirchmair, E.; Zipperle, J. miR-19a-3p containing exosomes improve function of ischemic myocardium upon shock wave therapy. Cardiovasc. Res. 2019, in press. [Google Scholar] [CrossRef]
- Sluijter, J.P.G.; Davidson, S.M.; Boulanger, C.M.; Buzas, E.I.; de Kleijn, D.P.V.; Engel, F.B.; Giricz, Z.; Hausenloy, D.J.; Kishore, R.; Lecour, S.; et al. Extracellular vesicles in diagnostics and therapy of the ischaemic heart: Position Paper from the Working Group on Cellular Biology of the Heart of the European Society of Cardiology. Cardiovasc. Res. 2018, 114, 19–34. [Google Scholar] [CrossRef]
- Kushnir, A.; Santulli, G.; Reiken, S.R.; Coromilas, E.; Godfrey, S.J.; Brunjes, D.L.; Colombo, P.C.; Yuzefpolskaya, M.; Sokol, S.I.; Kitsis, R.N.; et al. Ryanodine Receptor Calcium Leak in Circulating B-Lymphocytes as a Biomarker in Heart Failure. Circulation 2018, 138, 1144–1154. [Google Scholar] [CrossRef]
- Wang, X.; Morelli, M.B.; Matarese, A.; Sardu, C.; Santulli, G. Cardiomyocyte-derived exosomal microRNA-92a mediates post-ischemic myofibroblast activation both in vitro and ex vivo. ESC-Heart Fail. 2020, in press. [Google Scholar]
- Zhang, Y.; Kim, M.S.; Jia, B.; Yan, J.; Zuniga-Hertz, J.P.; Han, C.; Cai, D. Hypothalamic stem cells control ageing speed partly through exosomal miRNAs. Nature 2017, 548, 52–57. [Google Scholar] [CrossRef] [PubMed]
- Thomou, T.; Mori, M.A.; Dreyfuss, J.M.; Konishi, M.; Sakaguchi, M.; Wolfrum, C.; Rao, T.N.; Winnay, J.N.; Garcia-Martin, R.; Grinspoon, S.K.; et al. Adipose-derived circulating miRNAs regulate gene expression in other tissues. Nature 2017, 542, 450–455. [Google Scholar] [CrossRef] [PubMed]
- Lombardi, A.; Gambardella, J.; Du, X.L.; Sorriento, D.; Mauro, M.; Iaccarino, G.; Trimarco, B.; Santulli, G. Sirolimus induces depletion of intracellular calcium stores and mitochondrial dysfunction in pancreatic beta cells. Sci. Rep. 2017, 7, 15823. [Google Scholar] [CrossRef] [PubMed]
- Santulli, G.; Wronska, A.; Uryu, K.; Diacovo, T.G.; Gao, M.; Marx, S.O.; Kitajewski, J.; Chilton, J.M.; Akat, K.M.; Tuschl, T.; et al. A selective microRNA-based strategy inhibits restenosis while preserving endothelial function. J. Clin. Investig. 2014, 124, 4102–4114. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Wang, X. Prediction of functional microRNA targets by integrative modeling of microRNA binding and target expression data. Genome Biol. 2019, 20, 18. [Google Scholar] [CrossRef]
- Karagkouni, D.; Paraskevopoulou, M.D.; Chatzopoulos, S.; Vlachos, I.S.; Tastsoglou, S.; Kanellos, I.; Papadimitriou, D.; Kavakiotis, I.; Maniou, S.; Skoufos, G.; et al. DIANA-TarBase v8: A decade-long collection of experimentally supported miRNA-gene interactions. Nucleic Acids Res. 2018, 46, D239–D245. [Google Scholar] [CrossRef] [Green Version]





© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Morelli, M.B.; Shu, J.; Sardu, C.; Matarese, A.; Santulli, G. Cardiosomal microRNAs Are Essential in Post-Infarction Myofibroblast Phenoconversion. Int. J. Mol. Sci. 2020, 21, 201. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21010201
Morelli MB, Shu J, Sardu C, Matarese A, Santulli G. Cardiosomal microRNAs Are Essential in Post-Infarction Myofibroblast Phenoconversion. International Journal of Molecular Sciences. 2020; 21(1):201. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21010201
Chicago/Turabian StyleMorelli, Marco B., Jun Shu, Celestino Sardu, Alessandro Matarese, and Gaetano Santulli. 2020. "Cardiosomal microRNAs Are Essential in Post-Infarction Myofibroblast Phenoconversion" International Journal of Molecular Sciences 21, no. 1: 201. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21010201