Differential Expression of Genes at Panicle Initiation and Grain Filling Stages Implied in Heterosis of Rice Hybrids


 , ,
, , 
Abstract
:
1. Introduction
2. Results
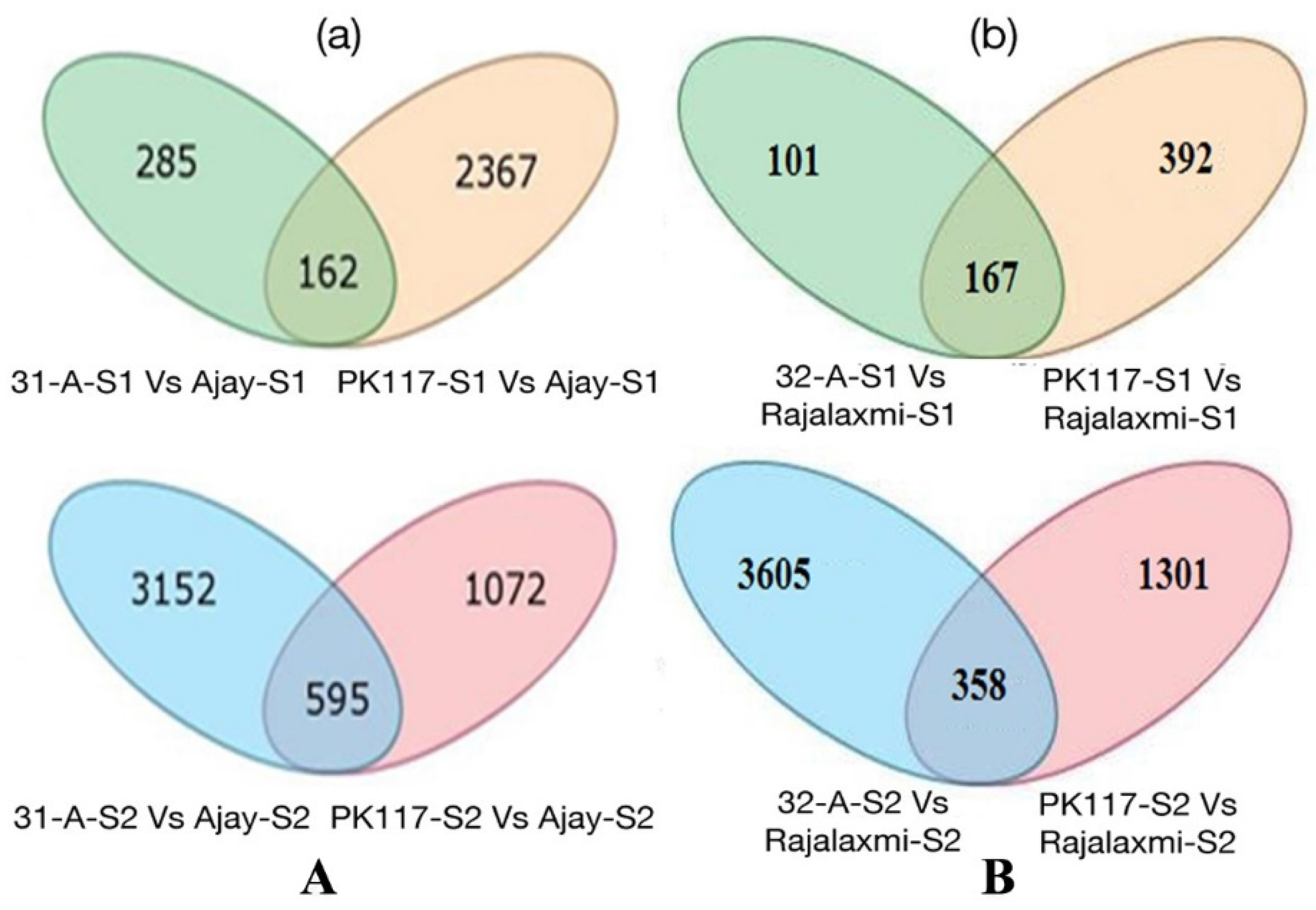
2.1. Differential Gene Expression among Hybrids and Its Parents
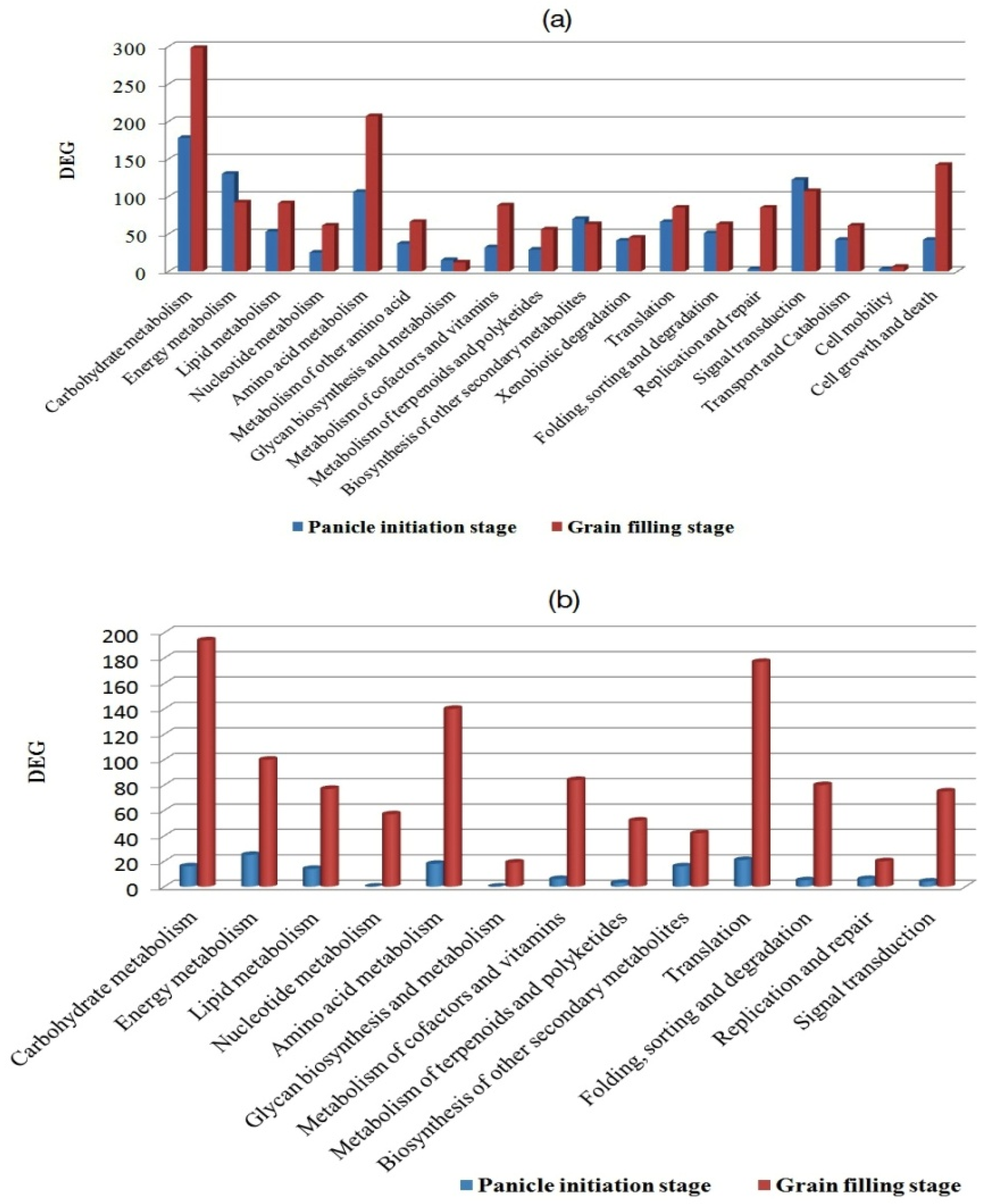
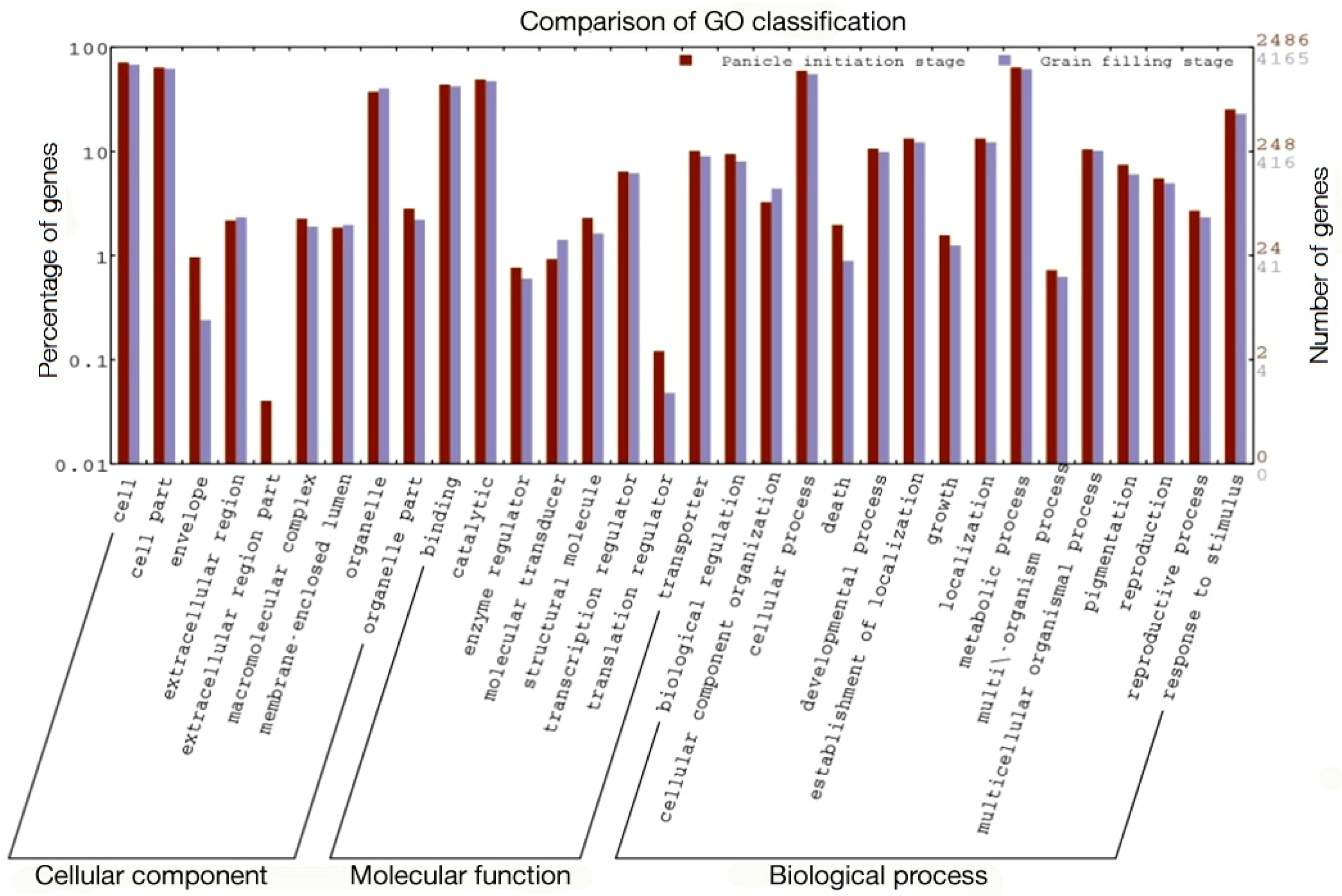
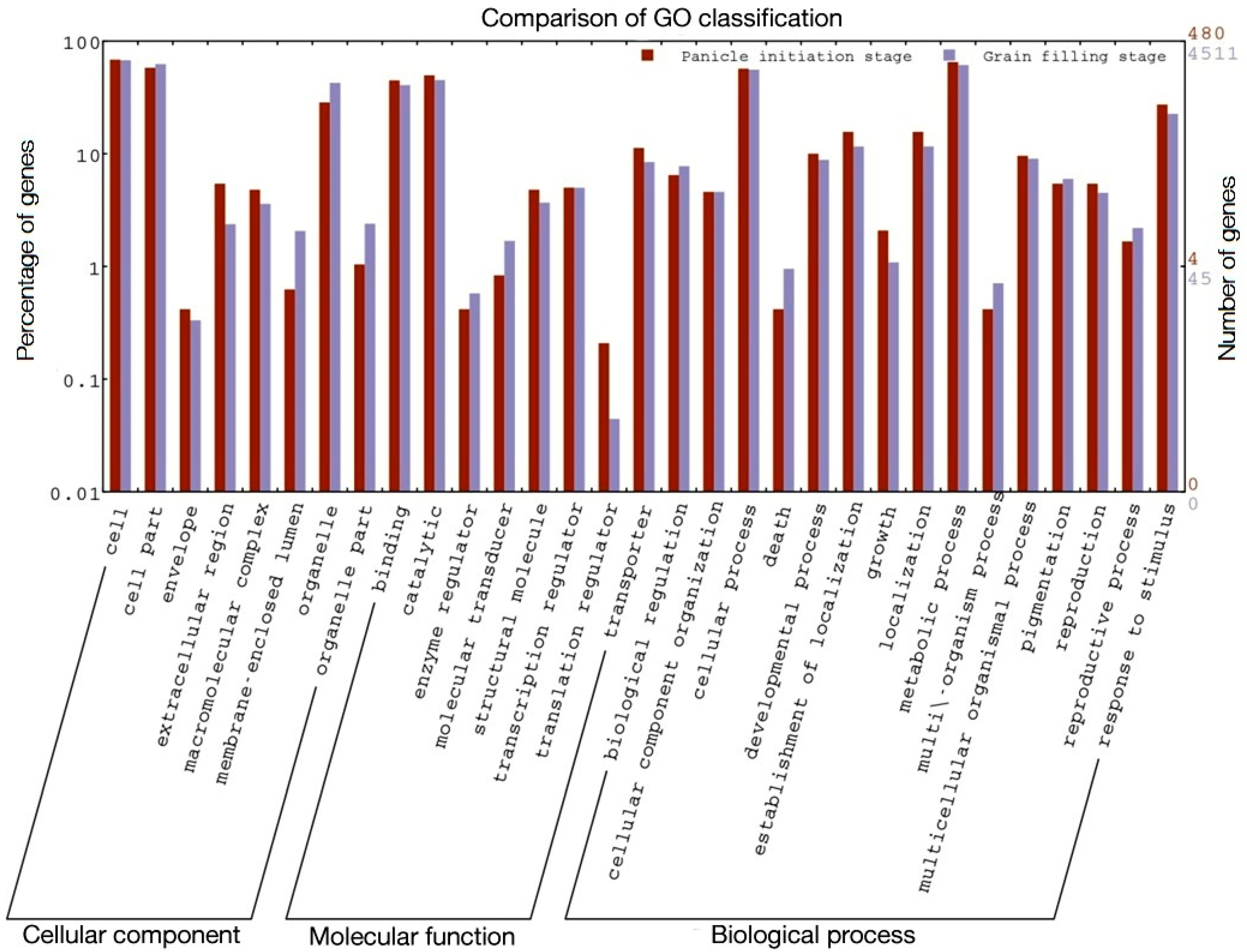
2.2. Functional Classes, Pathways Enriched, and Key Genes Involved in Heterosis
2.3. Validation of DEGs Identified in RNA-Seq Data Using qRT-PCR
2.4. Yield-Related Quantitative Trait Loci (QTLs) Identification by Mapping of DEGs
3. Discussion
3.1. Photosynthesis and Yield in Hybrid Rice
3.2. Role of Carbohydrate Metabolism in Heterosis
3.3. Stress Tolerance Potential of the Hybrids
3.4. Signal Transduction
3.5. DEGs Associated with Yield-Related QTL
3.6. TFs and Their Role in Heterosis
4. Materials and Methods
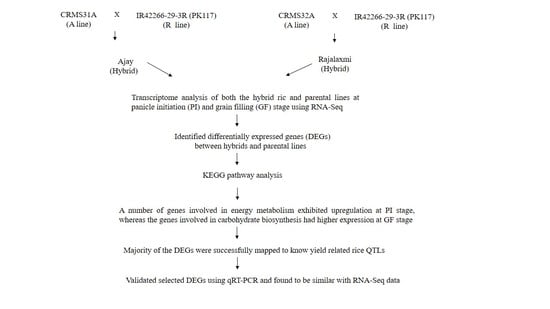
4.1. Plant Materials
4.2. RNA-Seq and Differential Gene Expression
4.3. RNA Extraction and Real-Time Quantitative PCR (qRT-PCR)
4.4. In-Silico Mapping the DEGs with the Known QTLs
4.5. TF Identification
5. Conclusions
6. Deposited Data
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| 31A | CRMS31A |
| 32A | CRMS32A |
| PK117 | IR42266-29-3R |
References
- Hossain, M.; Fischer, K. Rice research for food security and sustainable agricultural development in Asia: Achievements and future challenges. GeoJournal 1995, 35, 286–298. [Google Scholar] [CrossRef]
- Xing, Y.; Zhang, Q. Genetic and molecular bases of rice yield. Annu. Rev. Plant Biol. 2010, 61, 421–442. [Google Scholar] [CrossRef] [PubMed]
- Davenport, C.B. Degeneration, albinism and inbreeding. Science 1908, 28, 454–455. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- East, E.M. Inbreeding in corn. Rep. Conn. Agric. Exp. Stn. 1908, 1907, 419–428. [Google Scholar]
- Shull, G.H. The composition of a field of maize. J. Hered. 1908, 1, 296–301. [Google Scholar] [CrossRef]
- Virmani, S.S. Two-Line Hybrid Rice Breeding Manual; International Rice Research Institute: Los Banos, Philippines, 2003. [Google Scholar]
- Huang, Y.; Zhang, L.; Zhang, J.; Yuan, D.; Xu, C.; Li, X.; Zhou, D.; Wang, S.; Zhang, Q. Heterosis and polymorphisms of gene expression in an elite rice hybrid as revealed by a microarray analysis of 9198 unique ESTs. Plant Mol. Biol. 2006, 62, 579–591. [Google Scholar] [CrossRef]
- Bao, J.; Lee, S.; Chen, C.; Zhang, X.; Zhang, Y.; Liu, S.; Clark, T.; Wang, J.; Cao, M.; Yang, H. Serial analysis of gene expression study of a hybrid rice strain (LYP9) and its parental cultivars. Plant Physiol. 2005, 138, 1216–1231. [Google Scholar] [CrossRef] [Green Version]
- Song, G.-S.; Zhai, H.-L.; Peng, Y.-G.; Zhang, L.; Wei, G.; Chen, X.-Y.; Xiao, Y.-G.; Wang, L.; Chen, Y.-J.; Wu, B. Comparative transcriptional profiling and preliminary study on heterosis mechanism of super-hybrid rice. Mol. Plant 2010, 3, 1012–1025. [Google Scholar] [CrossRef]
- Wang, Z.; Gerstein, M.; Snyder, M. RNA-Seq: A revolutionary tool for transcriptomics. Nat. Rev. Genet. 2009, 10, 57. [Google Scholar] [CrossRef]
- Wei, G.; Tao, Y.; Liu, G.; Chen, C.; Luo, R.; Xia, H.; Gan, Q.; Zeng, H.; Lu, Z.; Han, Y. A transcriptomic analysis of superhybrid rice LYP9 and its parents. Proc. Natl. Acad. Sci. USA 2009, 106, 7695–7701. [Google Scholar] [CrossRef] [Green Version]
- Zhai, R.; Feng, Y.; Wang, H.; Zhan, X.; Shen, X.; Wu, W.; Zhang, Y.; Chen, D.; Dai, G.; Yang, Z. Transcriptome analysis of rice root heterosis by RNA-Seq. BMC Genom. 2013, 14, 19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ge, X.; Chen, W.; Song, S.; Wang, W.; Hu, S.; Yu, J. Transcriptomic profiling of mature embryo from an elite super-hybrid rice LYP9 and its parental lines. BMC Plant Biol. 2008, 8, 114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, S.; Zhang, Y.; Ge, L.; Wang, L. Genome-wide transcriptome profiles of rice hybrids and their parents. Int. J. Mol. Sci. 2014, 15, 20833–20845. [Google Scholar]
- Li, D.; Huang, Z.; Song, S.; Xin, Y.; Mao, D.; Lv, Q.; Zhou, M.; Tian, D.; Tang, M.; Wu, Q. Integrated analysis of phenome, genome, and transcriptome of hybrid rice uncovered multiple heterosis-related loci for yield increase. Proc. Natl. Acad. Sci. USA 2016, 113, E6026–E6035. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, L.; Li, X.; Huang, X.; Ma, T.; Liang, Y.; Ma, X.; Peng, X.; Jia, J.; Chen, S.; Chen, Y. Overexpression of sheepgrass R1-MYB transcription factor LcMYB1 confers salt tolerance in transgenic Arabidopsis. Plant Physiol. Biochem. 2013, 70, 252–260. [Google Scholar] [CrossRef] [PubMed]
- Boonchai, C.; Udomchalothorn, T.; Sripinyowanich, S.; Comai, L.; Buaboocha, T.; Chadchawan, S. Rice Overexpressing OsNUC1-S Reveals Differential Gene Expression Leading to Yield Loss Reduction after Salt Stress at the Booting Stage. Int. J.Mol. Sci. 2018, 19, 3936. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.; Jeong, H.; Lee, S.; Lee, J.; Kim, S.-J.; Park, J.-W.; Woo, H.R.; Lim, P.O.; An, G.; Nam, H.G. Molecular bases for differential aging programs between flag and second leaves during grain-filling in rice. Sci. Rep. 2017, 7, 8792. [Google Scholar] [CrossRef] [Green Version]
- Kholupenko, I.; Voronkova, N.; Burundukova, O.; Zhemchugova, V. Demand for assimilates determines the productivity of intensive and extensive rice crops in Primorskii krai. Russ. J. Plant Physiol. 2003, 50, 112–118. [Google Scholar] [CrossRef]
- Yoshida, S. Fundamentals of Rice Crop Science; International Rice Research Institute: Los Banos, Philippines, 1981. [Google Scholar]
- Ni, Z.; Kim, E.-D.; Ha, M.; Lackey, E.; Liu, J.; Zhang, Y.; Sun, Q.; Chen, Z.J. Altered circadian rhythms regulate growth vigour in hybrids and allopolyploids. Nature 2009, 457, 327. [Google Scholar] [CrossRef] [Green Version]
- Larièpe, A.; Mangin, B.; Jasson, S.; Combes, V.; Dumas, F.; Jamin, P.; Lariagon, C.; Jolivot, D.; Madur, D.; Fievet, J. The genetic basis of heterosis: Multiparental quantitative trait loci mapping reveals contrasted levels of apparent overdominance among traits of agronomical interest in maize (Zea mays L.). Genetics 2012, 190, 795–811. [Google Scholar] [CrossRef] [Green Version]
- Springer, N.M.; Stupar, R.M. Allelic variation and heterosis in maize: How do two halves make more than a whole? Genome Res. 2007, 17, 264–275. [Google Scholar] [CrossRef] [Green Version]
- Yu, S.; Li, J.; Xu, C.; Tan, Y.; Gao, Y.; Li, X.; Zhang, Q.; Maroof, M.S. Importance of epistasis as the genetic basis of heterosis in an elite rice hybrid. Proc. Natl. Acad. Sci. USA 1997, 94, 9226–9231. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Z.-K.; Luo, L.; Mei, H.; Wang, D.; Shu, Q.; Tabien, R.; Zhong, D.; Ying, C.; Stansel, J.; Khush, G. Overdominant epistatic loci are the primary genetic basis of inbreeding depression and heterosis in rice. I. Biomass and grain yield. Genetics 2001, 158, 1737–1753. [Google Scholar] [PubMed]
- Long, S.P.; Zhu, X.G.; Naidu, S.L.; Ort, D.R. Can improvement in photosynthesis increase crop yields? Plant Cell Environ. 2006, 29, 315–330. [Google Scholar] [CrossRef]
- Liang, F.; Lindberg, P.; Lindblad, P. Engineering photoautotrophic carbon fixation for enhanced growth and productivity. Sustain. Energy Fuels 2018, 2, 2583–2600. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.; Tan, G.; Yang, L.; Yang, J.; Zhang, J.; Zhao, B. Hormones in the grains and roots in relation to post-anthesis development of inferior and superior spikelets in japonica/indica hybrid rice. Plant Physiol. Biochem. 2009, 47, 195–204. [Google Scholar] [CrossRef]
- Yao, Y.; Ni, Z.; Zhang, Y.; Chen, Y.; Ding, Y.; Han, Z.; Liu, Z.; Sun, Q. Identification of differentially expressed genes in leaf and root between wheat hybrid and its parental inbreds using PCR-based cDNA subtraction. Plant Mol. Biol. 2005, 58, 367–384. [Google Scholar] [CrossRef]
- Erzberger, J.P.; Berger, J.M. Evolutionary relationships and structural mechanisms of AAA+ proteins. Annu. Rev. Biophys. Biomol. Struct. 2006, 35, 93–114. [Google Scholar] [CrossRef]
- Akula, R.; Ravishankar, G.A. Influence of abiotic stress signals on secondary metabolites in plants. Plant Signal. Behav. 2011, 6, 1720–1731. [Google Scholar] [CrossRef]
- Zhang, C.; Lin, C.; Fu, F.; Zhong, X.; Peng, B.; Yan, H.; Zhang, J.; Zhang, W.; Wang, P.; Ding, X. Comparative transcriptome analysis of flower heterosis in two soybean F1 hybrids by RNA-seq. PLoS ONE 2017, 12, e0181061. [Google Scholar] [CrossRef] [Green Version]
- Hutin, C.; Nussaume, L.; Moise, N.; Moya, I.; Kloppstech, K.; Havaux, M. Early light-induced proteins protect Arabidopsis from photooxidative stress. Proc. Natl. Acad. Sci. USA 2003, 100, 4921–4926. [Google Scholar] [CrossRef] [Green Version]
- Zhu, G.; Ye, N.; Yang, J.; Peng, X.; Zhang, J. Regulation of expression of starch synthesis genes by ethylene and ABA in relation to the development of rice inferior and superior spikelets. J. Exp. Bot. 2011, 62, 3907–3916. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahmadi, A.; Baker, D. Effects of abscisic acid (ABA) on grain filling processes in wheat. Plant Growth Regul. 1999, 28, 187–197. [Google Scholar] [CrossRef]
- Bhatia, S.; Singh, R. Phytohormone-mediated transformation of sugars to starch in relation to the activities of amylases, sucrose-metabolising enzymes in sorghum grain. Plant Growth Regul. 2002, 36, 97–104. [Google Scholar] [CrossRef]
- Tang, T.; Xie, H.; Wang, Y.; Lü, B.; Liang, J. The effect of sucrose and abscisic acid interaction on sucrose synthase and its relationship to grain filling of rice (Oryza sativa L.). J. Exp. Bot. 2009, 60, 2641–2652. [Google Scholar] [CrossRef] [Green Version]
- Akihiro, T.; Umezawa, T.; Ueki, C.; Lobna, B.M.; Mizuno, K.; Ohta, M.; Fujimura, T. Genome wide cDNA-AFLP analysis of genes rapidly induced by combined sucrose and ABA treatment in rice cultured cells. FEBS Lett. 2006, 580, 5947–5952. [Google Scholar] [CrossRef] [Green Version]
- Yang, J.; Wang, P.; Liu, L.; Wang, Z.; Zhu, Q. Evolution characteristics of grain yield and plant type for mid-season indica rice cultivars. Acta Agron. Sin. 2006, 32, 949. [Google Scholar]
- Yang, J.; Zhang, J. Grain-filling problem in ‘super’rice. J. Exp. Bot. 2009, 61, 1–5. [Google Scholar] [CrossRef] [Green Version]
- Panda, B.B.; Kariali, E.; Panigrahi, R.; Mohapatra, P.K. High ethylene production slackens seed filling in compact panicled rice cultivar. Plant Growth Regul. 2009, 58, 141–151. [Google Scholar] [CrossRef]
- Wuriyanghan, H.; Zhang, B.; Cao, W.-H.; Ma, B.; Lei, G.; Liu, Y.-F.; Wei, W.; Wu, H.-J.; Chen, L.-J.; Chen, H.-W. The ethylene receptor ETR2 delays floral transition and affects starch accumulation in rice. Plant Cell 2009, 21, 1473–1494. [Google Scholar] [CrossRef] [Green Version]
- Chung, H.S.; Howe, G.A. A critical role for the TIFY motif in repression of jasmonate signaling by a stabilized splice variant of the JASMONATE ZIM-domain protein JAZ10 in Arabidopsis. Plant Cell 2009, 21, 131–145. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cai, Q.; Yuan, Z.; Chen, M.; Yin, C.; Luo, Z.; Zhao, X.; Liang, W.; Hu, J.; Zhang, D. Jasmonic acid regulates spikelet development in rice. Nat. Commun. 2014, 5, 3476. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peng, B.; Kong, H.; Li, Y.; Wang, L.; Zhong, M.; Sun, L.; Gao, G.; Zhang, Q.; Luo, L.; Wang, G. OsAAP6 functions as an important regulator of grain protein content and nutritional quality in rice. Nat. Commun. 2014, 5, 4847. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, H.-Y.; He, H.; Chen, L.-B.; Li, L.; Liang, M.-Z.; Wang, X.-F.; Liu, X.-G.; He, G.-M.; Chen, R.-S.; Ma, L.-G. A genome-wide transcription analysis reveals a close correlation of promoter INDEL polymorphism and heterotic gene expression in rice hybrids. Mol. Plant 2008, 1, 720–731. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abogadallah, G.M.; Nada, R.M.; Malinowski, R.; Quick, P. Overexpression of HARDY, an AP2/ERF gene from Arabidopsis, improves drought and salt tolerance by reducing transpiration and sodium uptake in transgenic Trifolium alexandrinum L. Planta 2011, 233, 1265–1276. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, P.; Agarwal, P.K.; Joshi, A.J.; Sopory, S.K.; Reddy, M.K. Overexpression of PgDREB2A transcription factor enhances abiotic stress tolerance and activates downstream stress-responsive genes. Mol. Biol. Rep. 2010, 37, 1125. [Google Scholar] [CrossRef]
- Gao, F.; Chen, J.-M.; Xiong, A.-S.; Peng, R.-H.; Liu, J.-G.; Cai, B.; Yao, Q.-H. Isolation and characterization of a novel AP2/EREBP-type transcription factor OsAP211 in Oryza sativa. Biol. Plant. 2009, 53, 643–649. [Google Scholar] [CrossRef]
- Hu, J.; Barlet, X.; Deslandes, L.; Hirsch, J.; Feng, D.X.; Somssich, I.; Marco, Y. Transcriptional responses of Arabidopsis thaliana during wilt disease caused by the soil-borne phytopathogenic bacterium, Ralstonia solanacearum. PLoS ONE 2008, 3, e2589. [Google Scholar] [CrossRef]
- Dubos, C.; Stracke, R.; Grotewold, E.; Weisshaar, B.; Martin, C.; Lepiniec, L. MYB transcription factors in Arabidopsis. Trends Plant Sci. 2010, 15, 573–581. [Google Scholar] [CrossRef]
- Katiyar, A.; Smita, S.; Lenka, S.K.; Rajwanshi, R.; Chinnusamy, V.; Bansal, K.C. Genome-wide classification and expression analysis of MYB transcription factor families in rice and Arabidopsis. BMC Genom. 2012, 13, 544. [Google Scholar] [CrossRef] [Green Version]
- Du, Z.; Zhou, X.; Ling, Y.; Zhang, Z.; Su, Z. agriGO: A GO analysis toolkit for the agricultural community. Nucleic Acids Res. 2010, 38 (Suppl. 2), W64–W70. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ye, J.; Fang, L.; Zheng, H.; Zhang, Y.; Chen, J.; Zhang, Z.; Wang, J.; Li, S.; Li, R.; Bolund, L. WEGO: A web tool for plotting GO annotations. Nucleic Acids Res. 2006, 34 (Suppl. 2), W293–W297. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Goto, S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000, 28, 27–30. [Google Scholar] [CrossRef] [PubMed]
- Mao, X.; Cai, T.; Olyarchuk, J.G.; Wei, L. Automated genome annotation and pathway identification using the KEGG Orthology (KO) as a controlled vocabulary. Bioinformatics 2005, 21, 3787–3793. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Samples | Number of Reads Mapped on | Total Number of Reads Mapped | Percentage | ||||
|---|---|---|---|---|---|---|---|
| Exon | Intron | Intergenic | Exon | Intron | Intergenic | ||
| CRMS31A-PI | 40,218,002 | 17,052 | 10,391,484 | 51,094,480 | 78.7 | 0.0 | 20.3 |
| CRMS31A-GF | 65,833,226 | 87,087 | 8,165,337 | 75,026,355 | 87.7 | 0.1 | 10.9 |
| PK117-PI | 74,956,596 | 110,096 | 10,927,257 | 87,111,469 | 86.0 | 0.1 | 12.5 |
| PK117-GF | 40,239,045 | 27,816 | 13,489,140 | 54,046,852 | 74.5 | 0.1 | 25.0 |
| Ajay-PI | 31,177,458 | 45,592 | 5,292,987 | 36,815,969 | 84.7 | 0.1 | 14.4 |
| Ajay-GF | 41,685,048 | 52,409 | 7,598,708 | 49,937,045 | 83.5 | 0.1 | 15.2 |
| Samples | Number of Reads Mapped on | Total Number of Reads Mapped | Percentage | ||||
|---|---|---|---|---|---|---|---|
| Exon | Intron | Intergenic | Exon | Intron | Intergenic | ||
| CRMS32A-PI | 44,197,542 | 45,300 | 11,310,908 | 56,043,252 | 78.8 | 0.08 | 20.1 |
| CRMS32A-GF | 51,792,582 | 94,467 | 7,197,864 | 60,231,007 | 85.9 | 0.1 | 11.9 |
| PK117-PI | 74,956,596 | 110,096 | 10,927,257 | 87,111,469 | 86.0 | 0.1 | 12.5 |
| PK117-GF | 40,239,045 | 27,816 | 13,489,140 | 54,046,852 | 74.5 | 0.1 | 25.0 |
| Rajalaxmi-PI | 33,076,827 | 95,031 | 6,658,553 | 41,603,392 | 79.5 | 0.2 | 16.0 |
| Rajalaxmi-GF | 53,353,594 | 96,355 | 7,888,639 | 62,344,409 | 85.5 | 1.5 | 12.6 |
| STAGE | DEGPP | DEGHP | A/H | R/H | Total |
|---|---|---|---|---|---|
| Panicle Initiation (PI) of Ajay | 447 | 2814 | 447 | 2529 | 6237 |
| Grain filling (GF) of Ajay | 3924 | 4819 | 3747 | 1667 | 14,157 |
| Panicle Initiation (PI) of Rajalaxmi | 1375 | 660 | 268 | 559 | 2862 |
| Grain filling (GF) of Rajalaxmi | 3582 | 5264 | 3963 | 1659 | 14,468 |
| S. No | GO Term | Annotation | Ajay | Rajalaxmi | ||
|---|---|---|---|---|---|---|
| PI | GF | PI | GF | |||
| p-Value | p-Value | |||||
| 1 | GO:0009607 | Response to biotic stimulus | 1.9 × 10−12 | 0.0026 | 4.8 × 10−6 | 0.003 |
| 2 | GO:0015979 | Photosynthesis | 1.1 × 10−11 | 0.008 | NE * | 4 × 10−6 |
| 3 | GO:0006091 | Generation of precursor metabolites and energy | 2.6 × 10−10 | 0.0001 | 8.5 × 10−5 | 0.00019 |
| 4 | GO:0006810 | Transport | 3.9 × 10−10 | 3.30 × 10−10 | 4.8 × 10−5 | 1.5 × 10−7 |
| 5 | GO:0051234 | Establishment of localization | 3.9 × 10−10 | 3.30 × 10−10 | 4.8 × 10−5 | 1.5 × 10−7 |
| 6 | GO:0051179 | Localization | 3.9 × 10−10 | 3.30 × 10−10 | 4.8 × 10−5 | 1.5 × 10−7 |
| 7 | GO:0005975 | Carbohydrate metabolic process | 4.8 × 10−10 | 2.70 × 10−14 | 4.8 × 10−10 | 2.70 × 10−14 |
| 8 | GO:0009628 | Response to abiotic stimulus | 6.3 × 10−9 | 2.20 × 10−8 | NE * | 1.1 × 10−6 |
| 9 | GO:0050896 | Response to stimulus | 1.6 × 10−6 | 0.001 | 0.0032 | 0.0025 |
| 10 | GO:0009987 | Cellular process | 2.8 × 10−5 | 0.015 | NE * | 0.0027 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Katara, J.L.; Verma, R.L.; Parida, M.; Ngangkham, U.; Molla, K.A.; Barbadikar, K.M.; Mukherjee, M.; C, P.; Samantaray, S.; Ravi, N.R.; et al. Differential Expression of Genes at Panicle Initiation and Grain Filling Stages Implied in Heterosis of Rice Hybrids. Int. J. Mol. Sci. 2020, 21, 1080. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21031080
Katara JL, Verma RL, Parida M, Ngangkham U, Molla KA, Barbadikar KM, Mukherjee M, C P, Samantaray S, Ravi NR, et al. Differential Expression of Genes at Panicle Initiation and Grain Filling Stages Implied in Heterosis of Rice Hybrids. International Journal of Molecular Sciences. 2020; 21(3):1080. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21031080
Chicago/Turabian StyleKatara, Jawahar Lal, Ram Lakhan Verma, Madhuchhanda Parida, Umakanta Ngangkham, Kutubuddin Ali Molla, Kalyani Makarand Barbadikar, Mitadru Mukherjee, Parameswaran C, Sanghamitra Samantaray, Nageswara Rao Ravi, and et al. 2020. "Differential Expression of Genes at Panicle Initiation and Grain Filling Stages Implied in Heterosis of Rice Hybrids" International Journal of Molecular Sciences 21, no. 3: 1080. https://0-doi-org.brum.beds.ac.uk/10.3390/ijms21031080