Human-Induced Neurons from Presenilin 1 Mutant Patients Model Aspects of Alzheimer’s Disease Pathology


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
1.1. PS1 Mutant Human-Induced Neurons Model Aspects of AD Pathology
1.2. PS1 Mutations and AD Pathology
2. Results
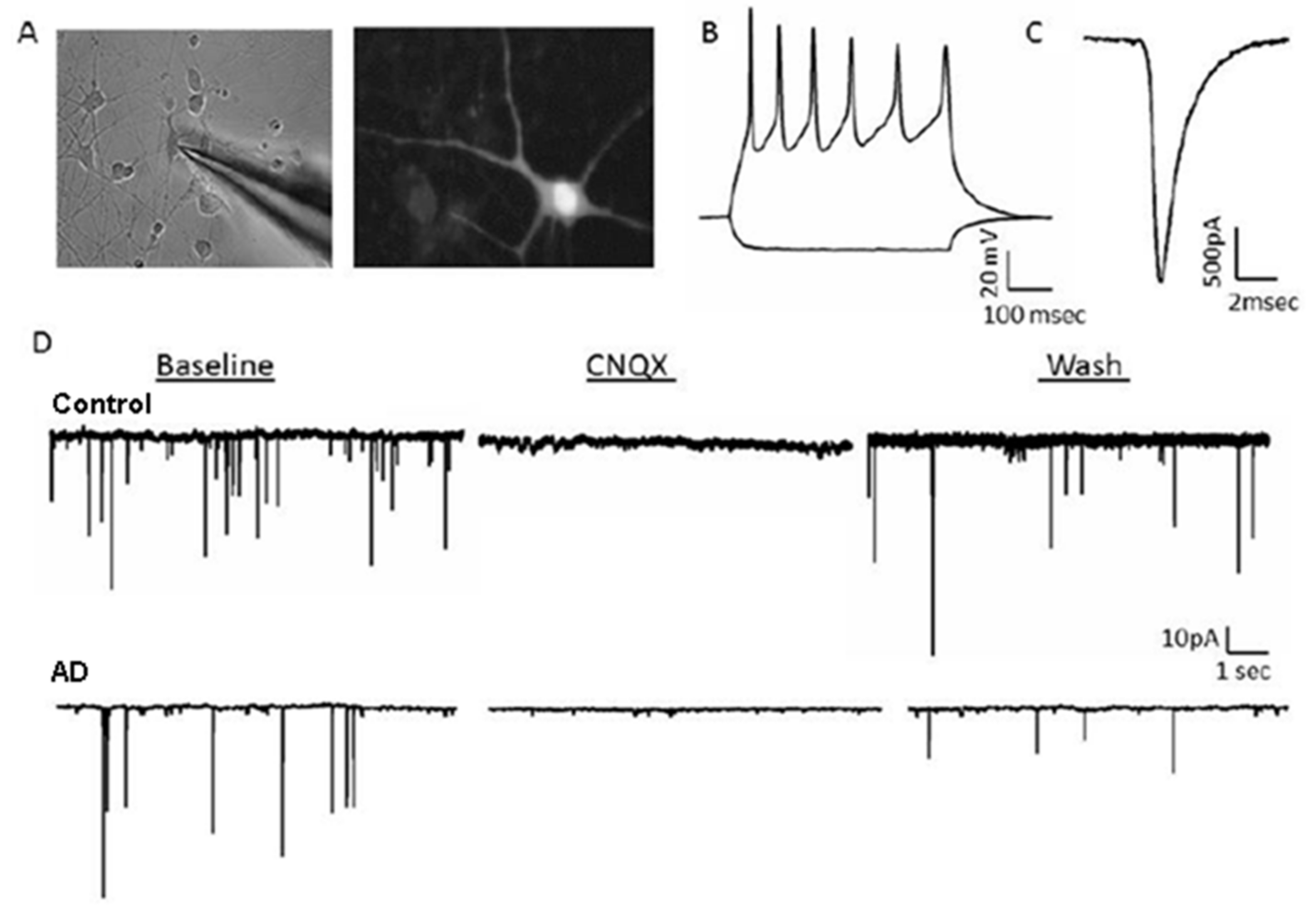
2.1. Generation of Human-Induced Neurons
2.2. Human-Induced Neurons Model Histopathological Hallmarks of AD
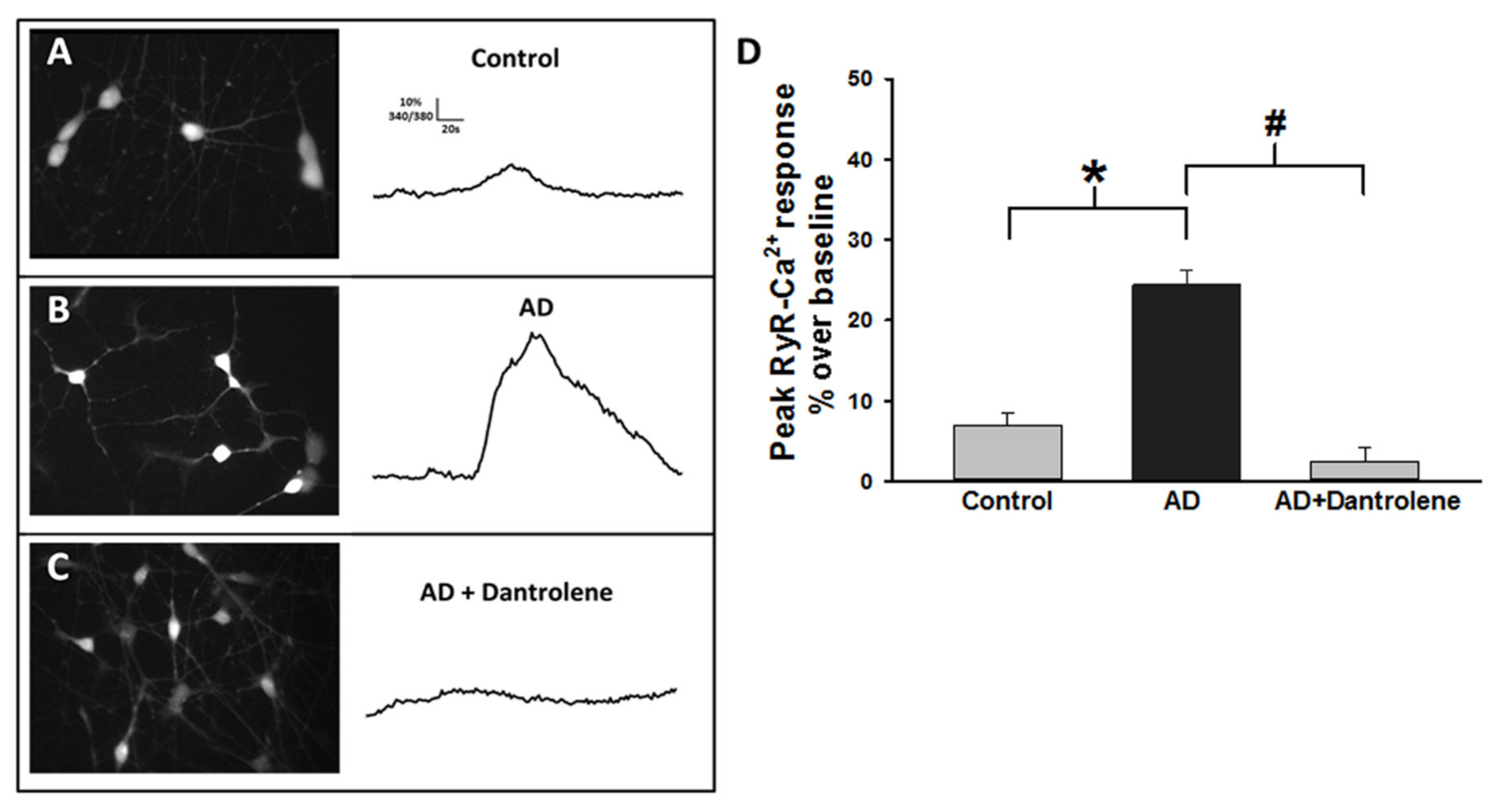
2.3. Human-Induced Neurons from AD Patients Exhibit Exaggerated ER Calcium Release
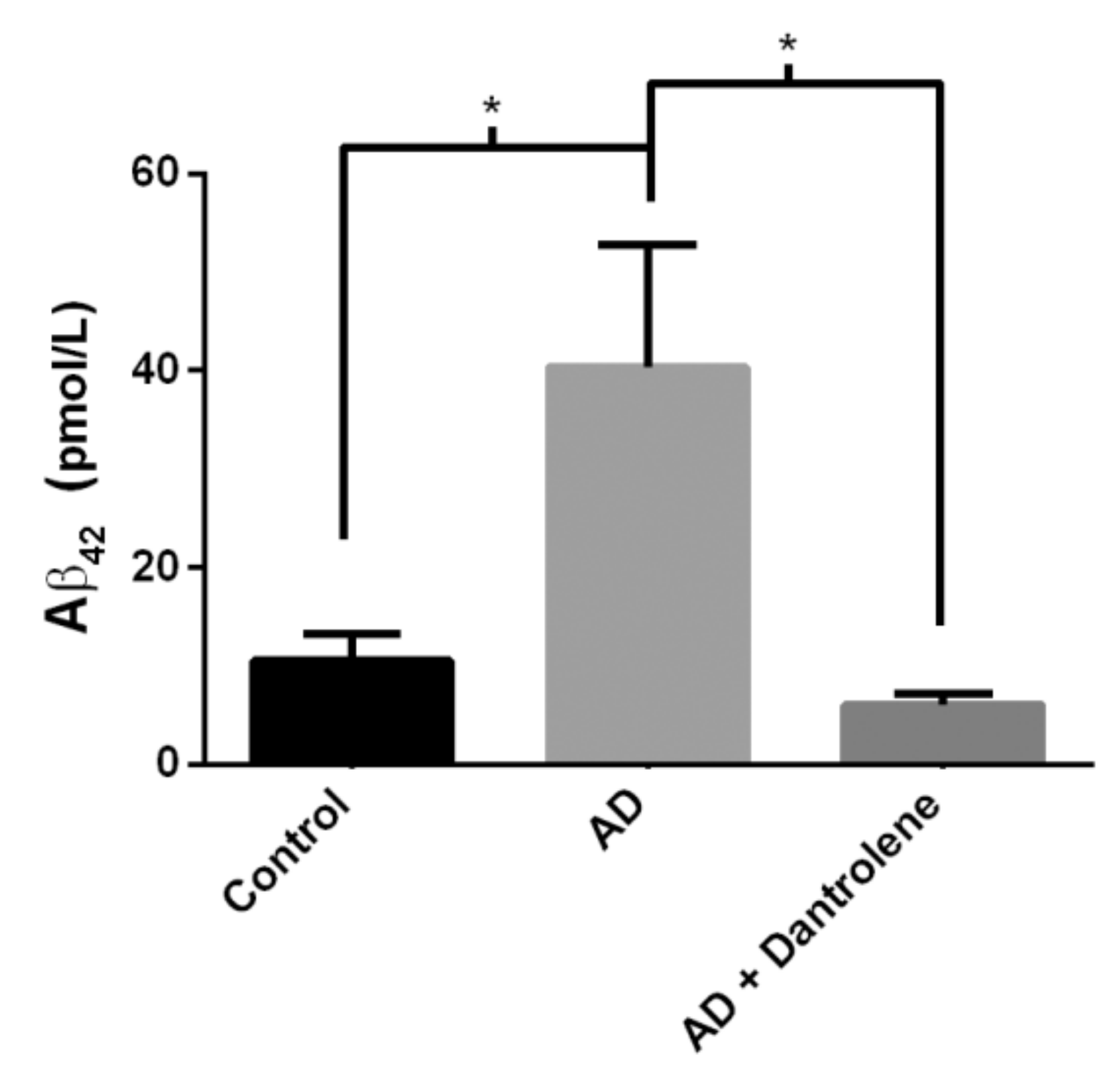
2.4. Normalizing RyR-Evoked Calcium Release Reduces Aβ42 Production in FAD HiNs
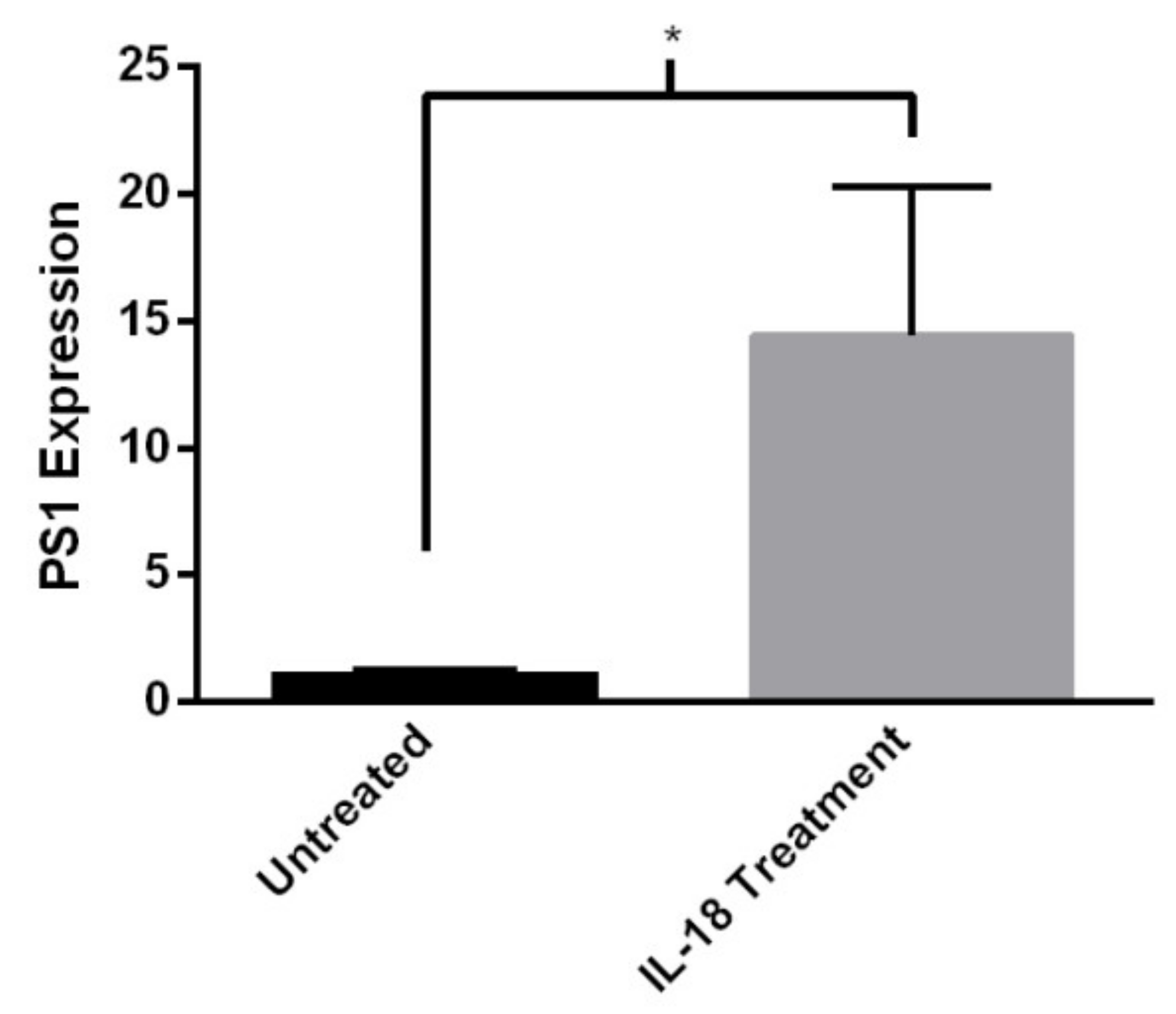
2.5. IL-18 Increases PS1 Expression in Human-Induced Neurons
3. Discussion
4. Methods
4.1. iPSC and HiN Generation
4.2. qRT-PCR
4.3. Immunohistochemistry
4.4. Enzyme Linked Immunosorbent Assays
4.5. Calcium Imaging
4.6. Statistics
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Alzheimer’s Association. Alzheimer’s Facts and Figures Report | Alzheimer’s Association. Available online: https://www.alz.org/alzheimers-dementia/facts-figures (accessed on 7 May 2019).
- Goate, A.; Chartier-Harlin, M.C.; Mullan, M.; Brown, J.; Crawford, F.; Fidani, L.; Giuffra, L.; Haynes, A.; Irving, N.; James, L.; et al. Segregation of a Missense Mutation in the Amyloid Precursor Protein Gene with Familial Alzheimer’s Disease. Nature 1991, 349, 704–706. [Google Scholar] [CrossRef] [PubMed]
- Hutton, M.; Busfield, F.; Wragg, M.; Crook, R.; Perez-Tur, J.; Clark, R.F.; Prihar, G.; Talbot, C.; Phillips, H.; Wright, K.; et al. Complete Analysis of the Presenilin 1 Gene in Early Onset Alzheimer’s Disease. Neuroreport 1996, 7, 801–805. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tanzi, R.E.; Gusella, J.F.; Watkins, P.C.; Bruns, G.A.P.; St. George-Hyslop, P.; Van Keuren, M.L.; Patterson, D.; Pagan, S.; Kurnit, D.M.; Neve, R.L. Amyloid β Protein Gene: CDNA, MRNA Distribution, and Genetic Linkage near the Alzheimer Locus. Science 1987, 235, 880–884. [Google Scholar] [CrossRef] [PubMed]
- Lanoiselée, H.M.; Nicolas, G.; Wallon, D.; Rovelet-Lecrux, A.; Lacour, M.; Rousseau, S.; Richard, A.C.; Pasquier, F.; Rollin-Sillaire, A.; Martinaud, O.; et al. APP, PSEN1, and PSEN2 Mutations in Early-Onset Alzheimer Disease: A Genetic Screening Study of Familial and Sporadic Cases. PLoS Med. 2017, 14, e1002270. [Google Scholar] [CrossRef] [Green Version]
- Wolk, A.; Dickerson, B. Clinical Features and Diagnosis of Alzheimer Disease. UpToDate 2019. Available online: https://www.uptodate.com/contents/clinical-features-and-diagnosis-of-alzheimer-disease (accessed on 7 June 2019).
- Hardy, J.A.; Higgins, G.A. Alzheimer’s Disease: The Amyloid Cascade Hypothesis. Science 1992, 256, 184–185. [Google Scholar] [CrossRef]
- Hemming, M.L.; Elias, J.E.; Gygi, S.P.; Selkoe, D.J. Identification of β-Secretase (BACE1) Substrates Using Quantitative Proteomics. PLoS ONE 2009, 4, e8477. [Google Scholar] [CrossRef] [Green Version]
- Martini, A.C.; Gomez-Arboledas, A.; Forner, S.; Rodriguez-Ortiz, C.J.; McQuade, A.; Danhash, E.; Phan, J.; Javonillo, D.; Ha, J.V.; Tram, M.; et al. Amyloid-Beta Impairs TOM1-Mediated IL-1R1 Signaling. Proc. Natl. Acad. Sci. USA 2019, 116, 21198–21206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, H.-K.; Kumar, P.; Fu, Q.; Rosen, K.M.; Querfurth, H.W. The Insulin/Akt Signaling Pathway Is Targeted by Intracellular Beta-Amyloid. Mol. Biol. Cell 2009, 20, 1533–1544. [Google Scholar] [CrossRef] [Green Version]
- Li, S.; Jin, M.; Koeglsperger, T.; Shepardson, N.E.; Shankar, G.M.; Selkoe, D.J. Soluble a β Oligomers Inhibit Long-Term Potentiation through a Mechanism Involving Excessive Activation of Extrasynaptic NR2B-Containing NMDA Receptors. J. Neurosci. 2011, 31, 6627–6638. [Google Scholar] [CrossRef]
- Qin, Z.; Zhou, X.; Gomez-Smith, M.; Pandey, N.R.; Lee, K.F.H.; Lagace, D.C.; Béïque, J.-C.; Chen, H.-H. LIM Domain Only 4 (LMO4) Regulates Calcium-Induced Calcium Release and Synaptic Plasticity in the Hippocampus. J. Neurosci. 2012, 32, 4271–4283. [Google Scholar] [CrossRef] [Green Version]
- Barucker, C.; Sommer, A.; Beckmann, G.; Eravci, M.; Harmeier, A.; Schipke, C.G.; Brockschnieder, D.; Dyrks, T.; Althoff, V.; Fraser, P.E.; et al. Alzheimer Amyloid Peptide Aβ42 Regulates Gene Expression of Transcription and Growth Factors. J. Alzheimer’s Dis. 2015, 44, 613–624. [Google Scholar] [CrossRef] [PubMed]
- Kayed, R.; Lasagna-Reeves, C.A. Molecular Mechanisms of Amyloid Oligomers Toxicity. J. Alzheimer’s Dis. 2013, 33, S67–S78. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Binder, L.I.; Guillozet-Bongaarts, A.L.; Garcia-Sierra, F.; Berry, R.W. Tau, Tangles, and Alzheimer’s Disease. Biochim. Biophys. Acta Mol. Basis Dis. 2005, 1739, 216–223. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brion, J.P. Neurofibrillary Tangles and Alzheimer’s Disease. Eur. Neurol. 1998, 40, 130–140. [Google Scholar] [CrossRef] [PubMed]
- DeTure, M.A.; Dickson, D.W. The Neuropathological Diagnosis of Alzheimer’s Disease. Mol. Neurodegener. 2019, 14, 32. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grundke-iqbal, I.; Iqbal, K.; Tung, Y.; Quinlan, M.; Wisniewski, H.M.; Bindert, L.I. Abnormal Phosphorylation of the Microtubule-Associated Protein Tau(Tau) in Alzheimer Cytoskeletal Pathology. Proc. Natl. Acad. Sci. USA 1986, 83, 4913–4917. [Google Scholar] [CrossRef] [Green Version]
- Melková, K.; Zapletal, V.; Narasimhan, S.; Jansen, S.; Hritz, J.; Škrabana, R.; Zweckstetter, M.; Jensen, M.R.; Blackledge, M.; Žídek, L. Structure and Functions of Microtubule Associated Proteins Tau and Map2c: Similarities and Differences. Biomolecules 2019, 9, 105. [Google Scholar] [CrossRef] [Green Version]
- Šimić, G.; Babić Leko, M.; Wray, S.; Harrington, C.; Delalle, I.; Jovanov-Milošević, N.; Bažadona, D.; Buée, L.; de Silva, R.; Di Giovanni, G.; et al. Tau Protein Hyperphosphorylation and Aggregation in Alzheimer’s Disease and Other Tauopathies, and Possible Neuroprotective Strategies. Biomolecules 2016, 6, 6. [Google Scholar] [CrossRef] [Green Version]
- LaFerla, F.M. Calcium Dyshomeostasis and Intracellular Signalling in Alzheimer’s Disease. Nat. Rev. Neurosci. 2002, 3, 862–872. [Google Scholar] [CrossRef]
- Leissring, M.A.; Paul, B.A.; Parker, I.; Cotman, C.W.; Laferla, F.M. Alzheimer’s Presenilin-1 Mutation Potentiates Inositol 1,4,5- Trisphosphate-Mediated Calcium Signaling in Xenopus Oocytes. J. Neurochem. 1999, 72, 1061–1068. [Google Scholar] [CrossRef]
- Leissring, M.A.; Yamasaki, T.R.; Wasco, W.; Buxbaum, J.D.; Parker, I.; LaFerla, F.M. Calsenilin Reverses Presenilin-Mediated Enhancement of Calcium Signaling. Proc. Natl. Acad. Sci. USA 2000, 97, 8590–8593. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berridge, M.J. Neuronal Calcium Signaling. Neuron 1998, 21, 13–26. [Google Scholar] [CrossRef] [Green Version]
- Bezprozvanny, I.; Mattson, M.P. Neuronal Calcium Mishandling and the Pathogenesis of Alzheimer’s Disease. Trends Neurosci. 2008, 31, 454–463. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stutzmann, G.E. The Pathogenesis of Alzheimers Disease—Is It a Lifelong “Calciumopathy”? Neuroscientist 2007, 13, 546–559. [Google Scholar] [CrossRef] [PubMed]
- Stutzmann, G.E.; Smith, I.; Caccamo, A.; Oddo, S.; Laferla, F.M.; Parker, I. Enhanced Ryanodine Receptor Recruitment Contributes to Ca2+ Disruptions in Young, Adult, and Aged Alzheimer’s Disease Mice. J. Neurosci. 2006, 26, 5180–5189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chakroborty, S.; Briggs, C.; Miller, M.B.; Goussakov, I.; Schneider, C.; Kim, J.; Wicks, J.; Richardson, J.C.; Conklin, V.; Cameransi, B.G.; et al. Stabilizing ER Ca2+ Channel Function as an Early Preventative Strategy for Alzheimer’s Disease. PLoS ONE 2012, 7, e52056. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shen, J. Function and Dysfunction of Presenilin. Neurodegener. Dis. 2013, 13, 61–63. [Google Scholar] [CrossRef] [Green Version]
- Karch, C.M.; Goate, A.M. Alzheimer’s Disease Risk Genes and Mechanisms of Disease Pathogenesis. Biol. Psychiatry 2015, 77, 43–51. [Google Scholar] [CrossRef] [Green Version]
- Bellinger, F.P.; Madamba, S.; Siggins, G.R. Interleukin 1β Inhibits Synaptic Strength and Long-Term Potentiation in the Rat CA1 Hippocampus. Brain Res. 1993, 628, 227–234. [Google Scholar] [CrossRef]
- Ghosh, S.; Wu, M.D.; Shaftel, S.S.; Kyrkanides, S.; LaFerla, F.M.; Olschowka, J.A.; Kerry O’Banion, M. Sustained Interleukin-1β Overexpression Exacerbates Tau Pathology despite Reduced Amyloid Burden in an Alzheimer’s Mouse Model. J. Neurosci. 2013, 33, 5053–5064. [Google Scholar] [CrossRef]
- Kaushik, D.K.; Thounaojam, M.C.; Kumawat, K.L.; Gupta, M.; Basu, A. Interleukin-1β Orchestrates Underlying Inflammatory Responses in Microglia via Krüppel-like Factor 4. J. Neurochem. 2013, 127, 233–244. [Google Scholar] [CrossRef] [PubMed]
- Monif, M.; Reid, C.A.; Powell, K.L.; Drummond, K.J.; O’brien, T.J.; Williams, D.A. Interleukin-1β Has Trophic Effects in Microglia and Its Release Is Mediated by P2X7R Pore. J. Neuroinflamm. 2016, 13, 173. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shaftel, S.S.; Griffin, W.S.T.; Kerry, K.M. The Role of Interleukin-1 in Neuroinflammation and Alzheimer Disease: An Evolving Perspective. J. Neuroinflammation 2008, 5, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heneka, M.T.; Kummer, M.P.; Stutz, A.; Delekate, A.; Schwartz, S.; Vieira-Saecker, A.; Griep, A.; Axt, D.; Remus, A.; Tzeng, T.-C.; et al. NLRP3 Is Activated in Alzheimer’s Disease and Contributes to Pathology in APP/PS1 Mice. Nature 2012, 493, 674–678. [Google Scholar] [CrossRef] [PubMed]
- Sutinen, E.M.; Pirttilä, T.; Anderson, G.; Salminen, A.; Ojala, J.O. Pro-Inflammatory Interleukin-18 Increases Alzheimer’s Disease-Associated Amyloid-β Production in Human Neuron-like Cells. J. Neuroinflamm. 2012, 9, 199. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Pak, C.H.; Han, Y.; Ahlenius, H.; Zhang, Z.; Chanda, S.; Marro, S.; Patzke, C.; Acuna, C.; Covy, J.; et al. Rapid Single-Step Induction of Functional Neurons from Human Pluripotent Stem Cells. Neuron 2013, 78, 785–798. [Google Scholar] [CrossRef] [Green Version]
- Chakroborty, S.; Goussakov, I.; Miller, M.B.; Stutzmann, G.E. Neurobiology of Disease Deviant Ryanodine Receptor-Mediated Calcium Release Resets Synaptic Homeostasis in Presymptomatic 3xTg-AD Mice. J. Neurosci. 2009, 29, 9458–9470. [Google Scholar] [CrossRef]
- Etcheberrigaray, R.; Hirashima, N.; Nee, L.; Prince, J.; Govoni, S.; Racchi, M.; Tanzi, R.E.; Alkon, D.L. Calcium Responses in Fibroblasts from Asymptomatic Members of Alzheimer’s Disease Families. Neurobiol. Dis. 1998, 5, 37–45. [Google Scholar] [CrossRef] [Green Version]
- Stutzmann, G.E.; Caccamo, A.; LaFerla, F.M.; Parker, I. Dysregulated IP3 Signaling in Cortical Neurons of Knock-In Mice Expressing an Alzheimer’s-Linked Mutation in Presenilin1 Results in Exaggerated Ca2+ Signals and Altered Membrane Excitability. J. Neurosci. 2004, 24, 508–513. [Google Scholar] [CrossRef] [Green Version]
- Goussakov, I.; Miller, M.B.; Stutzmann, G.E. NMDA-Mediated Ca(2+) Influx Drives Aberrant Ryanodine Receptor Activation in Dendrites of Young Alzheimer’s Disease Mice. J. Neurosci. 2010, 30, 12128–12137. [Google Scholar] [CrossRef] [Green Version]
- Mustaly-Kalimi, S.; Littlefield, A.M.; Stutzmann, G.E. Calcium Signaling Deficits in Glia and Autophagic Pathways Contributing to Neurodegenerative Disease. Antioxid. Redox Signal. 2018, 29, 1158–1175. [Google Scholar] [CrossRef]
- Chakroborty, S.; Hill, E.S.; Christian, D.T.; Helfrich, R.; Riley, S.; Schneider, C.; Kapecki, N.; Mustaly-Kalimi, S.; Seiler, F.A.; Peterson, D.A.; et al. Reduced Presynaptic Vesicle Stores Mediate Cellular and Network Plasticity Defects in an Early-Stage Mouse Model of Alzheimer’s Disease. Mol. Neurodegener. 2019, 14, 7. [Google Scholar] [CrossRef] [Green Version]
- Peng, J.; Liang, G.; Inan, S.; Wu, Z.; Joseph, D.J.; Meng, Q.; Peng, Y.; Eckenhoff, M.F.; Wei, H. Dantrolene Ameliorates Cognitive Decline and Neuropathology in Alzheimer Triple Transgenic Mice. Neurosci. Lett. 2012, 516, 274–279. [Google Scholar] [CrossRef] [Green Version]
- Potter, R.; Patterson, B.W.; Elbert, D.L.; Ovod, V.; Kasten, T.; Sigurdson, W.; Mawuenyega, K.; Blazey, T.; Goate, A.; Chott, R.; et al. Increased in Vivo Amyloid-B42 Production, Exchange, and Loss in Presenilin Mutation Carriers. Sci. Transl. Med. 2013, 5, 189ra77. [Google Scholar] [CrossRef] [Green Version]
- Smolarkiewicz, M.; Skrzypczak, T.; Wojtaszek, P. The Very Many Faces of Presenilins and the γ-Secretase Complex. Protoplasma 2013, 250, 997–1011. [Google Scholar] [CrossRef] [Green Version]
- De Strooper, B.; Iwatsubo, T.; Wolfe, M.S. Presenilins and γ-Secretase: Structure, Function, and Role in Alzheimer Disease. Cold Spring Harb. Perspect. Med. 2012, 2, a006304. [Google Scholar] [CrossRef]
- Verdile, G.; Gandy, S.E.; Martins, R.N. The Role of Presenilin and Its Interacting Proteins in the Biogenesis of Alzheimer’s Beta Amyloid. Neurochem. Res. 2007, 32, 609–623. [Google Scholar] [CrossRef] [Green Version]
- Yang, G.; Yu, K.; Kaitatzi, C.S.; Singh, A.; Labahn, J. Influence of Solubilization and AD-Mutations on Stability and Structure of Human Presenilins. Sci. Rep. 2017, 7, 1–12. [Google Scholar] [CrossRef]
- Ochalek, A.; Mihalik, B.; Avci, H.X.; Chandrasekaran, A.; Téglási, A.; Bock, I.; Lo Giudice, M.; Táncos, Z.; Molnár, K.; László, L.; et al. Neurons Derived from Sporadic Alzheimer’s Disease IPSCs Reveal Elevated TAU Hyperphosphorylation, Increased Amyloid Levels, and GSK3B Activation. Alzheimer’s Res. Ther. 2017, 9, 90. [Google Scholar] [CrossRef] [Green Version]
- Takashima, A.; Murayama, M.; Murayama, O.; Kohno, T.; Honda, T.; Yasutake, K.; Nihonmatsu, N.; Mercken, M.; Yamaguchi, H.; Sugihara, S.; et al. Presenilin 1 Associates with Glycogen Synthase Kinase-3β and Its Substrate Tau. Proc. Natl. Acad. Sci. USA 1998, 95, 9637–9641. [Google Scholar] [CrossRef] [Green Version]
- Mattson, M.P.; Chan, S.L. Dysregulation of Cellular Calcium Homeostasis in Alzheimer’s Disease: Bad Genes and Bad Habits. J. Mol. Neurosci. 2001, 17, 205–224. [Google Scholar] [CrossRef]
- Stutzmann, G.E.; Smith, I.; Caccamo, A.; Oddo, S.; Parker, I.; Laferla, F. Enhanced Ryanodine-Mediated Calcium Release in Mutant PS1-Expressing Alzheimer’s Mouse Models. Ann. N. Y. Acad. Sci. 2007, 1097, 265–277. [Google Scholar] [CrossRef]
- Briggs, C.A.; Chakroborty, S.; Stutzmann, G.E. Emerging Pathways Driving Early Synaptic Pathology in Alzheimer’s Disease. Biochem. Biophys. Res. Commun. 2017, 483, 988–997. [Google Scholar] [CrossRef] [PubMed] [Green Version]





© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Schrank, S.; McDaid, J.; Briggs, C.A.; Mustaly-Kalimi, S.; Brinks, D.; Houcek, A.; Singer, O.; Bottero, V.; Marr, R.A.; Stutzmann, G.E. Human-Induced Neurons from Presenilin 1 Mutant Patients Model Aspects of Alzheimer’s Disease Pathology. Int. J. Mol. Sci. 2020, 21, 1030. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21031030
Schrank S, McDaid J, Briggs CA, Mustaly-Kalimi S, Brinks D, Houcek A, Singer O, Bottero V, Marr RA, Stutzmann GE. Human-Induced Neurons from Presenilin 1 Mutant Patients Model Aspects of Alzheimer’s Disease Pathology. International Journal of Molecular Sciences. 2020; 21(3):1030. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21031030
Chicago/Turabian StyleSchrank, Sean, John McDaid, Clark A. Briggs, Sarah Mustaly-Kalimi, Deanna Brinks, Aiden Houcek, Oded Singer, Virginie Bottero, Robert A. Marr, and Grace E. Stutzmann. 2020. "Human-Induced Neurons from Presenilin 1 Mutant Patients Model Aspects of Alzheimer’s Disease Pathology" International Journal of Molecular Sciences 21, no. 3: 1030. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21031030